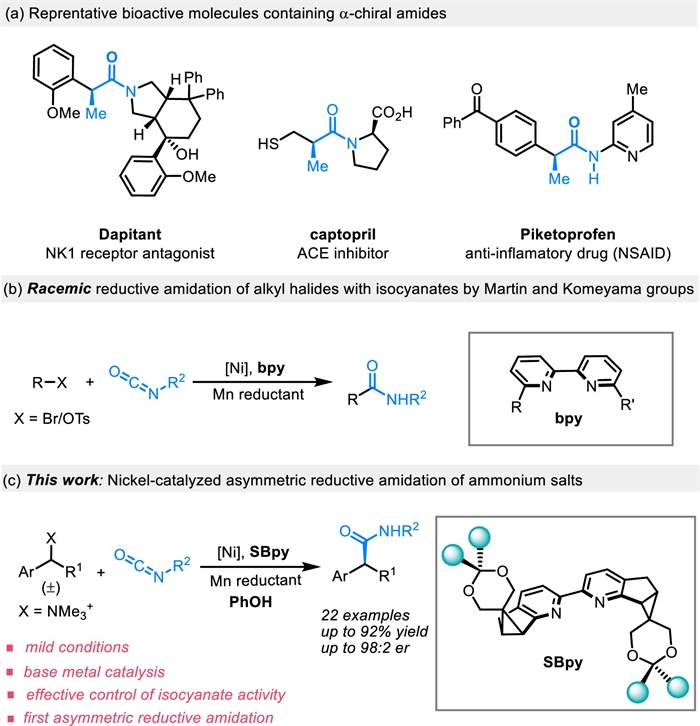
Scheme 1.
The occurrence and synthetic methods of α-chiral amides and the outline of this work.
Enantioconvergent reductive amidation of benzyl ammonium salts for synthesis of α-chiral amides
Saima Perveen , Xicheng Wang , Tao Li , Linghua Wang , Shuai Zhang , Yizhao Ouyang , Xue Zhao , Liang Xu , Pengfei Li

Amides are among the most essential scaffolds in both natural and synthetic chemicals, and they are vital in biological systems as well as in the design of pharmaceuticals [1-9]. Specifically, α-chiral amides are highly prized due to their prevalence in natural products, medicinal agents and agrochemicals, and are crucial in forming chiral ligands for asymmetric catalysis (Scheme 1a) [10,11]. Consequently, devising new and efficient strategies for the asymmetric synthesis of α-chiral amide has been long desirable. In this context, the direct union of amines with α-chiral carboxylic acids is a traditional method. However, the preparation of α-chiral carboxylic acids already poses a challenge. Therefore, direct synthesis of α-chiral amides from readily accessibe achiral starting materials would represent more streamlined approach, such as enantioselective alkylation, hydroaminocarbonylation, carbamoylation, hydrofunctionalization, hydrogenation and similar processes [12-19].
Recently, isocyanates have emerged as promising amidating reagent due to their low cost, good availability, and ease of modification [20-22]. In this context, Mazet and co-workers have introduced a Cu-catalyzed asymmetric borylative carboxamidation of alkenes and alkynes to obtain a range of β-borylated secondary amides [23]. Additionally, Nevado group has disclosed a dual Ni/photo-catalyzed asymmetric carbamoylation reaction with isocyanates for the efficient synthesis of α-chiral amides, which involves a dual cycle mechanism that generates benzyl radicals [24]. Despite the progress in asymmetric amidation reactions, the use of isocyanates as amidating reagents for synthesizing α-chiral amides faces two main challenges: (1) Isocyanates are susceptible to self-polymerization or overreaction with nucleophiles, which can complicate product isolation and purity; (2) Most asymmetric reactions are only compatible with aryl isocyanates, resulting a limited substrate scope and reduced applications. Over the past decade, there has been significant progress in nickel-catalyzed asymmetric reductive coupling reactions, marking a new era in asymmetric C—C bond formation [25-37]. These reactions are known for their mild conditions and ease of execution, offering a more accessible route to C–C bond formation without the need of organometallic reagents that are often necessary in conventional coupling processes [38].
The groups of Martin and Komeyama, have independently developed innovative racemic reductive amidation reactions with isocyanates, using alkyl halides or tosylates as the substrates (Scheme 1b) [39-43]. The success of these reactions in terms of reactivity and regioselectivity is largely influenced by the choice of specific ligand architectures. Nonetheless, the limited availability of chiral ligands, especially 2,2′-bipyridine ligands, has posed a challenge in developing an enantioselective variant of the reductive coupling reaction that includes isocyanates.
Over the past few years, our team has brought to life an innovative chiral 2,2′-bipyridine ligand named as SBpy, characterized by its [6-5-3] rigid fused-ring structure and adjustable properties [44]. This development has successfully overcome the balancing act between reactivity and selectivity. The SBpy ligand has proven to be highly effective and precise in directing stereochemistry across a range of reactions, such as the asymmetric Ullmann coupling [45], the asymmetric addition to aryl aldehydes [44], and asymmetric carboxylation reactions [46] with CO2 [47]. Drawing inspiration from the electronic similarities between isocyanates and CO2, and capitalizing on the adjustable nature of SBpy, we now introduce the nickel-catalyzed asymmetric reductive amidation of quaternary ammonium salts with alkyl isocyanates. This work paves the way for the production of chiral amides with both good yields and high enantioselectivity (Scheme 1c). It is important to highlight that the inclusion of phenol is pivotal in managing the reactivity of isocyanates throughout the reaction, effectively minimizing unwanted side reactions.
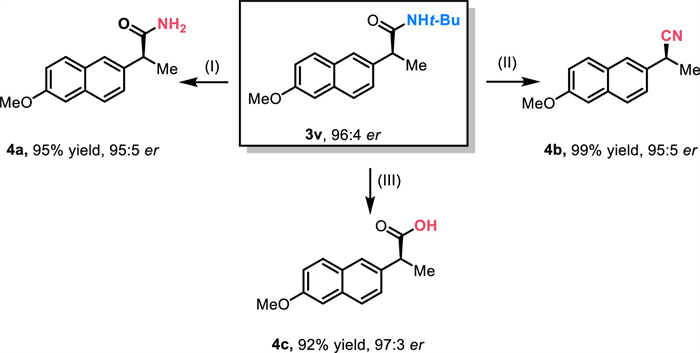
To begin our investigations, we chose trimethyl 1-(naphthalen-2-yl)ethyl ammonium trifluoromethanesulfonate (1a) and tert‑butyl isocyanate (2a) as the benchmark substrates to test our envisioned asymmetric reductive amidation. Pleasingly, after thorough experimentation, we established an optimal set of conditions. These involved using NiCl2[P(mesityl)3]2 as the precatalyst, Ph-SBpy (L1) as the chiral ligand, Mn as the reductant, PhOH as an additive, and phenyl acetate as the solvent at 70 ℃ for 20 h, yielding the target product 3a with 90% yield and 96:4 enantiomeric ratio (er) (Table 1, entry 1). As depicted in Table 1, alterations in the acetal substitution in SBpy markedly influence the reaction’s activity and stereoselectivity. For example, utilizing Me-SBpy (L2), the chiral amides were obtained with 81% yield and 93:7 er (Table 1, entry 2). Employing DTB-SBpy (L4), which offers greater steric hindrance, resulted in poor outcome of 51% yield and 72:28 er (Table 1, entry 4). Moreover, the use of the bulkier i-Pr-SBpy (L3) completely inhibited the reaction (Table 1, entry 3). Additionally, we evaluated the impact of several readily available dinitrogen chiral ligands, Pyox (L5) and BOX (L6), and found their incompatibility with this reaction (entries 5 and 6). Furthermore, the comparatively high rigidity of Z-bpy (L7) did not aid in the amidation reaction (Table 1, entry 7). Additionally, it is plausible that molecular interactions between the aryl group at the ligand’s distal end and the substrate could enhance the reaction. Subsequently, we discovered that the selection of the Ni catalyst played a crucial role in the reaction’s outcome. For instance, NiCl2 and Ni(PPh3)2Cl2 were less effective in terms of yield, though they did not significantly impact stereoselectivity (Table 1, entries 8 and 9). This might be because of the stability of the ligands on the nickel center can affect the rate of ligand exchange processes. More stable complexes might undergo slower ligand exchange, leading to more controlled reaction conditions and higher yields [48]. Moreover, polar solvents with good solubility, such as 2-MeTHF and ethyl formate, appeared to be suitable for the reaction but yielded slightly inferior results (Table 1, entries 10 and 11). Additionally, control experiments underscored the significance of strategies for regulating isocyanate activity. When the reaction was carried out without phenol, there was a significant increase in side reactions involving isocyanates, which led to a reduction in the yield of the desired product to 63%. However, adding phenol did not affect the enantioselectivity of the reaction, indicating that the presence of phenol helped to reduce unwanted byproducts without changing the preference for producing one enantiomer over the other (Table 1, entry 12). Furthermore, low temperatures were not favorable for the reaction to occur (Table 1, entry 13).
 DownLoad:
CSV
DownLoad:
CSV
 |
|||
| Entry | Deviation from standard conditions | Yield (%) b | erc |
| 1 | None | 90 | 97:3 |
| 2 | L2 instead of L1 | 81 | 93:7 |
| 3 | L3 instead of L1 | N.R | - |
| 4 | L4 instead of L1 | 51 | 72:28 |
| 5 | L5 instead of L1 | N.R | - |
| 6 | L6 instead of L1 | N.R | - |
| 7 | L7 instead of L1 | N.R | - |
| 8 | NiCl2 instead of NiCl2[P(mesityl)3]2 | 60 | 95:5 |
| 9 | NiCl2(PPh3)2 instead of NiCl2[P(mesityl)3]2 | 65 | 95:5 |
| 10 | 2-MeTHF as solvent | 51 | 92:8 |
| 11 | Ethyl formate as solvent | 75 | 96:4 |
| 12 | w/o PhOH | 63 | 95:5 |
| 13 | 30 ℃ instead of 70 ℃ | N.R | - |
| a Reaction conditions: 1a (0.10 mmol), 2a (0.20 mmol), NiCl2[P(mesityl)3]2 (0.010 mmol), Ligand (0.012 mmol), Mn (0.40 mmol), PhOH (0.15 mmol), phenyl acetate (0.20 mL), 20 h at 70 ℃. b Isolated yield after silica gel column chromatography. c The enantiomeric ratio was determined on a chiral HPLC OJ-H column. |
|||
With the viable reaction conditions in hand, we explored the scope of the asymmetric amidation reaction, focusing initially on quaternary ammonium salts (Scheme 2). Delightfully, the reaction was amenable to a variety of quaternary ammonium salts with polycyclic arenes, including those with naphthalene, anthracene, and phenanthrene (3a-3d), as well as those with heterocyclic fused rings (3e, 3f), all of which were efficiently converted to products with good yields and high enantiomeric ratios. Additionally, quaternary ammonium salts with electron-donating and electron-withdrawing groups at the meta and/or para positions also participated in the asymmetric amidation reaction, yielding chiral amides in good yields (63%−70%) and enantiomeric ratios (84:16–97:3) (3g-3j). Biphenyl substrates, both unsubstituted (3k) and those with various electronic substituents like t‑butyl (3l) and trifluoromethyl (3m), were compatible with the reaction, albeit with moderate yields (64%−75%) and enantiomeric ratios (84:16–94:6). Notably, biphenyl substrates substituted at the meta position (3n) resulted in 70% and 81:19 er. Given the significance of fluorinated compounds in pharmaceuticals and agrochemicals, we tested ammonium salt with fluorine substitution. Encouragingly, biphenyl-containing difluoro benzene-substituted ammonium salt produced fluorinated chiral amide product (3o) with moderate yield and er. Furthermore, heterocyclic substrate i.e., 3-phenylthiophene (3p), resulted 60% yield and 83:17 er. Replacing the benzyl methyl ammonium salt with a benzyl ethyl ammonium salt resulted the product (3q) with 70% yield and 86:14 er. We then investigated the effect of isocyanates in this asymmetric amidation reaction. Interestingly, alkyl isocyanates, which were challenging to couple in previous studies, could be transformed into the desired amides (3r and 3s) in 81% and 84% yield and er 96:4 and 94:6, respectively. A lower er was observed for isocyanates with primary carbon center, such as n‑butyl isocyanate (3t), yielding chiral amides with 70% yield and 84:16 er. Ultimately, this reaction was also applicable for the synthesis of amide derivatives of two important drugs, flurbiprofen and naproxen. Under standard conditions, the flurbiprofen amide derivative (3u) was successfully prepared with 81% yield and 97:3 er, and the naproxen amide derivative (3v) was generated with 80% yield and 96:4 er. The absolute stereochemistry of (S)-2-([1,1′-biphenyl]−4-yl)-N-(tert‑butyl)propanamide (3k) was unambiguously confirmed to be the opposite configuration of the reported product, enabling the configuration assignment of all other products by analogy [49].
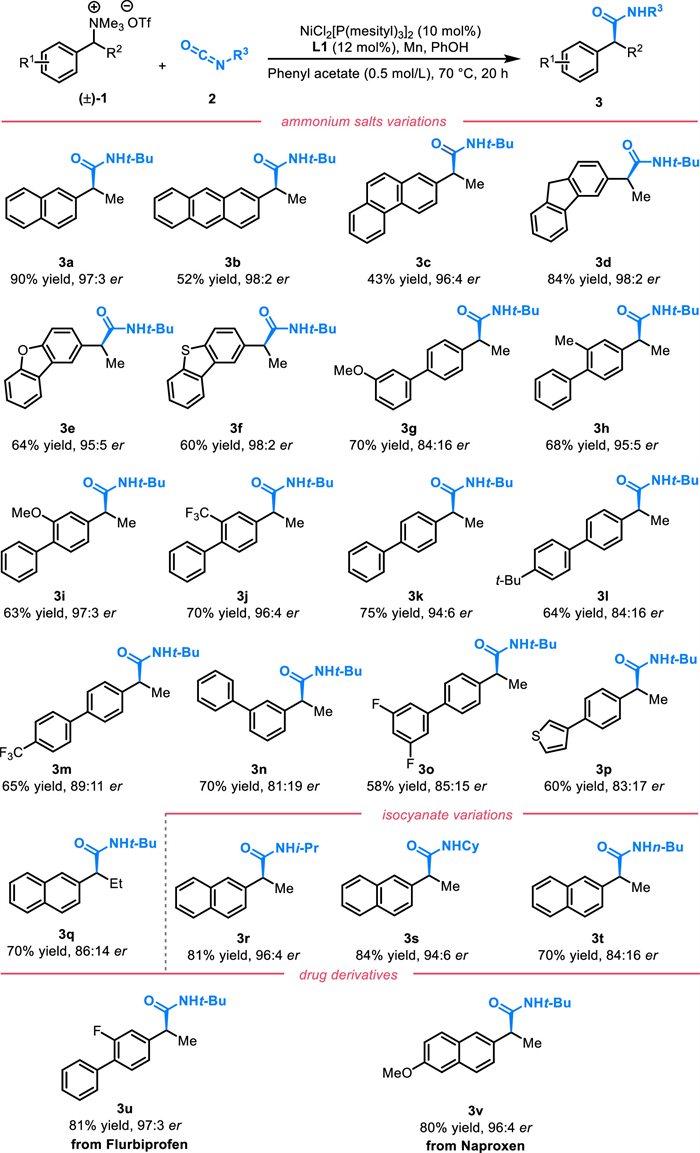
The practicality of the nickel-catalyzed asymmetric amidation was demonstrated through post-catalytic transformations without erosion of enantioselectivity (Scheme 3). α-Chiral amide (3v) was transformed to amide 4a via an N-dealkylation reaction of the amide using trifluoromethanesulfonic acid, in 95% yield. Additionally, cyanide, a highly useful chemical intermediate 4b, was produced with 99% yield under the influence of trifluoromethanesulfonic anhydride and 2-fluoropyridine. Lastly, the synthesis of the nonsteroidal anti-inflammatory drug (S)-naproxen 4c was accomplished through a gentle hydrolysis process in 92% yield.
Preliminary mechanistic investigations were performed to elucidate the pathway of the nickel-catalyzed asymmetric reductive amidation of isocyanates with benzyl ammonium salts. First, radical-trapping experiments were performed by adding one equivalent of either 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) or diphenyl disulfide (PhSSPh) as a radical scavenger. The presence of these scavengers inhibited the amidation reaction, suggesting that benzyl radicals were generated during the reaction cycle, contributing to side reactions (Fig. 1a). Additionally, we found that the reaction outcome of triethylamine-derived quaternary ammonium salt was comparable to that of trimethylamine-derived one under the standard conditions, yielding the desired product 3a in 92% yield and 93% ee (Fig. 1b). The amidation reaction did not proceed when phenyl carbamate (5) was used instead of isocyanate (2a), demonstrating its inertness towards amidation. However, when additional triethylamine was added under identical conditions, the amidation product 3a was obtained in 60% yield and 94% ee (Fig. 1c). This result suggested that trialkylamines, particularly triethylamine, can regulate the release of isocyanates from phenyl carbamate under the reaction conditions. Reversibility of carbamate formation during reaction process was confirmed by the reaction involving phenylcarbamate and p-cresol in the presence of triethylamine. The presence of triethylamine likely facilitates the deprotonation of p-cresol, promoting the formation of the carbamate intermediate (Fig. 1d). NMR analysis confirmed the generation of both phenylcarbamate (5) and 4-methylphenylcarbamate (6) in the reaction mixture, with approximately 20% of the latter (see Supporting information for detail). To further confirm these hypotheses, the profiles of the key species over reaction time was monitored. The results indicated that under asymmetric amidation conditions, the release of trimethylamine from quaternary ammonium salts has a regulatory effect similar to that of triethylamine. In the early stages of the reaction, phenyl carbamate (5) was formed with isocyanates (2a) and phenol. After an induction period of approximately 12 h, during which the active nickel intermediate and trialkylamine gradually form, phenyl carbamate 5 begins to release isocyanate at 16 h and rapidly forms the chiral amide 3a, completing the reaction in 20 h. Based on this observation, we performed a reaction in the presence of triethylamine. As indicated in the graph, the reaction proceeds faster in the presence of triethylamine, which is likely due to the rapid release of isocyanate from carbamate, completing the reaction in 16 h (Fig. 1e).

To investigate the origin of enantiocontrol in the reductive amidation reaction, we conducted a series of experiments using enantioenriched starting materials and ligands, as outlined in Fig. 2. Initially, we examined the effect of the chiral and racemic ligand on the reaction outcome using racemic substrate 1a (Fig. 2a). Under standard conditions, the use of optically pure (–)-Ph-SBpy and (+)-Ph-SBpy ligands afforded (S)-3a and (R)-3a, respectively, in high yields and enantiomeric ratios. In contrast, the use of racemic (±)-Ph-SBpy resulted in a completely racemic product, (±)-3a, indicating that the stereochemical outcome is solely controlled by the ligand and not influenced by kinetic resolution (Fig. 2a). Next, we evaluated the impact of the substrate’s absolute configuration by performing the reaction with enantioenriched (S)-1a. When (–)-SBpy (matched pair) is used the (S)-3a obtained in 90% yield and 95:5 er. In contrast, using (+)-SBpy (mismatched pair) resulted in reducing both selectivity and yield (40%, 87:13 er). With rac-SBpy, both matched and mismatched pathways compete, causing poor overall performance (32% yield, 40:60 er), with a slight bias toward the (S)-3a, suggesting that the matched pathway with (–)-SBpy is slightly more competitive, or that the S-ammonium salt interacts more favorably with the (–)-SBpy ligand and the substrate’s configuration can either reinforce or oppose that preference, leading to matched or mismatched reactivity (Fig. 2b).
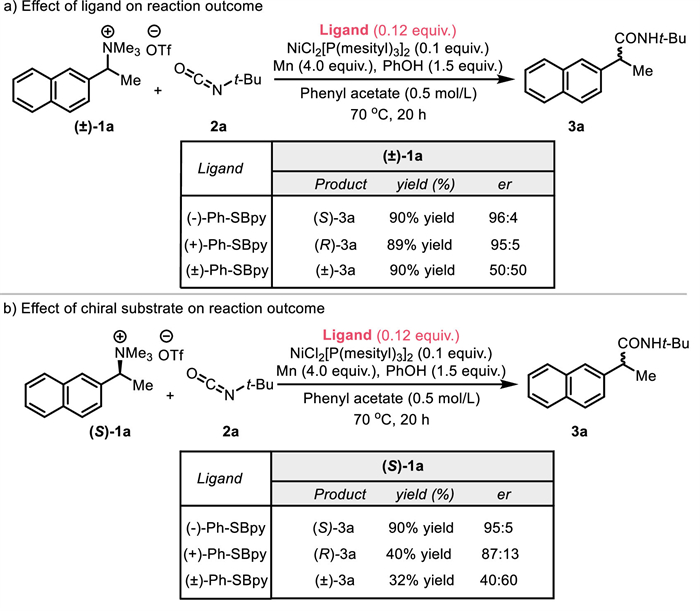
Based on the above experimental results and relevant reports [50-55], we have formulated a likely catalytic cycle for the nickel-catalyzed reductive amidation reaction (Scheme 4a). Initially, the nickel precursor is activated through ligand exchange with Ph-SBpy and subsequent reduction by Mn0, yielding the active Ni0L* complex A. This complex A then engages in oxidative addition with the quaternary ammonium salt substrate, resulting in the formation of the benzyl NiⅡ intermediate B and the release of trimethylamine, which plays a role in controlling the release of isocyanates. The highly reactive intermediate B may dissociate into NiⅠXL* and the benzyl radical F, with the latter prone to escaping the solvent cage and engaging in self-coupling side reactions. The intermediate B is reduced to the NiⅠ species C by Mn0, a critical step in the amidation process. Insertion occurs more readily from monovalent nickel complexes, which have a lower energy barrier compared to divalent nickel complexes, resulting increased reaction activity [56]. Moreover, phenyl acetate might participate in this reaction as both a solvent cage and a source of phenol to facilitate carbamate formation. So, the solvent itself can be involved in the reaction mechanism, providing a dual role that can enhance reaction efficiency. Following this, isocyanates insert to form the NiⅠ intermediate D. This step will determine the enantioselectivity of the process. Throughout this stage, the trimethylamine formed in situ modulates the protective effect of phenol on isocyanates, giving rise to the formation of inert phenyl carbamate and preventing the self-polymerization of isocyanates. Concurrently, the release of phenyl carbamate is dynamically regulated to liberate isocyanates. Ultimately, the chiral amide salt D is reduced further by Mn0 to regenerate A and E. Protonation of E then delivers final product 3.
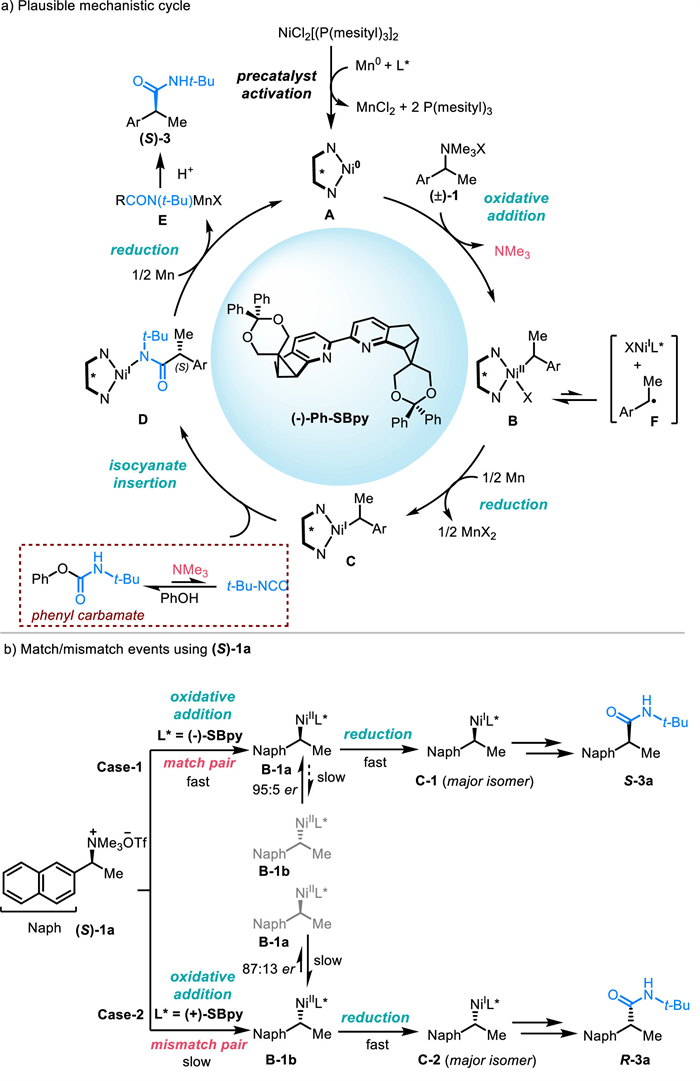
Consistent with the mechanism shown in Scheme 4a and the enantiocontrol data in Fig. 2b, the oxidative addition step involving the (S)-1a/(–)-SBpy “matched” pair (Case-1) rapidly establishes a well-aligned chiral environment, contributing to the formation of the thermodynamically stable intermediate B-1a as the major enantiomer. This intermediate B-1a is reluctant to isomerize to the less favorable enantiomer B-1b, and instead proceeds through a fast reduction to C-1, ultimately delivering the S-3a with high rate and selectivity. In contrast, the (S)-1a/(+)-SBpy “mismatched” pair (Case-2) experiences a slow oxidative addition due to the lack of a well-aligned chiral environment, resulting in the formation of B-1b, which can partially isomerize to the enantiomer B-1a. Both enantiomers B-1b (major isomer) and B-1a (minor isomer) then undergo fast reduction to C-2, ultimately yielding the R-3a with lower rate and selectivity. The slower oxidative addition also leads to increased side reactions in this case, resulting in reduced yield and enantioselectivity. These results confirm that the stereochemical outcome is governed by the ability of the chiral ligand to form a well-aligned chiral environment and generate thermodynamically stable intermediates, as illustrated in Scheme 4b.
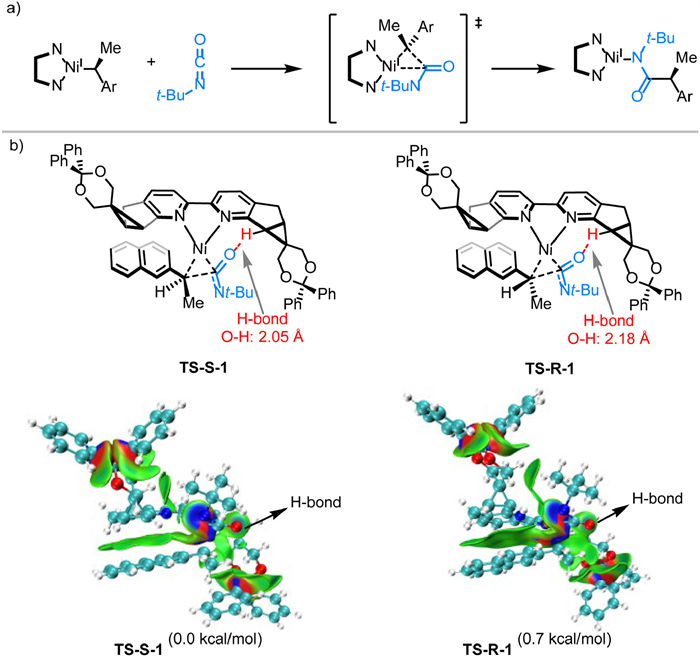
The plausible transition state of the enantio‑determining step in the reductive amidation process is depicted in Fig. 3a. To provide a more intuitive visual understanding of enantioselectivity using Ph-SBpy, the structural model of the stereo-determining step (C to D) was generated through density functional theory (DFT) calculations, as illustrated in Fig. 3b (For the computational details, see Supporting information). Among the obtained insertion transition states corresponding to different configurations/conformations, the transition state TS-S-1, which led to the (S)-enantiomer, was calculated to have the lowest activation barrier. In contrast, TS-R-1, which was the energetically lowest conformation for the formation of the other enantiomer, was 0.7 kcal/mol more energy-demanding than TS-S-1. This is consistent with the experimental observations that (S)-enantiomer was generated favorably. Furthermore, to elucidate the origin of the free energy difference between TS-S-1 and TS-R-1, Independent Gradient Model based on Hirshfeld partition (IGMH) analysis was conducted using Multiwfn software to probe the non-covalent interactions between the chiral pocket and substrates. As shown in Fig. 3b, for both TS-S-1 and TS-R-1, the naphthyl was preferably positioned in parallel with pyridyl unit of Ph-SBpy, which resulted in π-π stacking interaction. Also, the H-bond interaction between the carbonyl oxygen and bridgehead hydrogen between 3- and 5-membered ring of ligand was observed in both cases. For TS-S-1, the methyl was positioned outward the chiral pocket, which obviated the steric repulsion between methyl and the bulky pyridyl substituents. In TS-R-1, methyl was in a reversed direction, making the repulsion inevitable. This steric effect should also be responsible for the enlengthened H-bond distance (0.13 Å longer) in TS-R-1. Overall, TS-R-1 contained decreased favorable non-covalent interaction and increased steric repulsion than TS-S-1, which raised its free energy and made it difficult to achieve relatively.
In summary, we have successfully developed an enantioselective reductive amidation of quaternary ammonium salts employing readily accessible isocyanates as the practical amidating agents, providing a direct approach to α-chiral amides that are frequently found in bioactive molecules. With mild nickel catalysis and the use of chiral bipyridine ligand SBpy, a variety of substrates have been smoothly converted to α-chiral amides with high yields and excellent enantioselectivities. Furthermore, the inclusion of phenol in the reaction mixture effectively controls the reactivity of isocyanates, thereby minimizing side reactions and enhancing the overall yield of the desired products. Moreover, the stereochemical outcome is confirmed to be governed by the chiral ligand’s ability, as indicated by enantiocontrol experiments and DFT computational studies. These findings should provide valuable tools and inspirations for the synthesis of relevant chiral functional molecules and development of other base-metal catalyzed asymmetric reactions.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Saima Perveen: Writing – review & editing, Writing – original draft, Validation, Supervision, Funding acquisition. Xicheng Wang: Formal analysis, Data curation. Tao Li: Formal analysis, Data curation. Linghua Wang: Investigation, Formal analysis, Data curation. Shuai Zhang: Investigation. Yizhao Ouyang: Formal analysis, Data curation. Xue Zhao: Formal analysis. Liang Xu: Writing – review & editing, Supervision. Pengfei Li: Supervision, Project administration, Funding acquisition, Conceptualization.
The financial support was provided by the National Natural Science Foundation of China (Nos. 22150410339, W2432012, 22301233 and 22171218), the Ministry of Science and Technology China (No. wgxz2022188). The authors thank Pei Zhou and Lu Bai at the Instrumental Analysis Center of XJTU for the assistance with NMR and high-resolution mass spectrometry analysis.
Supplementary material associated with this article can be found, in the online version, at doi:
D.G. Brown, J. Boström, J. Med. Chem. 59 (2015) 4443–4458.
M.T. Sabatini, L.T. Boulton, H.F. Sneddon, T.D. Sheppard, Nat. Catal. 2 (2019) 10–17. doi: 10.1038/s41929-018-0211-5
J. Pitzer, K. Steiner, J. Biotechnol. 235 (2016) 32–46. doi: 10.1016/j.jbiotec.2016.03.023
S. Sun, Q. Jia, Z. Zhang, Bioorg. Med. Chem. Lett. 29 (2019) 2535–2550. doi: 10.1016/j.bmcl.2019.07.033
X. Wang, Nat. Catal. 2 (2019) 98–102. doi: 10.1038/s41929-018-0215-1
A. Greenberg, C.M. Breneman, J.F. Liebman, Biochem. Mater. Sci, Wiley-Interscience, New York, 2000.
S. Ponra, B. Boudet, P. Phansavath, V.R. Vidal, Synthesis 53 (2020) 193–214.
X. Fu, Y. Zhang, J. Liao, et al., Chin. Chem. Lett. 35 (2024) 109688. doi: 10.1016/j.cclet.2024.109688
C.D. Zhao, H. Yao, S.Y. Li, et al., Chin. Chem. Lett. 35 (2024) 108879. doi: 10.1016/j.cclet.2023.108879
N.A. Magnus, T.M. Braden, J.Y.B. DeBaillie, et al., Org. Process Res. Dev. 16 (2012) 830–835. doi: 10.1021/op300053a
E.A. Iardi, A. Zakarian, Chem. Asian J. 6 (2011) 2260–2263. doi: 10.1002/asia.201100338
S. Feng, Y. Dong, S.L. Buchwald, Angew. Chem. Int. Ed. 61 (2022) e202206692. doi: 10.1002/anie.202206692
B. Li, T. Li, M.A. Aliyu, Z.H. Li, W. Tang, Angew. Chem. Int. Ed. 58 (2019) 11355–11359. doi: 10.1002/anie.201905174
C.L. Allen, J.M.J. Williams, Chem. Soc. Rev. 40 (2011) 3405–3415. doi: 10.1039/c0cs00196a
J. Wang, Y. Chen, S. Miao, C. Yao, K. Zhang, New J. Chem. 48 (2024) 15287–15291. doi: 10.1039/d4nj03410d
X.G. Zhang, Z.C. Yang, J.B. Pan, X.H. Liu, Q.L. Zhou, Nat. Commun. 15 (2024) 4793. doi: 10.1007/s11071-024-09285-5
J.B. Pan, Z.C. Yang, X.G. Zhang, M.L. Li, Q.L. Zhou, Angew. Chem. Int. Ed. 62 (2023) e202308122. doi: 10.1002/anie.202308122
C. Pei, S. Han, H. Wu, B. Li, B. Wang, Chin. J. Chem. 43 (2025) 1255–1262. doi: 10.1002/cjoc.202401208
M. Feng, A.J. Fernandes, R. Meyrelles, N. Maulide, Chem 9 (2023) 1538–1548. doi: 10.1016/j.chempr.2023.03.002
G. Schäfer, C. Matthey, J.W. Bode, Angew. Chem. Int. Ed. 51 (2012) 9173–9175. doi: 10.1002/anie.201204481
V. Pace, L. Castoldi, W. Holzer, Chem. Commun. 49 (2013) 8383–8385. doi: 10.1039/c3cc44255a
H. Zhou, P. Lu, X. Gu, P. Li, Org. Lett. 15 (2013) 5646–5649. doi: 10.1021/ol402573j
D. Fiorito, Y. Liu, C. Besnard, C. Mazet, J. Am. Chem. Soc. 142 (2019) 623–632.
S.C. Galisteo, J. Schörgenhumer, C. Hervieu, C. Nevado, Angew. Chem. Int. Ed. 63 (2024) e202313717. doi: 10.1002/anie.202313717
X. Wang, Y. Dai, H. Gong, Topp. Curr. Chem. 374 (2016) 43. doi: 10.1007/s41061-016-0042-2
C. Tran, A. Abdallah, V. Duchemann, G. Lefèvre, A. Hamze, Chin. Chem. Lett. 34 (2023) 107758. doi: 10.1016/j.cclet.2022.107758
Y.Z. Yang, G.F. Lv, M. Hu, Y.L. Li, J.H. Li, Chin. Chem. Lett. 34 (2023) 108590. doi: 10.1016/j.cclet.2023.108590
J. Liu, X. Tao, Z. Zou, et al., Chin. Chem. Lett. 36 (2025) 110461. doi: 10.1016/j.cclet.2024.110461
J. Li, C. Chen, Y. Dong, et al., Chin. Chem. Lett. 35 (2024) 109732. doi: 10.1016/j.cclet.2024.109732
C.E.I. Knappke, S. Grupe, D. Gärtner, et al., Chem. Eur. J. 20 (2014) 1–16. doi: 10.1002/chem.201390210
X.B. Liu, R.M. Liu, X.D. Bao, et al., Chin. Chem. Lett. 35 (2024) 109783. doi: 10.1016/j.cclet.2024.109783
S. Perveen, G. Zhang, P. Li, Org. Biomol. Chem. 23 (2025) 4006–4023. doi: 10.1039/d5ob00392j
T.Z. Wang, L.Y. Tang, Y.Q. Guan, et al., Chin. Chem. Lett. 36 (2025) 111050. doi: 10.1016/j.cclet.2025.111050
R. Wang, J. Xu, J.X. Li, et al., Chin. Chem. Lett. 34 (2023) 108490. doi: 10.1016/j.cclet.2023.108490
L. Wan, Y. Tong, X. Lu, Y. Fu, Chin. Chem. Lett. 35 (2024) 109283. doi: 10.1016/j.cclet.2023.109283
X.W. Chen, J.P. Yue, K. Wang, et al., Angew. Chem. Int. Ed. 60 (2021) 14068–14075. doi: 10.1002/anie.202102769
C. Li, X.W. Chen, L.L. Liao, et al., Angew. Chem. Int. Ed. 64 (2025) e202413305. doi: 10.1002/anie.202413305
X. Hu, Chem. Sci. 2 (2011) 1867–1886. doi: 10.1039/c1sc00368b
E. Serrano, R. Martin, Angew. Chem. Int. Ed. 55 (2016) 11207–11211. doi: 10.1002/anie.201605162
T. Michiyuki, I. Osaka, K. Komeyama, Chem. Commun. 56 (2020) 1247–1250. doi: 10.1039/c9cc09377j
S. Zheng, D.N. Primer, G.A. Molander, ACS Catal. 7 (2017) 7957–7961. doi: 10.1021/acscatal.7b02795
A. Correa, R. Martin, J. Am. Chem. Soc. 136 (2014) 7253–7256. doi: 10.1021/ja5029793
J.C. Hsieh, C.H. Cheng, Chem. Commun. (2005) 4554–4556. doi: 10.1039/b506903c
S. Zhang, S. Perveen, Y. Ouyang, et al., Angew. Chem. Int. Ed. 61 (2022) e202117843. doi: 10.1002/anie.202117843
S. Perveen, S. Zhang, L. Wang, et al., Angew. Chem. Int. Ed. 61 (2022) e202212108. doi: 10.1002/anie.202212108
L. Wang, T. Li, S. Perveen, et al., Angew. Chem. Int. Ed. 61 (2022) e202213943. doi: 10.1002/anie.202213943
S. Zhang, Y. Ouyang, Y. Gao, P. Li, Acc. Chem. Res. 57 (2024) 957–970. doi: 10.1021/acs.accounts.3c00808
M.M. Xu, Q. Chen, L.H. Xie, J.R. Li, Coord. Chem. Rev. 213421 (2020) 1–27.
Z. Li, G. Zhang, Y. Song, et al., Org. Lett. 25 (2023) 3023–3028. doi: 10.1021/acs.orglett.3c00816
M. Kalek, G.C. Fu, J. Am. Chem. Soc. 139 (2017) 4225–4229. doi: 10.1021/jacs.7b01826
W. Guo, L. Zuo, M. Cui, B. Yan, S. Ni, J. Am. Chem. Soc. 143 (2021) 7629–7634. doi: 10.1021/jacs.1c03182
P. Maity, D.M.S. McAtee, G.P.A. Yap, E.R. Sirianni, M.P. Watson, J. Am. Chem. Soc. 135 (2013) 280–285. doi: 10.1021/ja3089422
R. Yuan, Z. Lin, Organometallics 33 (2014) 7147–7156. doi: 10.1021/om500962a
N.W.M. Michel, A.D.M. Jeanneret, H. Kim, S.A.L. Rousseaux, J. Org. Chem. 83 (2018) 11860–11872. doi: 10.1021/acs.joc.8b01763
R.D. He, C.L. Li, Q.Q. Pan, et al., J. Am. Chem. Soc. 141 (2019) 12481–12486. doi: 10.1021/jacs.9b05224
H. Chen, L. Huang, H. Chen, J. Li, Mol. Catal. 559 (2024) 114088.

Scheme 1 The occurrence and synthetic methods of α-chiral amides and the outline of this work.

Scheme 2 Scope of the asymmetric reductive amidation. Reaction conditions: 1a (0.10 mmol), 2a (0.20 mmol), NiCl2[P(mesityl)3]2 (0.010 mmol), ligand (0.012 mmol), Mn (0.40 mmol), PhOH (0.15 mmol), phenyl acetate (0.20 mL), 20 h at 70 ℃. Isolated yield after silica gel column chromatography. The enantiomeric ratio was determined on a chiral HPLC OJ-H column.

Scheme 3 Demonstrative transformations of α-chiral amide 3v, (Ⅰ) TfOH, toluene, (Ⅱ) Tf2O, 2-F-Py, DCM, (Ⅲ) LiOH, H2O2, THF/H2O.

Scheme 4 Proposed catalytic cycle of nickel-catalyzed asymmetric reductive amidation and explanation of match/mismatch events in case of chiral substrate.

Figure 3 (a) The plausible transition state of enantio‑determining step. (b) IGMH analysis of the two energetically lowest conformations in the enantio‑determining step.
Table 1. Optimization of reaction conditions.a
|
|||
| Entry | Deviation from standard conditions | Yield (%) b | erc |
| 1 | None | 90 | 97:3 |
| 2 | L2 instead of L1 | 81 | 93:7 |
| 3 | L3 instead of L1 | N.R | - |
| 4 | L4 instead of L1 | 51 | 72:28 |
| 5 | L5 instead of L1 | N.R | - |
| 6 | L6 instead of L1 | N.R | - |
| 7 | L7 instead of L1 | N.R | - |
| 8 | NiCl2 instead of NiCl2[P(mesityl)3]2 | 60 | 95:5 |
| 9 | NiCl2(PPh3)2 instead of NiCl2[P(mesityl)3]2 | 65 | 95:5 |
| 10 | 2-MeTHF as solvent | 51 | 92:8 |
| 11 | Ethyl formate as solvent | 75 | 96:4 |
| 12 | w/o PhOH | 63 | 95:5 |
| 13 | 30 ℃ instead of 70 ℃ | N.R | - |
| a Reaction conditions: 1a (0.10 mmol), 2a (0.20 mmol), NiCl2[P(mesityl)3]2 (0.010 mmol), Ligand (0.012 mmol), Mn (0.40 mmol), PhOH (0.15 mmol), phenyl acetate (0.20 mL), 20 h at 70 ℃. b Isolated yield after silica gel column chromatography. c The enantiomeric ratio was determined on a chiral HPLC OJ-H column. |
|||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们


 下载:
下载: