Received Date:
22 May 2025 Accepted Date:
21 August 2025 Revised Date:
18 August 2025 Available Online:
15 July 2026
Abstract:
Extracellular vesicles (EVs), mainly comprising microvesicles (MiV) and exosomes (Exo), were successfully isolated from normal human embryonic kidney cells (HEK293T) and demonstrated specific uptake by human tongue squamous cell carcinoma cells (SCC-9). Initial mechanistic investigations revealed that both MiV and Exo were mainly internalized via endocytic pathways and predominantly relied on the surface proteins of SCC-9 cells for specific uptake. Furthermore, Exo, with better stability and uptake efficiency, were chosen as the carriers of the clinical drug cisplatin (CDDP) for the treatment of tongue cancer. Comprehensive in vitro and in vivo evaluations demonstrated that the Exo-CDDP system exhibited remarkable biocompatibility, mitigated drug-related toxicity and minimized CDDP efflux from tumor cells, and displayed potent anti-tumor efficacy. These findings collectively indicated that HEK293T-derived Exo represent a highly promising drug delivery platform for tongue cancer therapy, while simultaneously offering innovative ideas for the development of versatile Exo-based therapeutic delivery systems.
Extracellular vesicles (EVs), spherical membranous vesicles released by diverse cell types, are primarily classified into three subtypes: microvesicles (MiV), exosomes (Exo) and apoptotic bodies (ApB). Functioning as natural intercellular messengers, EVs demonstrate intrinsic tropism for recipient cells and play crucial roles in mediating tumor-stroma communication, thereby regulating multiple aspects of tumorigenesis and malignant progression [1-6]. The multifaceted therapeutic potential of EVs spans their utility as diagnostic biomarkers, therapeutic agents, and drug delivery carriers [7,8]. Particularly, EVs-based drug carriers have attracted significant research attention due to their capacity for delivery of diverse drugs while enhancing drug stability, evading reticuloendothelial clearance, exhibiting superior biocompatibility and minimal immunogenicity, and promoting drug delivery via innate tissue tropism and cellular uptake mechanisms [9-13].
Currently, researches on EVs as drug delivery carriers primarily focus on their targeting properties, including natural targeting and engineered targeting [14-17]. EVs derived from special cells, such as tumor and stem cells, exhibit intrinsic tumor-natural targeting [18-21]. However, significant concerns remain regarding the direct therapeutic application of these EVs due to their cargo of tumor-promoting molecules, and the stem cell EVs production faces complex culture requirements and low yields [22-25]. Moreover, the engineering and modification processes required to enhance the targeting capabilities of EVs inevitably elevate both technical complexity and production costs [26,27]. Therefore, it is critically important to select EVs that are readily obtainable and devoid of tumorigenic molecular constituents, and exhibit inherent targeting capabilities as clinical drug carriers for tumors to achieve efficient drug delivery.
SCC-9 cells, a type of squamous cell carcinoma (SCC) cell line, are characterized by poor prognosis, high recurrence and metastasis rates, and low survival rates. In clinical practice, cisplatin (CDDP) is the first-line chemotherapeutic agent for tongue cancer treatment [28].
CDDP, a DNA synthesis inhibitor, induces damage to healthy tissues while exerting antitumor effects [29-31]. Furthermore, prolonged CDDP administration triggers the resistance in cancer cells, substantially compromising treatment outcomes. A key mediator of this resistance is P-glycoprotein (P-gp), which promotes cellular efflux of CDDP [32,33]. The severe toxic side effects and acquired drug resistance are major clinical limitations for CDDP monotherapy, often leading to suboptimal chemotherapeutic efficacy. These issues underscore the critical need for rationally designed nanocarrier systems to overcome these therapeutic barriers.
In this study, our findings revealed that MiV and Exo derived from HEK293T cells were specifically internalized by SCC-9 cells. We conducted preliminary investigation into the internalization and specific uptake mechanisms and selected Exo as optimal carriers for CDDP in tongue cancer therapy. The therapeutic effect of this system was systematically evaluated through the in vitro and in vivo experimental analyses.
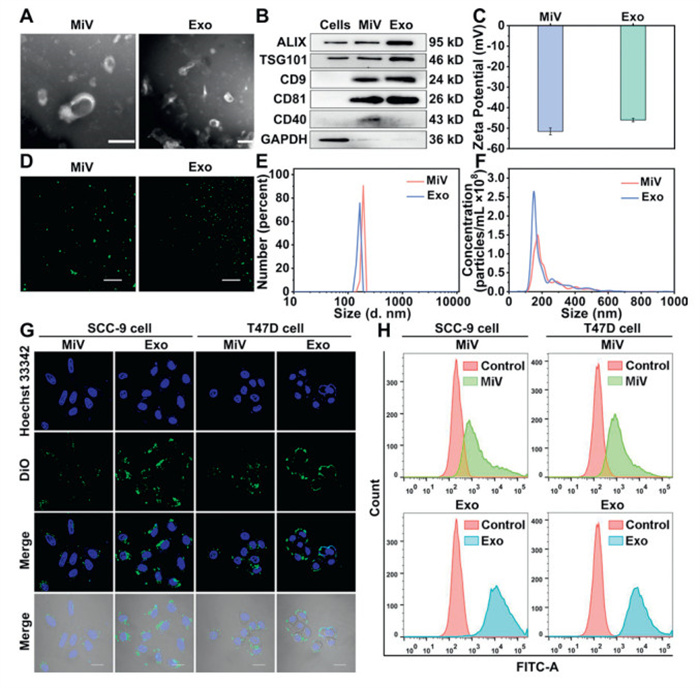
The isolated MiV and Exo were characterized via multiple analytical techniques. Transmission electron microscopy (TEM) analysis revealed the characteristic irregular saucer-like morphologies of the two vesicles, with diameters between 50 nm and 200 nm (Fig. 1A). Both MiV and Exo expressed typical EVs markers, including CD9, CD81, ALIX and TSG101. Notably, the expression of CD40 in MiV was higher than that in Exo (Fig. 1B) [34]. MiV and Exo exhibited negative charges, which originated from their membrane compositions (Fig. 1C). Confocal laser scanning microscopy (CLSM) visualization revealed the circular morphology of both vesicles, with MiV exhibiting larger diameters than Exo (Fig. 1D). Size distribution measured by dynamic light scattering (DLS) and nanoparticle tracking analysis (NTA) consistently also showed that MiV possessed larger diameters compared to Exo (Figs. 1E and F). Among them, the polydispersity index (PDI) of MiV was 0.31 ± 0.07, and that of Exo was 0.23 ± 0.04, suggesting a relatively uniform size distribution for both isolated vesicles. Collectively, these comprehensive characterization data verified the successful isolation and purification of MiV and Exo populations.
Figure 1
Figure 1.
(A) The TEM images of MiV and Exo. Scale bar: 200 nm. (B) Western blot analysis of ALIX, TSG101, CD9, CD81 and CD40 in HEK293T cells, MiV and Exo. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. (C) The zeta potential of MiV and Exo. (D) The CLSM images of DiO-labeled MiV and Exo. Scale bar: 10 µm. (E) The DLS spectra and (F) the NTA spectra of MiV and Exo. (G) The CLSM images of the uptake of MiV and Exo by SCC-9 and T47D cells. Scale bar: 10 µm. (H) The FCM results of MiV and Exo in SCC-9 and T47D cells. Values represent the means ± SD, the experiment was repeated three times.
We evaluated the stability of MiV and Exo at 4 ℃ to inform the selection of optimal drug carriers. Following 7-day storage at 4 ℃, Exo demonstrated significantly less variation in morphologies, zeta potential and size compared to MiV, indicating superior stability of Exo under 4 ℃ (Fig. S1 in Supporting information).
To investigate whether normal cell-derived EVs possess similar specificity to tumor or stem cell EVs, we co-cultured MiV and Exo with ten types of tumor cell lines (MDA-MB-231, MCF-7, T47D, HepG2, PC3, SCC-9, HeLa, A549, BXPC3, SW480). The cellular distribution of DiO labeled EVs was analyzed by CLSM and flow cytometry (FCM). Both MiV and Exo displayed specificity for SCC-9 and T47D cell lines (Figs. 1G and H), with MiV showing preferential accumulation in T47D cells while Exo in SCC-9 cells. It may be related to the surface molecules of EVs and target cells, and the uptake mechanism of target cells [35,36]. Notably, at same concentrations, Exo showed higher internalization than MiV (Figs. S2 and S3 in Supporting information). Therefore, SCC-9 cells were selected for subsequent investigations. In addition, we performed parallel experiments with human normal oral keratinocytes (HOK), the normal counterpart of SCC-9 cells. Quantitative analysis revealed higher EVs uptake in tumor cells compared to normal cells (Fig. S4 in Supporting information), providing compelling evidence for the specific targeting of SCC-9 cells by HEK293T-derived EVs.
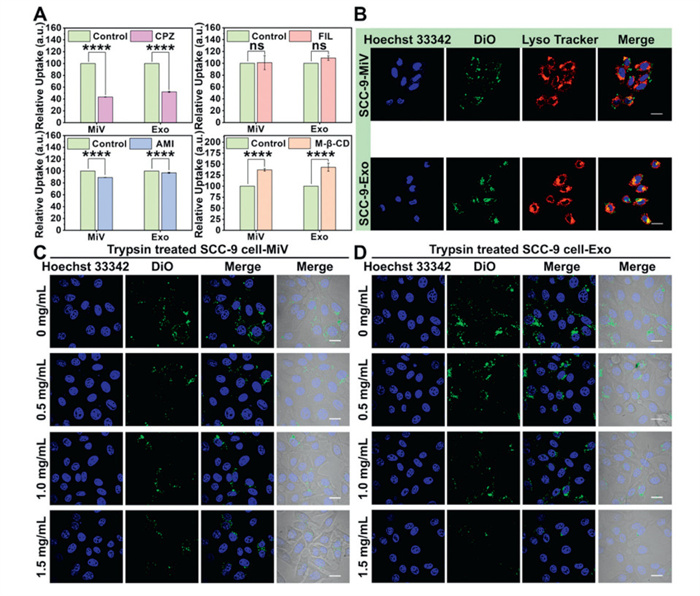
Previous studies have reported the multiple internalization pathways of EVs [37,38]. To elucidate the internalization mechanisms of MiV and Exo, we treated SCC-9 cells with multiple inhibitors. Chlorpromazine hydrochloride (CPZ) and amiloride (AMI) caused reduction in vesicle uptake. In contrast, neither filipin (FIL) nor methyl-β-cyclodextrin (M-β-CD) showed any appreciable effect on internalization (Fig. 2A). These findings suggested that clathrin-mediated endocytosis and macropinocytosis served as the predominant uptake routes for both MiV and Exo. In addition, we observed significant colocalization of internalized MiV and Exo with lysosomes in SCC-9 cells (Fig. 2B), providing additional evidence for an endocytosis-dependent internalization mechanism.
Figure 2
Figure 2.
(A) The relative uptake of MiV and Exo by SCC-9 cells treated with CPZ, AMI, FIL and M-β-CD. (B) MiV and Exo colocalized with lysosomes in SCC-9 cells. Scale bar: 10 µm. The CLSM images of the uptake of (C) MiV and (D) Exo by SCC-9 cells treated with different concentrations of trypsin. Scale bar: 10 µm. Values represent the means ± SD, the experiment was repeated three times. ****P < 0.0001.
Numerous studies have demonstrated that the proteins (integrins, tetra transmembrane protein superfamily, fibronectin) on either cells or EVs can influence EVs uptake specificity [39,40]. To investigate the protein-dependence of MiV and Exo uptake by SCC-9 cells, we performed trypsin digestion experiments targeting SCC-9 cell surface proteins. Treatment with varying trypsin concentrations resulted in substantial reductions in vesicle uptake. The relative fluorescence intensity of MiV decreased from 100% to 59.20%, and that of Exo decreased from 100% to 55.30% (Figs. 2C and D, Fig. S5B in Supporting information), demonstrating the involvement of SCC-9 cell surface proteins in mediating specific EVs internalization. To further elucidate this mechanism, we conversely digested surface proteins on MiV and Exo. Interestingly, trypsin treatment of the vesicles did not significantly impair their cellular uptake (Figs. S5C–E in Supporting information), suggesting that vesicle surface proteins played a minimal role in this specific recognition process. Collectively, these findings indicated that recipient cell surface proteins exerted a more pronounced influence on specific EVs uptake than do vesicle surface proteins.
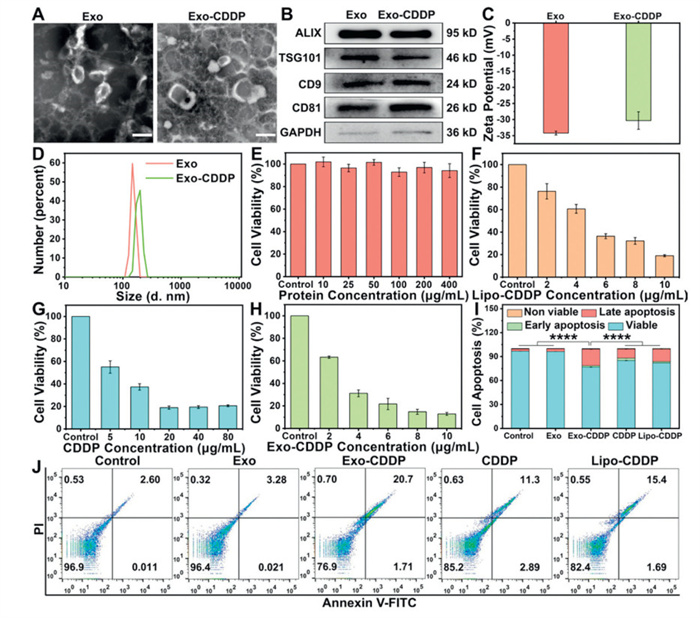
Based on stability and cellular uptake efficiency analysis, Exo were selected as the optimal drug delivery carriers for subsequent investigations. Following CDDP loading via sonication, Exo retained their characteristic vesicular morphology (Fig. 3A). The expression of typical EVs marker proteins (Fig. 3B), and the maintenance of negative surface charge confirmed membrane integrity post-loading (Fig. 3C). Size distribution analysis revealed that Exo-CDDP exhibited an average diameter of 197.6 ± 1.6 nm with a PDI of 0.28 ± 0.03 (Fig. 3D), representing a predictable increase relative to unloaded Exo. These comprehensive characterization data unequivocally demonstrated the successful encapsulation of CDDP within Exo.
Figure 3
Figure 3.
(A) The TEM images of Exo and Exo-CDDP. Scale bar: 200 nm. (B) The expression levels of marker proteins of Exo and Exo-CDDP. (C) The zeta potential and (D) The DLS spectra of Exo and Exo-CDDP. The cytotoxicity of (E) Exo, (F) Lipo-CDDP, (G) CDDP and (H) Exo-CDDP. (I, J) The cell apoptosis analysis of SCC-9 cells after 24 h exposure to different drugs. The equivalent concentrations of Exo and CDDP were 4.81 µg/mL and 2.50 µg/mL, respectively. Values represent the means ± SD, the experiment was repeated three times. ****P < 0.0001.
The cytotoxic effects of each sample on SCC-9 cells were evaluated by the cell counting kit-8 (CCK-8) assay. Exo demonstrated negligible cytotoxicity and excellent biocompatibility, while Lipo-CDDP, free CDDP and Exo-CDDP showed dose-dependent cytotoxicity, with Exo-CDDP exhibiting significantly enhanced growth inhibition (Figs. 3E–H). To further validate the cytotoxicity, we quantified apoptosis rates in SCC-9 cells following treatment. At equivalent concentrations, Exo-CDDP induced markedly higher apoptosis rate than other drugs (Figs. 3I and J), demonstrating the superior pro-apoptotic activity. Complementary calcein acetoxymethyl ester/propidium iodide (calcein-AM/PI) double staining experiments revealed extensive apoptosis in the Exo-CDDP group (Fig. S7B in Supporting information), providing visual confirmation of the enhanced therapeutic efficacy achieved through Exo delivery.
Western blot analysis was conducted to elucidate the protein-level molecular mechanisms underlying Exo-CDDP-induced apoptosis in SCC-9 cells. Exo-CDDP significantly downregulated the anti-apoptotic protein Bcl-2 while upregulating the pro-apoptotic protein Bax. This imbalance in Bcl-2/Bax ratio initiates the mitochondrial apoptotic cascade, leading to caspase-3 activation and subsequent cleavage of its substrate PARP, ultimately resulting in cell death [41,42]. These results confirmed that Exo-CDDP triggered apoptosis through both the intrinsic mitochondrial pathway and caspase-dependent signaling pathways (Fig. S7C in Supporting information).
Drug efflux and the migration/invasion behavior of tumor cells are major factors contributing to ineffective drug therapy and poor clinical outcomes [43]. The overexpression of P-gp in tumor cells can impair drug uptake, leading to increased drug efflux and diminished therapeutic efficacy [44]. Compared with other treatments, Exo-CDDP significantly reduced P-gp expression (Fig. S7D in Supporting information), and the accumulation of CDDP in the cells of this group was the highest (Fig. S8 in Supporting information), confirming the ability of Exo delivery to overcome CDDP efflux and improve anti-tumor efficacy. Exo-CDDP potently inhibited SCC-9 cell migration, achieving a remarkably low migration rate of just 4.53% (Figs. S7E and F in Supporting information). Furthermore, Exo-CDDP demonstrated strong anti-invasive properties (Figs. S7G and H in Supporting information). Collectively, Exo-CDDP effectively inhibited cell migration and invasion without inducing drug efflux.
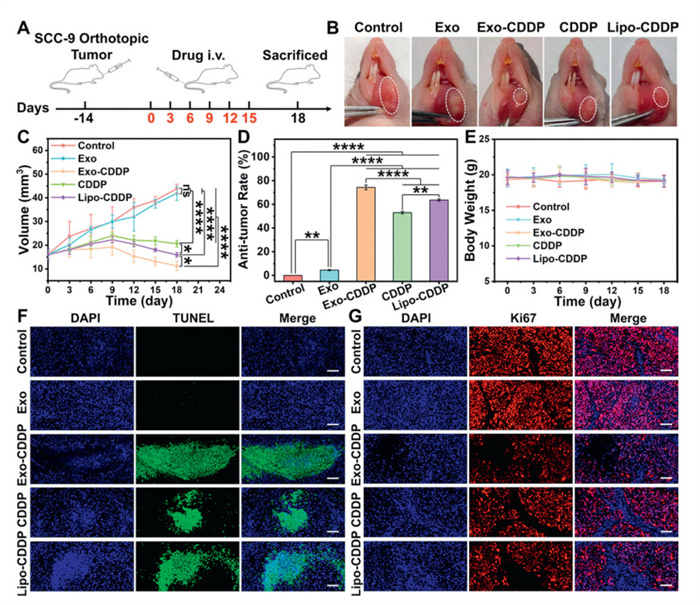
The in vivo anti-tumor activity of Exo-CDDP was evaluated via orthotopic tongue cancer xenograft models. All animal experiments were approved by the Animal Ethical and Welfare Committee of Nanjing Normal University (the ethical approval code: IACUC-20,240,702). During the treatment period, no significant body weight fluctuations were observed in nude mice. Comparative assessment of tumor growth inhibition revealed that Exo-CDDP treatment resulted in significantly reduced tumor size and volume compared to other treatment groups, confirming the potent tumor-suppressive effects (Figs. 4A–E). Histopathological examination via hematoxylin and eosin (HE) staining revealed characteristic apoptotic features in Exo-CDDP-treated tumors, including nuclear condensation, fragmentation, and abnormal chromatin staining (Fig. S9A in Supporting information). TdT-mediated dUTP nick-end labeling (TUNEL) staining further validated the pronounced pro-apoptotic activity of Exo-CDDP (Fig. 4F), while Ki67 staining showed the suppressed tumor cell proliferation (Fig. 4G). These complementary findings provided compelling evidence for the robust in vivo anti-tumor activity of Exo-CDDP.
Figure 4
Figure 4.
(A) The establishment of the orthotopic xenograft tumor models and the administration plan. (B) Tongue tumor images of mice in each treatment group. (C) The changes of tumor volume in each group of mice during treatment. (D) Anti-tumor rate in each treatment group (compared with control group). (E) Body weight changes of mice in each group during treatment. (F) TUNEL staining and (G) Ki67 staining results of tumor tissue in each treatment group of orthotopic xenograft tumor models. Scale bar: 100 µm. Each experimental group contains 4 mice. The equivalent concentration of CDDP was 4 mg/kg. Values represent the means ± SD (n = 4). P < 0.05, **P < 0.01, ****P < 0.0001.
The biocompatibility profile of Exo-CDDP was assessed via organ index measurements and histopathological analysis. CDDP exhibited minimal impact on the organs except the spleen, likely due to the immunosuppressive effect of CDDP [45]. Importantly, Exo-CDDP caused significantly less spleen weight reduction compared to free CDDP (Table S2 in Supporting information), indicating that Exo attenuated the toxicity of CDDP. Histological examination via HE staining confirmed the absence of pathological alterations in Exo-CDDP-treated organs (Fig. S9B in Supporting information). These collective findings validated the excellent biocompatibility of Exo-CDDP.
In this study, we found HEK293T-derived MiV and Exo exhibiting specific uptake by SCC-9 cells via comprehensive co-culture experiments with various tumor cell lines. Preliminary mechanistic studies revealed that both MiV and Exo were internalized by SCC-9 cells primarily through endocytic pathways and achieved specific uptake mainly through interactions with surface proteins of SCC-9 cells. Among the two vesicle types, Exo demonstrated superior characteristics as nanocarriers and were subsequently used to deliver the clinical drug CDDP for tongue cancer therapy. Exo-CDDP system effectively mitigated CDDP-associated toxicity while significantly reducing drug efflux from tumor cells. Both in vitro and in vivo evaluations consistently demonstrated the system's potent anti-tumor activity. These findings collectively establish a safe and efficient drug delivery platform for tongue cancer therapy and significantly advance the clinical applicability of Exo-based delivery systems.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
S. Banerjee, K. Sinha, S. Chowdhury, et al., Chem. Biol. Interact. 279 (2018) 159–170. doi: 10.1016/j.cbi.2017.11.019
Figure 1
(A) The TEM images of MiV and Exo. Scale bar: 200 nm. (B) Western blot analysis of ALIX, TSG101, CD9, CD81 and CD40 in HEK293T cells, MiV and Exo. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. (C) The zeta potential of MiV and Exo. (D) The CLSM images of DiO-labeled MiV and Exo. Scale bar: 10 µm. (E) The DLS spectra and (F) the NTA spectra of MiV and Exo. (G) The CLSM images of the uptake of MiV and Exo by SCC-9 and T47D cells. Scale bar: 10 µm. (H) The FCM results of MiV and Exo in SCC-9 and T47D cells. Values represent the means ± SD, the experiment was repeated three times.
Figure 2
(A) The relative uptake of MiV and Exo by SCC-9 cells treated with CPZ, AMI, FIL and M-β-CD. (B) MiV and Exo colocalized with lysosomes in SCC-9 cells. Scale bar: 10 µm. The CLSM images of the uptake of (C) MiV and (D) Exo by SCC-9 cells treated with different concentrations of trypsin. Scale bar: 10 µm. Values represent the means ± SD, the experiment was repeated three times. ****P < 0.0001.
Figure 3
(A) The TEM images of Exo and Exo-CDDP. Scale bar: 200 nm. (B) The expression levels of marker proteins of Exo and Exo-CDDP. (C) The zeta potential and (D) The DLS spectra of Exo and Exo-CDDP. The cytotoxicity of (E) Exo, (F) Lipo-CDDP, (G) CDDP and (H) Exo-CDDP. (I, J) The cell apoptosis analysis of SCC-9 cells after 24 h exposure to different drugs. The equivalent concentrations of Exo and CDDP were 4.81 µg/mL and 2.50 µg/mL, respectively. Values represent the means ± SD, the experiment was repeated three times. ****P < 0.0001.
Figure 4
(A) The establishment of the orthotopic xenograft tumor models and the administration plan. (B) Tongue tumor images of mice in each treatment group. (C) The changes of tumor volume in each group of mice during treatment. (D) Anti-tumor rate in each treatment group (compared with control group). (E) Body weight changes of mice in each group during treatment. (F) TUNEL staining and (G) Ki67 staining results of tumor tissue in each treatment group of orthotopic xenograft tumor models. Scale bar: 100 µm. Each experimental group contains 4 mice. The equivalent concentration of CDDP was 4 mg/kg. Values represent the means ± SD (n = 4). P < 0.05, **P < 0.01, ****P < 0.0001.

 DownLoad:
DownLoad:

 下载:
下载:





 下载:
下载:
