The Key Lab of Pollution Control and Ecosystem Restoration in Industry Clusters, Ministry of Education, School of Environment and Energy, South China University of Technology, Guangzhou 510006, China
b.
State Key Laboratory of Urban Water Resource and Environment, School of Environment, Harbin Institute of Technology, Harbin 150090, China
c.
State Key Laboratory of Urban Water Resource and Environment, School of Ecology and Environment, Harbin Institute of Technology, Shenzhen 518055, China
d.
School of Chemical Engineering, The University of Adelaide, SA 5005, Australia
Received Date:
26 March 2025 Accepted Date:
04 September 2025 Revised Date:
03 July 2025 Available Online:
15 May 2026
Abstract:
Benzohydroxamic acid (BHA) occurs as recalcitrant organic pollutant discharged from mining industry. While Fenton-like oxidation based on peroxymonosulfate (PMS) has been extensively applied for organic contamination mitigation, its conventional reaction pathway dependent on free radicals needs high energy input with elevated carbon emission. Here, we meticulously developed a novel single-atom catalyst featuring Co-N4 coordination (Cox@NC) to initiate a non-radical Fenton-like oxidation for BHA treatment. Results showed single-atom Co-N4 with the considerable Co content (>2 wt%) and quantitative N coordination displayed exceptional reactivity to activate PMS for BHA degradation with a turnover frequency > 16 min−1. Such single-atom Co-N4 formed a surface-reactive complexes with mild oxidation potential by coordinating with PMS to mediate electron transfer for oxidation of BHA. The mediated ETP further triggered polymerization transformation pathway of BHA through formation and coupling of phenoxy-like radicals, resulting in a considerable recovery yield of BHA polymers (~43%) and superior utilization efficiency of PMS (~434%). Combined with ultrahigh-resolution mass analysis, the identified polymerized products illustrated the related polymerization mechanisms of BHA including hydroxylation, monomer radical generation, dimerization, and chain extension. Such Fenton-like catalysis of single-atom Co-N4 exhibited more remarkable application potentials in mineral processing wastewater treatment compared to traditional Fenton reaction, reducing oxidant consumption and increasing organic carbon recovery. This study enhances development of resource-efficient Fenton-like oxidation technologies for mineral processing wastewater treatment.
Mining industry faces escalating challenges in addressing mineral processing wastewater (MPW) treatment, during which the reuse of MPW emerges as a promising strategy [1,2]. Currently, the MPW extensively includes residual recalcitrant organic flotation reagents, becoming a primary obstacle limiting the MPW recycling. Benzohydroxamic acid (BHA), a widely used organic flotation agent for sulfide and oxide minerals [3], presents particular concern due to its persistence and toxicity, which stem from benzene ring structure and oxime group. Consequently, development of effective strategies for removing BHA in MPW is highly necessary for sustainable mining practices and ecological safety.
Fenton-like oxidation based on peroxymonosulfate (PMS) have been extensively applied in wastewater treatment for refractory organic pollutant breakdown [4–7]. Conventional PMS-based oxidation largely relies on the formation of highly oxidative radicals (e.g., sulfate radical (SO4•−) and hydroxy radical (•OH)) via energy or chemical activation of PMS [8,9]. Such traits of radical-induced oxidation potentially result in formation of toxic intermediates, increasing treatment period and costs, and more importantly, elevated carbon emission owing to strong mineralization efficacy [10]. To relieve this problem, oxidative polymerization pathway is recently developed for Fenton-like oxidation to enhance organic carbon recovery with lower energy input [11–13]. Following a polymerization pathway, organic compounds will first form monomer organic radicals, converting into recyclable polymers on catalyst surface through coupling reactions [12]. Compared to radical-induced oxidation, the polymerization pathway mainly involves electron transfer process (ETP) capable of removing electron-rich organic pollutants more selectively, during which surface metastable species formed with mild oxidation capacity [14,15]. Notably, BHA as electron-rich phenolic compound shows feasibility for suffering from polymerization pathway [15]. Therefore, it is of environmental significance to develop an ETP-mediated oxidation technology to selectively recover the BHA from MPW.
Single atom catalysts (SACs) with nitrogen-coordination serves as an exceptional candidate for Fenton-like oxidation, facilitating rapid decontamination through ETP-mediated mechanism [6,16,17]. Metal-organic frameworks (MOFs) satisfies structural needs as substates for designing the SACs due to tunability of metal sites [17–22], in which their reactivities are intrinsically linked to electronic structures [11,23]. For example, Ni and Cu characterized by electronic configurations of 3d8 and 3d10 holds a lower D-band center and deficiency of unpaired electrons, probably weakening catalytic efficiency [11,24]. On the other hand, Fe with the higher D-band center exhibits the greater affinity and reactivity for PMS decomposition, however, such higher reactivity may also promote the radical-induced oxidation, potentially causing over-oxidation of pollutants and a consequent decrease in polymerization yield [11,24–26]. Liu et al. [11] also confirmed the role of Fe-N4 sites in enhancing the activation efficacy of PMS, yet arising a synergistic effect of PMS* and high-valence Fe-oxo species on suppressing the polymerization of phenolics. Additionally, Zeng et al. [25] demonstrated that the Fe-N4 sites promoted chemisorption and electron transfer of PMS, facilitating inner electron shuttling and shifting the oxidation pathway from ETP to radical-induced oxidation. In comparison, Co featuring with a moderate d-band center could be expected to exhibit the satisfactory efficiency in the ETP mechanism. This characteristic might not only prevent the over-oxidation of BHA, but also possibly facilitate the production of a significant yield of polymerization products.
Herein, we constructed a Co-containing SAC (Co0.5@NC) with the precise Co-N4 coordination. Combined with electron paramagnetic resonance (EPR), in situ Raman spectroscopy, and electrochemical characterizations, it confirmed that ETP dominated the oxidative abatement of BHA. The ETP mechanism initiated the polymerization of BHA in solution and on Co0.5@NC surface, resulting in an outstanding utilization efficiency of PMS (434%). Based on ultraperformance liquid chromatography-quadrupole time-of-flight premier mass spectrometry (UPLC-QTOF-MS/MS), the polymerization products and transformation pathways of BHA were comprehensively elucidated. Moreover, such Co0.5@NC exhibited superior anti-interference properties and stability, achieving > 80% removal of chemical oxygen demand (COD) in real beneficiation wastewater. This study provides inspiration for designing the ETP-dominated oxidation for resource-effective treatment of organic wastewater with low carbon emission. Details of materials and methods were shown in Texts S1-S8 (Supporting information).
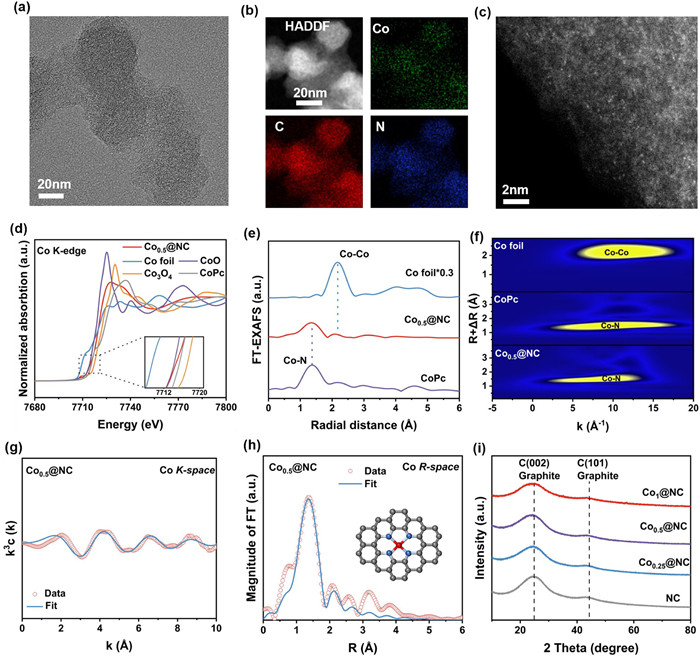
Synthesis strategy of Cox@NC was illustrated in Fig. S1 (Supporting information). A spatial encapsulation method was employed to embed Co(NO3)2 into Zn-MOFs to form ZnCox−MOFs precursor, then the obtained ZnCox−MOFs was pyrolyzed to produce Cox@NC. Especially, the Zn could be vaporized during pyrolysis process [18], further producing abundant free nitrogen sites to anchor Co atoms and effectively hindering the aggregation of Co atoms. All prepared NC, Co0.25@NC, Co0.5@NC, and Co1@NC samples exhibited similar dodecahedral structures with an average size of 30–60 nm (Fig. S2 in Supporting information). N2 adsorption-desorption curves revealed that Co addition increased specific surface area and pore volume/size of NC (Fig. S4 and Table S7 in Supporting information). High-resolution transmission electron (HR-TEM) rarely detected the apparent nanoparticles (Fig. 1a and Figs. S3a and c in Supporting information). In addition, energy-dispersive-spectroscopy (EDS) mapping verified the uniform distributions of Co, C, and N elements across the samples (Fig. 1b). In particular, aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC HAADF-STEM) showed brilliant and spread spots of Co atoms in the Cox@NC (Fig. 1c), identifying potential occurrence and even dispersion of single-atom Co [27].
Figure 1
Figure 1.
Properties of nitroeeegen-coordinated single-atom Co-N4. (a) HR-TEM image, (b) HAADF-STEM image and EDS mapping spectra. (c) AC-HAADF-STEM image of Co0.5@NC. XANES spectra at the (d) Co K-edge, (e) Co R-space of Co0.5@NC and their reference samples. (f) Wavelet transform of Co0.5@NC and their reference samples. EXAFS spectra fitting curves of Co0.5@NC at the (g) Co K-edge and (h) Co R-space (inset: structural model of Co0.5@NC). (i) XRD spectra of Cox@NC.
To probe structural properties of Cox@NC catalysts, elemental composition and valence were compared using X-ray photoelectron spectroscopy (XPS) spectra (Fig. S5 in Supporting information). From Fig. S7 (Supporting information), Co 2p3/2 spectra could be deconvoluted into Co3+ (780.3 eV), Co2+ (781.7 eV), and satellite peaks (786.5 eV) [28], demonstrating the oxidation states of single-atom Co instead of metallic states. The N 1s spectra could be deconvoluted at 398.4, 399.4, 400.5, and 401.5 eV, corresponding to pyridinic-N, Co-N, pyrrolic-N, and graphitic-N, respectively (Fig. S6 in Supporting information) [29]. Notably, increasing content of Co led to enhancement in Co-N sites and decreases in pyridinic-N (Table S3 in Supporting information), indicating that isolated Co atoms were mostly anchored onto pyridinic-N sites [16]. Inductively coupled plasma atomic emission spectroscopy (ICP-OES) analysis further suggested that the Co loadings accounted for 0.55, 2.06, 2.18 wt% in Co0.25@NC, Co0.5@NC, and Co1@NC, respectively (Table S2 in Supporting information). In addition, all the catalysts exhibited two obvious peaks at about 1350 cm−1 and 1580 cm−1, which were assigned to the D-band and G-band, respectively. Fig. S19 (Supporting information) displayed the value of IG/ID followed by 0.846, 0.857, 0.864 and 0.876 for NC, Co0.25@NC, Co0.5@NC, and Co1@NC, indicating that Co addition modestly facilitated the graphitization of the catalysts.
The coordination environment and chemical state of single-atom Co were further identified using X-ray absorption fine structure (XAFS) spectra [30]. Based on X-ray absorption near edge structure (XANES) profiles (Fig. 1d), near-edge energy absorption intensity for Co0.5@NC was higher than that for CoO, but lower than that for Co3O4. Fourier transform extended X-rays absorption fine structure (FT-EXAFS) spectra of R-space showed a prominent peak at approximately 1.5 Å, deviating from the peak of Co-Co (about 2.2 Å) in Co foil (Fig. 1e). The results proved that the isolated distribution of Co atoms on the Co0.5@NC and their valence states ranged between +2 and +8/3. The wavelet transform (WT) contour plots were conducted to clarify the max intensity related to Co-N (Fig. 1f), while EXAFS fitting results also confirmed the quantitative N coordination configuration of Co on the Co0.5@NC (Figs. 1g and h and Table S1 in Supporting information). The bond length and coordination number of Co were further calculated as 1.93 ± 0.015 Å and 4.09 ± 0.15, respectively. Based on X-ray diffraction (XRD) patterns, two broad characteristic peaks at ~26° and 44° suggested the formation of graphitic carbonaceous layers without crystal minerals (Fig. 1i)
The optimal removal efficiency of BHA could be achieved with 0.3 g/L Cox@NC and 1 mmol/L PMS (Text S9 in Supporting information). The Fenton-like catalysis of Cox@NC with varying Co contents for oxidation were compared. The pseudo-first-order rate constant (kobs) of BHA in Cox@NC/PMS systems ranged from 0.02 min−1 to 0.38 min−1, increasing as elevating the Co content (Fig. S8b in Supporting information). As is shown in Fig. 2a, over ~99% removal efficiency of BHA could be observed in Co0.5@NC/PMS system. In comparison, only ~1% of BHA could be removed using the PMS alone, along with ~23% removal in NC/PMS system. While Co0.5@NC could adsorb ~20% BHA (Fig. S10 in Supporting information), desorbed amounts of BHA (the details shown in Text S6 in Supporting information) from Co0.5@NC/PMS system (0.14 µmol/L) was much lower than that from NC/PMS system (70 µmol/L). The observations further indicated that the Co-N4 sites might enhance the Fenton-like catalysis through promoting both the adsorption and oxidation of pollutants [34]. To ascertain active sites involved in the Cox@NC, potassium thiocyanate (KSCN) was employed due to its strong chelation ability with metal-N4 sites [31,32]. From Fig. 2b and Fig. S12 (Supporting information), the presence of 5 mmol/L KSCN led to a significant inhibition on removal efficiency of BHA, confirming the dominant role of atomically dispersed Co-N4 as reactive sites. To evaluate mass-specific activity of Co-N4 sites, turnover frequency (TOF) values, capable of quantifying catalytic activity of Co-N4 [33], were calculated using kobs and amounts of Co atoms. The calculated TOF values of Co0.25@NC, Co0.5@NC and Co1@NC reached 16.3, 18.0, and 17.6 min−1, respectively (Fig. S9 in Supporting information), also highlighting the superior reactivity of Co-N4 sites.
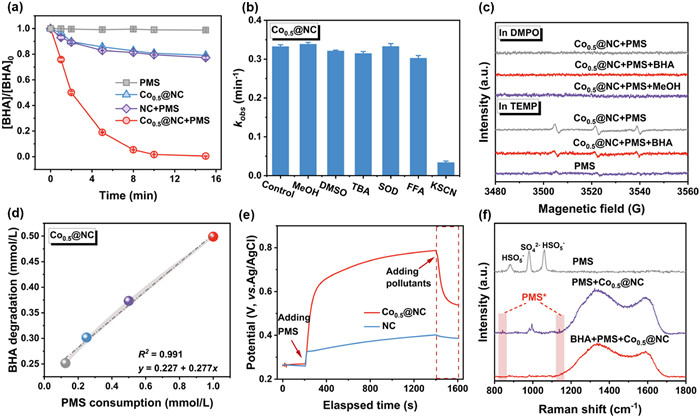
Figure 2
Figure 2.
Electron transfer of single-atom Co-N4 for Fenton-like catalysis. (a) The degradation of BHA by the different catalysts activated PMS system. (b) Correlation between BHA degradation and PMS consumption within the Co0.5@NC/PMS system. (c) The circuit potential on Co0.5@NC-GCE and NC-GCE electrode. (d) In situ Raman spectra on the catalyst surface to identify surface reactive species. (e) Quenching effect on BHA degradation by various scavengers in the Co0.5@NC/PMS system. (f) EPR signals were captured on DMPO and TEMP adducts during PMS activation. Reaction conditions: [Catalyst] = 0.3 g/L, [PMS] = 1 mmol/L, [BHA] = 0.5 mmol/L, pH 7.00.
The role of ROS in enhancing the Fenton-like oxidation was comprehensively elucidated. Methanol (MeOH) and tert‑butyl alcohol (TBA) served as SO4•−/ •OH scavengers [35,36], while superoxide dismutase (SOD), dimethyl sulfoxide (DMSO), and furfuryl alcohol (FFA) could efficiently quench O2•−, Co(Ⅳ), and 1O2, respectively [37,38]. The addition of TBA (500 mmol/L) and MeOH (500 mmol/L) in the Co0.5@NC/PMS system barely influenced the degradation of BHA (Fig. 2b and Fig. S12 in Supporting information), supporting the minor contribution of SO4•−/ •OH. Such proposal was also evidenced by the absence of electron spin resonance (EPR) signals for •OH and SO4•− (Fig. 2c). As O2•−displayed a minimal reactivity in water owing to its extensive solvation [39], potential generation of O2•−was tested using EPR measurement in MeOH [11]. Since both the sextet peak in EPR system and the inhibition of SOD on removal of BHA were negligible (Figs. 2b and c), the occurrence and impact of O2•−could be excluded. While 1O2 could mediate the formation of TEMPO adducts from TEMP, the emerging triplet peaks of TEMPO adducts in EPR system (Fig. 2c) did not sufficiently testify the contribution of 1O2 to oxidation [16,36,40,41]. This is because the self-decomposition of PMS at pH < 9.4 might also generate weak triplet peaks in EPR system (kPMS self-decomposition = ~1.3 × 10−2 L mol−1 s−1) [42]. Other 1O2-independent pathways might also promote transformation of TEMP to be TEMP•+ [43], further producing as peroxide radicals for TEMPO formation [41]. The inhibition of 10 mmol/L FFA on removal of BHA degradation was extremely limited (Fig. 2b). Meanwhile, the solvent substitution of deuterium oxide (D2O), which was reported to prolong the lifetime of 1O2 by ~18 times [11], did not enhance the oxidation of BHA (Fig. S14 in Supporting information). The results collective suggested a false positive result of 1O2 with its minimal contribution to Fenton-like oxidation of Cox@NC/PMS systems. In addition, the effect of typical high-valence metals, previously reported to be important in PMS-based oxidation process [11], could be excluded due to the much weak inhibition of 10 mmol/L DMSO (Fig. 2b and Fig. S12).
Since the minor contribution of ROS, a mediated ETP as typical non-radical oxidation might be dominant in the Cox@NC/PMS systems. From Fig. 2d, the oxidation of BHA showed a well positive correlation to the consumption of PMS, demonstrating simultaneous decomposition of both the BHA and PMS, which was characterized as one of the traits of a ETP mechanism [44]. To discern this proposal, a possible electron flow of ETP experiencing between pollutants and PMS was continuously monitored (Fig. S13 in Supporting information) [15,24]. As shown in Fig. 2e, open circuit potential (OCP) of Co0.5@NC-GCE with injection of PMS was significantly higher than that of NC-GCE, supporting the formation of a surface-activated PMS complex (PMS*) as electron acceptor on Cox@NC. Previous finding declared that the PMS could be rapidly absorbed onto single atom-N4 sites (Fig. S13), forming the surface PMS* with mild oxidation potential through intense charge distribution [6,25] Upon the addition of BHA, a subsequent decrease in OCP for Co0.5@NC-GCE was observed, substantiating the ETP from BHA as electron donor toward the newly formed Co0.5@NC-PMS*. In addition, in-situ Raman spectroscopy analysis showed the emerging peaks at ~840 cm⁻¹ and 1130 cm⁻¹ (Fig. 2f), previously identified as the characteristic signals of PMS* [11]. Notably, the intensities of Raman peaks of PMS* disappeared as introducing the BHA, further confirming the involvement of PMS* in the ETP for pollutant oxidation.
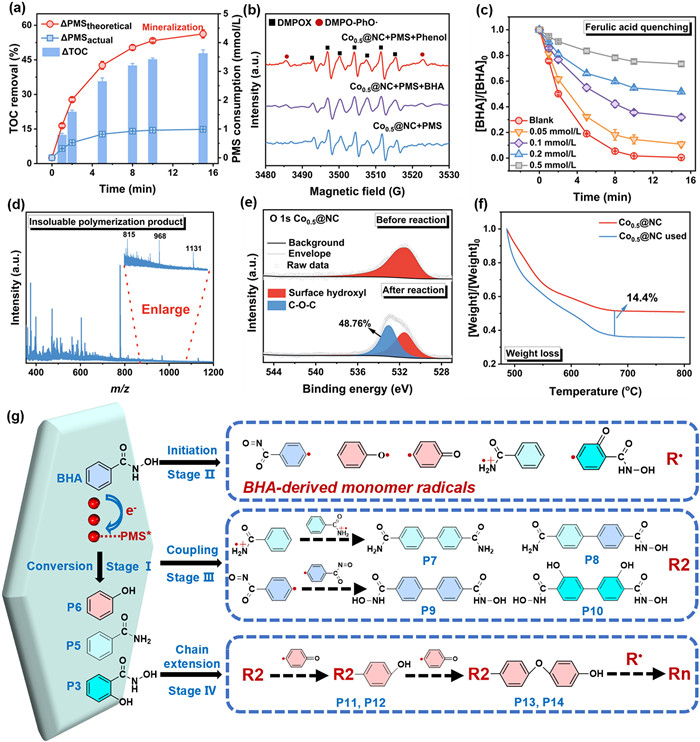
Traditional AOPs mainly rely on generation of highly reactive radicals (e.g., •OH and SO4•−) to mineralize organic pollutants through a 28-electron consumption of PMS (Eq. 1). In comparison, a mediated ETP might raise the transformation of organic pollutants into organic radicals (e.g., phenoxy radicals) via single-electron transfer (only consuming one electron of PMS), subsequently undergoing the cross-coupling to form polymerized products (Eq. 2) [12]. To testify the potential polymerization pathway, changes in total organic carbon (TOC) was evaluated and employed to compare with the PMS consumption (Fig. 3a and Table S4 in Supporting information). Noting that the practical consumptions of PMS in the Cox@NC/PMS systems were significantly lower than that needed for complete mineralization pathway against same TOC removal, thus, the utilization efficiency of PMS by Cox@NC was as high as ~434% (Table S4). The observations indicated that the loss of aqueous TOC likely resulted from the accumulation of polymerized products of BHA on Cox@NC surface through crossing-linking pattern [11]. Especially, the EPR system identified the obvious characteristic signals of phenoxy-like radicals (PhO•) in the Co0.5@NC/PMS system (Fig. 3b), supporting that the PhO• acted as the important reactive intermediates to induce polymerization of BHA [45]. The role of PhO• in governing the polymerization could be further verified by the inhibition of ferulic acid (FA), which was treated as an efficient scavenger of PhO• [11]. The results showed that the increasing concentration of FA led to a more significant inhibition on degradation efficiency of BHA (Fig. 3c), eventually reaching ~80% maximum inhibition ratio. Furthermore, the polymerized products could be verified by the analyses of matter-assisted laser desorption/ionization time-of-flight mass spectrometer (MALDI-TOF-MS) (Fig. 3d). Moreover, the O 1s spectra of XPS measurement showed a distinct transition from the hydroxyl group to C-O-C (Fig. 3e). Thermogravimetric analysis (TGA), enabling detection of polymerization products on solid surface [11,12], revealed more significant pyrolysis decomposition (14.4%) for Co0.5@NC after Fenton-like oxidation compared to pristine Co0.5@NC (Fig. 3f). Furthermore, Fig. S18 (Supporting information) demonstrated the polymeric products derived from BHA achieved a recovery yield of 43%. These observations further demonstrated a substantial cumulation of polymerized products on the Co0.5@NC surface.
Figure 3
Figure 3.
Characteristics of the BHA polymerization products. (a) In the Co0.5@NC/PMS/BHA system TOC removal efficiency, actual PMS consumption, and theoretical PMS consumption in complete mineralization pathway. (b) EPR signals of phenoxy radical (PhO•). ([Co0.5@NC] = 1 g/L, [PMS] = 100 mmol/L, [Phenol/BHA] = 50 mmol/L, [DMPO] = 100 mmol/L). (c) The effect of different concentrations of ferulic acid on the BHA removal efficiency. (d) XPS O 1s of Co0.5@NC before and after the reaction. (e) MALDI-TOF-MS of solid-phase polymerization product of BHA on catalyst surface (the isolation and characterization procedures for the polymeric products derived from BHA are provided in Text S5 in Supporting information). (f) TGA curves of Co0.5@NC before and after the reaction. (g) Possible polymerization pathways of BHA in the Co0.5@NC/PMS system. Reaction conditions: [Co0.5@NC] = 0.3 g/L, [PMS] = 1 mmol/L, [BHA] = 0.5 mmol/L, pH 7.00.
The findings demonstrated that the exceptional Fenton-like reactivity of single-atom Co-N4 triggered a mediated ETP for the polymerized transformation of BHA. This is because (1) single-atom Co-N4 enhanced formation of surface-reactive PMS* complexes and (2) superior electron transfer dynamics from BHA to PMS* for efficient generation of monomer radicals and following polymerization chain reactions. The potential intermediate products of BHA in the Cox@NC/PMS system were further analyzed based on HPLC-QTOF-MS. The possible structures of intermediate products were detected by the corresponding mass spectra of precursor ions (Figs. S20-S33 and Table S5 in Supporting information). The molecular weight of oxidation products was mostly located between 200 Da and 500 Da (Fig. S16b in Supporting information), identified as the polymerized oligomers (n = 2, 3, and 4) (P7-P14 in Table S5). As a result, the potential polymerization pathways of BHA may be systematically categorized into four stages (Fig. 3g) as following. (1) BHA transformation. Initial substrate BHA (m/z 138) was first decomposed by the ETP, leading to hydroxylation to generate hydroxybenzohydroxamic acid (P3, m/z = 154) and cleavage of N-O to produce benzamide (P5, m/z 122) [8]. Simultaneously, the oxidative hydroxylation of BHA resulted in formation of phenolic structure (P6, m/z 95) [7]. (2) Generation of monomer radicals (R•). Enhanced formation of reactive PMS* would effectively transform the BHA and its hydroxylated products into PhO• as intermediate forms [12]. (3) Formation of dimeric products (R2). The monomer radicals served as precursors for oligomeric products in the radical chain reactions, further undergoing C-C coupling to generate R2 with various structures (P7, P8, P9, and P10). (4) Chain extension. The aforementioned dimers continuously donated electrons to PMS* for generation of dimeric radical, further binding with PhO• to produce further trimers (R3, P11 and P12) by C-C coupling. Particularly, trimers and other products may also lose electrons to form larger organic radicals, coupling with other organic radicals to increase polymerization degree with a large quantity of high-molecular-weight polymers (Rn) adhered onto Co0.5@NC surface.
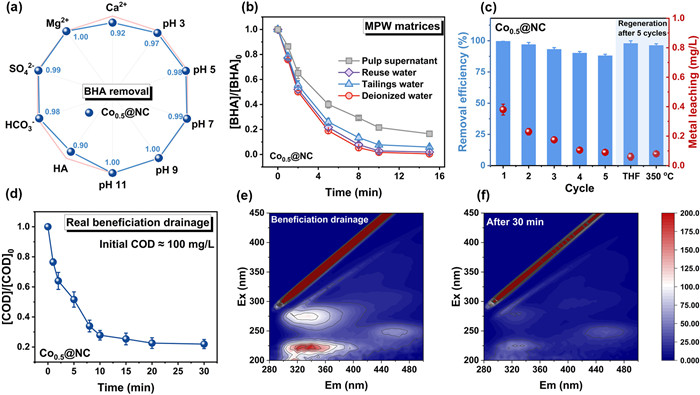
The adaptability of Co0.5@NC/PMS system in the practical MPW treatment was evaluated based on the water quality of MPW (Table S6 in Supporting information). As illustrated in Figs. 4a and b and Figs. S11c and d (Supporting information), Co0.5@NC/PMS system exhibited the remarkable anti-interference capabilities in practical MPW matrices and exceptional stability during five cycles of treatment, still maintaining a 90% removal efficiency over consecutive cycles and low leaching of Co (<0.4 mg/L, below established regulatory thresholds for safe drinking water quality) (Fig. 4c) [6]. Furthermore, the catalytic activity would be restored through 350 ℃ calcination or THF elution (Fig. 4c and Fig. S18). The results suggested the high application potential of Cox@NC in Fenton-like oxidation with extremely limited secondary pollution. The water purification performance of Co0.5@NC/PMS system was also compared to that of the Fe2+/H2O2 system, which is widely used as radical-based AOP in MPW treatment (Fig. S15a in Supporting information). Notably, Co0.5@NC/PMS showed a significantly lower oxidant consumption (PMS, 1 mmol/L vs. 10 mmol/L) and a higher TOC removal efficiency (40% vs. 24%) than the traditional Fenton system (Fig. S15b in Supporting information), further confirming the characteristics of polymerization pathway induced by Co0.5@NC/PMS system. Additionally, Co0.5@NC/PMS exhibited superior removal efficiency of chemical oxygen demand (COD) in real beneficiation drainage samples (Fig. 4d). After a 30-min treatment, excitation-emission matrix (EEM) spectroscopy (Figs. 4e and f) revealed a substantial reduction in fluorescence intensity, indicating the effective removal of soluble microbial byproducts and phenolic humic acid-like organic compounds. These results collectively indicated the great adaptability and stability of single-atom Co-N4 in water purification during the real MPW treatment.
Figure 4
Figure 4.
Assessment of the adaptability and stability of Co0.5@NC in real mineral processing wastewater (MPW). (a) Co0.5@NC catalytic removal efficiency of BHA with the interference of 10 mmol/L salts, 5 mmol/L HA and different pH. (b) The BHA removal efficiency and metal leaching in cyclic experiments and regeneration by solvent extraction or calcination. (c) Treatment of BHA in real mineral processing water matrices. (d) The COD removal of real beneficiation drainage ([Co0.5@NC] = 0.3 g/L, [PMS] = 2 mmol/L, without pH adjusting). (e, f) EEM spectra of collected real beneficiation drainage before (e) and after (f) 30 min treatment. Reaction conditions: [Co0.5@NC] = 0.3 g/L, [PMS] = 1 mmol/L, [BHA] = 0.5 mmol/L.
The principal findings of this investigation are delineated as follows: (1) We successfully developed a single-atom Co-N4 with considerable atomically dispersed Co content, demonstrating its exceptional Fenton-like catalysis performance for BHA treatment with a high TOF > 16 min-1 and great utilization efficiency of PMS (>434%). (2) The PMS activation by single-atom Co-N4 barely produced conventional reactive oxygen species, revealing that single-atom Co-N4 dominantly coordinated with PMS to form a surface-reactive PMS* complexes with mild oxidation potential to initiate the electron transfer for oxidation of BHA. (3) In particular, the mediated ETP further triggered polymerization transformation pathway of BHA through formation and coupling of phenoxy-like radicals, resulting in low carbon emission. Combined with HPLC-QTOF-MS, the polymerized products were identified and the involved mechanisms could be verified to include hydroxylation of BHA, generation of monomer radicals, formation of dimeric products, and chain extension. (4) The polymeric products derived from BHA achieved a recovery yield of 43%. (5) The single-atom Co-N4 exhibited remarkable stability in cyclic experiments, maintaining a consistent BHA removal efficiency exceeding 90%, with metal leaching levels well below the permissible limits for drinking water standards. The Co0.5@NC catalyst demonstrated superior performance in simulated mineral processing wastewater (MPW) matrices, showing significantly reduced oxidant consumption (1 mmol/L vs. 10 mmol/L in Fenton process) and enhanced total organic carbon (TOC) removal efficiency (40% vs. 24% in Fenton process). When applied to real beneficiation wastewater from MPW, the Co0.5@NC/PMS system achieved approximately 80% chemical oxygen demand (COD) removal within 30 min using only 0.3 g/L catalyst and 2 mmol/L PMS, underscoring its potential for practical MPW treatment applications. These findings provide valuable insights for designing efficient single-atom Co catalysts for MPW treatment, offering a promising approach with low oxidant consumption and reduced carbon emissions.
Declaration of competing interest
The authors declare that they have no known competing financial interest or personal relationships that could have appeared to influence the work reported in this paper.
Y. Yang, G. Banerjee, G.W. Brudvig, et al., Environ. Sci. Technol. 52 (2018) 5911–5919. doi: 10.1021/acs.est.8b00735
[43]
Y. Wen, C.H. Huang, D.C. Ashley, et al., Environ. Sci. Technol. 56 (2022) 2626–2636. doi: 10.1021/acs.est.1c06696
[44]
Q. Tang, B. Wu, X. Huang, et al., Nat. Commun. 15 (2024) 9549.
[45]
X. Gao, Z. Yang, W. Zhang, et al., Nat. Commun. 15 (2024) 2808.
Figure 1
Properties of nitroeeegen-coordinated single-atom Co-N4. (a) HR-TEM image, (b) HAADF-STEM image and EDS mapping spectra. (c) AC-HAADF-STEM image of Co0.5@NC. XANES spectra at the (d) Co K-edge, (e) Co R-space of Co0.5@NC and their reference samples. (f) Wavelet transform of Co0.5@NC and their reference samples. EXAFS spectra fitting curves of Co0.5@NC at the (g) Co K-edge and (h) Co R-space (inset: structural model of Co0.5@NC). (i) XRD spectra of Cox@NC.
Figure 2
Electron transfer of single-atom Co-N4 for Fenton-like catalysis. (a) The degradation of BHA by the different catalysts activated PMS system. (b) Correlation between BHA degradation and PMS consumption within the Co0.5@NC/PMS system. (c) The circuit potential on Co0.5@NC-GCE and NC-GCE electrode. (d) In situ Raman spectra on the catalyst surface to identify surface reactive species. (e) Quenching effect on BHA degradation by various scavengers in the Co0.5@NC/PMS system. (f) EPR signals were captured on DMPO and TEMP adducts during PMS activation. Reaction conditions: [Catalyst] = 0.3 g/L, [PMS] = 1 mmol/L, [BHA] = 0.5 mmol/L, pH 7.00.
Figure 3
Characteristics of the BHA polymerization products. (a) In the Co0.5@NC/PMS/BHA system TOC removal efficiency, actual PMS consumption, and theoretical PMS consumption in complete mineralization pathway. (b) EPR signals of phenoxy radical (PhO•). ([Co0.5@NC] = 1 g/L, [PMS] = 100 mmol/L, [Phenol/BHA] = 50 mmol/L, [DMPO] = 100 mmol/L). (c) The effect of different concentrations of ferulic acid on the BHA removal efficiency. (d) XPS O 1s of Co0.5@NC before and after the reaction. (e) MALDI-TOF-MS of solid-phase polymerization product of BHA on catalyst surface (the isolation and characterization procedures for the polymeric products derived from BHA are provided in Text S5 in Supporting information). (f) TGA curves of Co0.5@NC before and after the reaction. (g) Possible polymerization pathways of BHA in the Co0.5@NC/PMS system. Reaction conditions: [Co0.5@NC] = 0.3 g/L, [PMS] = 1 mmol/L, [BHA] = 0.5 mmol/L, pH 7.00.
Figure 4
Assessment of the adaptability and stability of Co0.5@NC in real mineral processing wastewater (MPW). (a) Co0.5@NC catalytic removal efficiency of BHA with the interference of 10 mmol/L salts, 5 mmol/L HA and different pH. (b) The BHA removal efficiency and metal leaching in cyclic experiments and regeneration by solvent extraction or calcination. (c) Treatment of BHA in real mineral processing water matrices. (d) The COD removal of real beneficiation drainage ([Co0.5@NC] = 0.3 g/L, [PMS] = 2 mmol/L, without pH adjusting). (e, f) EEM spectra of collected real beneficiation drainage before (e) and after (f) 30 min treatment. Reaction conditions: [Co0.5@NC] = 0.3 g/L, [PMS] = 1 mmol/L, [BHA] = 0.5 mmol/L.

 DownLoad:
DownLoad:

 下载:
下载:





 下载:
下载:
