Citation:
Jiadong Li, Yanduo Liu, Yang Qu. Highly efficient methane-to-low alcohols conversion via ZnO based photocatalysis in aqueous medium[J]. Chinese Chemical Letters,
2026, 37(1): 111741.
doi:
10.1016/j.cclet.2025.111741
Highly efficient methane-to-low alcohols conversion via ZnO based photocatalysis in aqueous medium
English
Highly efficient methane-to-low alcohols conversion via ZnO based photocatalysis in aqueous medium
School of New Energy, Ningbo University of Technology, Ningbo 315336, China
b.
School of Chemistry and Chemical Engineering, Harbin Normal University, Harbin 150025, China
c.
Key Laboratory of Functional Inorganic Materials Chemistry (Ministry of Education), School of Chemistry and Materials Science, International Joint Research Center for Catalytic Technology, Heilongjiang University, Harbin 150080, China
Received Date:
02 April 2025 Accepted Date:
18 August 2025 Revised Date:
28 June 2025 Available Online:
15 January 2026
Abstract:
The photocatalytic oxidation of methane (CH4) to valuable chemicals like low alcohols (CH3OH and C2H5OH) represents a significant technological advancement with implications for energy conversion and environmental purification. A major challenge in this field is the chemical inertness of methane and the strong oxidizing nature of photogenerated holes, which can lead to over-oxidation and reduced selectivity and efficiency. To address these issues, we have developed a sodium-doped zinc oxide (Na-ZnO) modified with cobalt oxide (CoO) catalyst. This catalyst has demonstrated excellent performance in converting methane to low alcohols, achieving a yield of 130 µmol g−1 h−1 and a selectivity of up to 96 %. The doping of Na in ZnO significantly enhances methane adsorption, while the surface-modified CoO effectively captures photogenerated holes, activates water molecules, and uses hydroxyl radicals to activate methane, thus controlling the dehydrogenation degree of methane and preventing the formation of over-oxidized products. This strategy has successfully improved the efficiency and selectivity of photocatalytic methane oxidation to low alcohols, offering a new perspective for the application of photocatalytic technology in energy and environmental fields.
The photocatalytic oxidation of methane (CH4) to high-value low alcohols (CH3OH and C2H5OH), represents a transformative approach for energy conversion and environmental remediation [1–3]. This process not only enhances the utilization efficiency of methane, a rich energy source, but also reduces its emission as a greenhouse gas, having a profound impact on the sustainable development of the chemical industry [4–6]. The photocatalytic process involves multiple steps including light absorption, generation of electron-hole pairs, surface reactions, and product desorption, each of which directly affects the overall catalytic performance [7,8].
The main challenges in photocatalytic oxidation of methane lie in the chemical inertness of methane molecules and the strong oxidizing nature of photogenerated holes. Methane molecules have high C—H bond energy, making them difficult to activate at room temperature and pressure, which requires catalysts with high adsorption capacity to enhance the concentration of methane on the catalyst surface [9–11]. Additionally, the direct involvement of photogenerated holes often leads to over-oxidation of methane, producing non-target products such as carbon dioxide, which limits the selectivity and efficiency of the photocatalytic process [12,13]. Theoretically, by modulating the electronic structure and surface properties of the catalyst, the adsorption capacity of methane and the selectivity of photogenerated holes can be optimized, thereby improving the yield and selectivity of target products.
Alkaline and alkaline earth metal doping is a widely researched and effective strategy for enhancing methane adsorption capacity. From a chemical theory perspective, according to the Hard-Soft Acid-Base (HSAB) theory, alkaline and alkaline earth metals belong to the category of soft bases, while methane molecules have soft acid characteristics [14,15]. There is a natural affinity between them, and this interaction between soft acids and soft bases can promote the formation of relatively strong chemical bonds or mutual attraction, thereby significantly enhancing the adsorption capacity of methane on specific materials or systems. Moreover, the regulation of metal sites can profoundly affect the intrinsic electronic structure of the catalyst [16–19]. During the photocatalytic reaction process, photogenerated electron-hole pairs are generated within the catalyst, and the presence of doped metal ions can effectively regulate the separation efficiency of these photogenerated electron-hole pairs [20,21]. By optimizing the separation process of electrons and holes and reducing their recombination probability, more photogenerated electrons and holes can participate in subsequent chemical reaction steps, greatly enhancing the activity of the entire photocatalytic system and providing more efficient catalytic performance assurance for methane conversion and utilization and other photocatalytic reactions.
In conventional methane conversion reactions, especially in gas-solid systems, the direct hole oxidation mechanism is common. Holes have strong oxidation ability to surpass methane dehydrogenation barriers and activate methane efficiently. However, this mechanism leads to non-selective methane dehydrogenation, easily forming undesired by-products, reducing product selectivity and the practical value of methane conversion. Using water as a reactant and hydroxyl radicals (·OH) as activators in the indirect hole oxidation mechanism is an extremely effective strategy for methane activation [22–24]. Compared with methane dehydrogenation, water is more easily oxidized by holes to generate ·OH, blocking the reaction pathway between methane and holes. The newly formed ·OH has milder oxidation ability than holes, enabling stepwise methane dehydrogenation and ensuring the reaction proceeds as desired. Moreover, by adjusting the electronic structure of the photocatalyst surface and reaction conditions to control the generation rate or amount of ·OH, the activation degree of methane can be effectively regulated, providing a more convenient route for controllable methane activation and the stepwise formation of low alcohols.
Herein, we prepared sodium-doped zinc oxide (Na-ZnO) modified with cobalt oxide (CoO), leveraging its stability and high photocatalytic activity. Experimental results show that this catalyst exhibits excellent performance in the photocatalytic oxidation of methane to low alcohols, with a product yield of 130 µmol g−1 h−1 and a selectivity of up to 96 %. The main advantages of this achievement lie in the doping of Na in the ZnO, which significantly promotes methane adsorption. Meanwhile, the surface-modified CoO can effectively capture photogenerated holes, activate water molecules, and promote methane activation by hydroxyl radicals, thereby effectively controlling the dehydrogenation degree of methane and avoiding the generation of over-oxidized products.
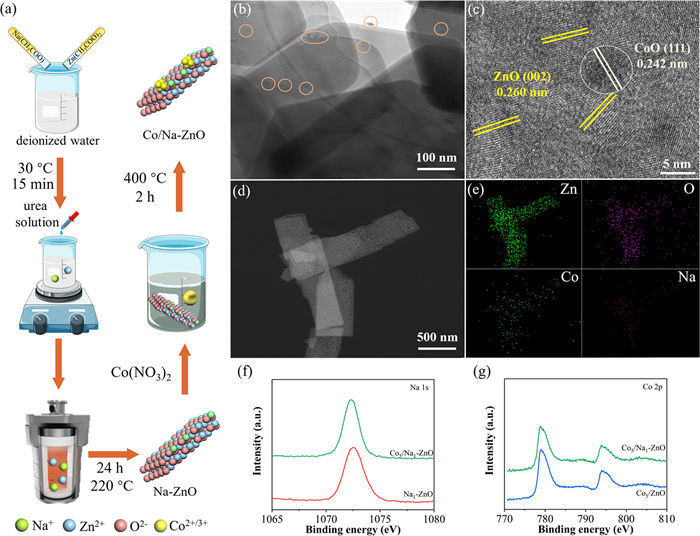
The two-stage fabrication process of Co5/Na1-ZnO through solvent heating followed by calcination is illustrated in Fig. 1a. The X-ray diffraction (XRD) patterns (Fig. S1a in Supporting information) reveal the hexagonal wurtzite structure of ZnO with characteristic (100), (002), and (101) peaks observed at 31.8°, 34.5°, and 36.4°, respectively [25]. The absence of additional secondary phase peaks in the modified ZnO samples (Na1-ZnO, Co5/ZnO, and Co5/Na1-ZnO) suggests that the CoO nanoparticles and Na species are well-dispersed within the ZnO matrix. The concentrations of Na, Co by ICP-MS results were 1.75 mg/L, 8.22 mg/L in Co5/Na1-ZnO (100 mg/L), respectively. The concentrations of Zn by ICP-OES results were 6.86 mg/L in Co5/Na1-ZnO (10 mg/L), respectively. The actual contents percentages of Na, Co and Zn in Na1-ZnO were 1.75 %, 8.22 % and 68.59 %, respectively. UV–vis DRS (diffuse reflectance spectroscopy) (Fig. S1b in Supporting information) show that the light absorption properties of modified ZnO catalysts do not significantly affect, confirming structural stability and homogeneity.
Figure 1
Figure 1.
(a) Schematic diagram of Co5/Na1-ZnO sample preparation process. (b) TEM image, (c) HRTEM image, (d) HAADF-STEM images and (e) The corresponding EDX elemental mapping images of Co5/Na1-ZnO. (f) Na 1s XPS spectra and (g) Co 2p XPS spectra of Na1-ZnO, Co5/ZnO and Co5/Na1-ZnO.
Compared to pristine ZnO and Na-doped ZnO (Na1-ZnO), the scanning electron microscope (SEM) images (Fig. S2 in Supporting information) of the Co5/Na1-ZnO catalysts maintain similar morphology and thickness, indicating homogeneous Na species distribution within the ZnO lattice and uniform dispersion of CoO nanoparticles on the surface. Fig. 1b displays ultrathin interconnected ZnO nanosheets uniformly decorated with CoO nanoparticles of a few nanometers, indicating enhanced dispersion and suppressed agglomeration. The high-resolution transmission electron microscope (HRTEM) image in Fig. 1c reveals lattice fringes matching ZnO (002) and CoO (111) planes, confirming the successful preparation and preserved crystallinity of the Co-Na-ZnO catalysts [26,27]. The absence of visible aggregates in transmission electron microscope (TEM) image indicates homogeneous incorporation of sodium species into the ZnO lattice. The homogeneous elemental distribution of Zn, O, Co, and Na demonstrated by high-angle annular dark-field scanning transmission electron microscope (HAADF-STEM) (Fig. 1d) and energy-dispersive X-ray spectroscopy (EDX) mapping (Fig. 1e) verifies successful sodium incorporation and CoO dispersion.
We conducted X-ray photoelectron spectroscopy (XPS) analysis to investigate the interaction between CoO nanoparticles and Na species within the ZnO system. From the Fig. S3a (Supporting information), the Zn 2p peaks of the ZnO samples modified with Na (Na1-ZnO and Co5/Na1-ZnO) shifted to a higher binding energy compared to the unmodified ZnO, suggesting an alteration in the local coordination environment around the ZnO by incorporated Na [28]. The substitution of Zn2+ by Na+ induced the formation of oxygen vacancies to maintain charge balance, as depicted in Fig. S3b (Supporting information). Furthermore, the presence of Na 1s and Co 2p signals in the XPS spectra (Figs. 1f and g) confirms the successful incorporation of CoO nanoparticles and Na species into the ZnO [29,30].
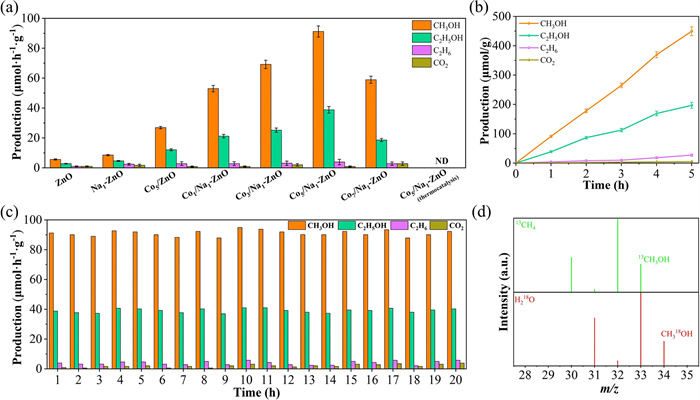
The activity of methane oxidation was evaluated using water as the oxidizing agent. As depicted in Fig. 2, Fig. S4 and Table S1 (Supporting information), the formation of oxidized alcohols (CH3OH and C2H5OH), alkanes (C2H6), and over-oxidation products (CO2) for pure ZnO is minimal. With increasing amounts of Na, the generated products gradually increase. Particularly, Na1-ZnO delivered the optimal photocatalytic activity of CH4 oxidation. Interestingly, the introduction of Na dopants and CoO nanoparticles significantly enhances alcohol production yield and selectivity. The optimized Co5/Na1-ZnO catalyst exhibits methanol and ethanol yields of 91.2 and 38.8 µmol h-1 g-1 respectively, with a combined selectivity of 96.44 % for these two alcohol products. It is noteworthy that no oxidation products are detected under thermal conditions without light, confirming the aqueous oxidation of CH4 is entirely photo-driven and independent of thermal effects. The Na-doped ZnO nanosheets with CoO surface modification exhibit significant advantages in both alcohol yield and selectivity compared to other analogous materials, as detailed in Table S2 (Supporting information). The optimized Co5/Na1-ZnO photocatalyst demonstrated sustained efficiency in CH4 oxidation, as evidenced by time-dependent photocatalytic performance (Fig. 2b and Table S3 in Supporting information) and long-term multi-cycle stability (Fig. 2c and Table S4 in Supporting information). Moreover, as shown in Figs. S5 and S6 (Supporting information), the XRD, DRS and XPS spectra of the Co5/Na1-ZnO sample show no obvious changes before and after reaction, confirming its structural stability and uniformity throughout the reaction process.
Figure 2
Figure 2.
(a) Photocatalytic activity of CH4 conversion over different samples. (b) Time-dependent photocatalytic activity over Co5/Na1-ZnO under light. (c) Production yields in the cyclic tests of Co5/Na1-ZnO. (d) GC–MS spectra of CH318OH and 13CH3OH produced over Co5/Na1-ZnO using an equivalent amount of H218O and 13CH4 as the reactant.
To verify the origin of methanol products, isotope-labeling experiments using 13CH4 and H218O tracers were conducted (Fig. 2d). The signals for isotopically labeled at m/z = 34 and 33 correspond to CH318OH and 13CH3OH, respectively, which confirms that CH4 and water directly transform into methanol during the reaction, excluding interference from other potential carbon and oxygen sources.
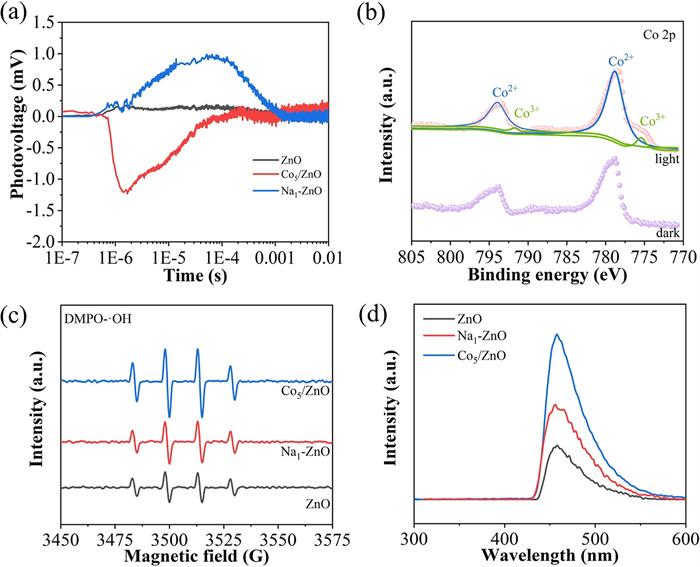
Charge separation is critical for achieving excellent photocatalytic performance over ZnO-based photocatalysts in CH4 oxidation using water as the activator, depending on efficient hole capture and water activation. The time-resolved surface photovoltage (TR-SPV) responses (Fig. 3a) display negligible photocurrent signal for pure ZnO under N2 due to charge recombination in the inert atmosphere [31]. In contrast, the Na-ZnO sample facilitates efficient migration of photogenerated electrons to the Na species surface, prolonging hole residence time on ZnO surface and generating a positive electrical signal. Conversely, the Co/ZnO sample exhibits a distinct negative electrical signal, indicating that photogenerated electrons preferentially accumulate on the ZnO surface. This observation suggests CoO nanoparticles actively capture photogenerated holes during migration, and the prolonged hole lifetimes enable their direct participation in water activation, thereby boosting hydroxyl radical generation. Furthermore, in situ NAP-XPS analysis of Co₅/ZnO (Fig. 3b) reveals the emergence of Co3+species after 5 min of light irradiation, which are not observed under dark conditions. Combined with the TR-SPV results demonstrate that photogenerated holes are effectively captured by CoO nanoparticles, driving their oxidation and subsequent formation of new Co3+ species [32].
Figure 3
Figure 3.
(a) TR-SPV responses of ZnO, Na1-ZnO and Co5/ZnO. (b) In situ NAP-XPS spectra of Co 2p over Co5/ZnO under light irradiation. (c) EPR spectra and (d) fluorescence spectra related to the amounts of hydroxyl radical formed after irradiation for 1 h of ZnO, Na1-ZnO and Co5/ZnO.
Hydroxyl radical trapping tests using DMPO reveal the water activation by photogenerated holes in ZnO-based catalysts (Fig. 3c). ZnO and Na1-ZnO show similar DMPO-·OH electron paramagnetic resonance (EPR) signals, suggesting ·OH generation primarily originates from ZnO surface-accumulated holes [33]. The slightly stronger signal for Na1-ZnO stems from enhanced electron migration via Na doping, extending hole lifetimes. Notably, the DMPO-·OH EPR signal on Co5/ZnO is significantly enhanced, which is ascribed to the prolonged lifetime of photogenerated holes and the boosted activation ability of water by CoO. And the fluorescence spectra related to the amounts of hydroxyl radicals (Fig. 3d) also confirm the effect of CoO in hole capture and hole-mediated water activation.
Steady-state surface photovoltage spectroscopy (SS-SPS) characterizes photogenerated charge dynamics by probing their separation and migration in semiconductors [34]. From Fig. S7a (Supporting information), the SPS signal in ZnO samples modified with Na species and CoO nanoparticles has been significantly improved, further confirming the effectiveness of modification in enhancing the charge separation capability of ZnO. The I-V spectrum reveals that incorporating Na species and CoO nanoparticles enhances the conductivity and charge transport of ZnO (Fig. S7b in Supporting information), which is consistent with the TR-SPV and SS-SPS results.
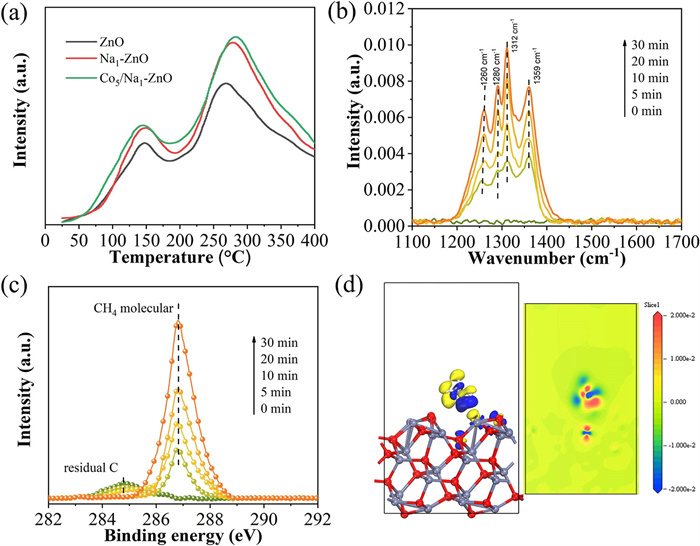
The CH4-TPD curve (Fig. 4a) reveals that Na-doped ZnO (Na1-ZnO) significantly boosts CH4 adsorption capacity compared to pure ZnO, as evident by a distinct desorption peak above 250 ℃. This can be attributed to alkaline nature of Na, which enhances surface affinity through Lewis acid interactions and generates new sites. Additionally, CoO nanoparticle-modified samples (Co5/Na1-ZnO) exhibit CH4 adsorption similar to that of Na1-ZnO, indicating negligible influence of CoO nanoparticles on CH4 adsorption.
Figure 4
Figure 4.
(a) CH4-TPD spectra of Na1-ZnO. (b) In-situ DRIFTS spectra at room temperature with CH4 adsorption in dark for different time Na1-ZnO. (c) C 1s near ambient pressure XPS spectra over Na1-ZnO in Ar purging and upon exposure to 0.45 mbar of CH4 in dark. (d) Different dimensional plots of electron density differences for CH4 adsorbed on Na1-ZnO.
In-situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) analysis further verified the effect of Na species on CH4 adsorption capacity. As seen in Fig. S8 (Supporting information), the characteristic peaks of pure ZnO at 1260, 1280, 1312 and 1359 cm-1 correspond to C—H bonds in adsorbed CH4 molecules [35–37]. Notably, Na₁-ZnO exhibits markedly intensified C—H vibrational signals compared to pure ZnO (Fig. 4b), indicating Na-induced adsorption enhancement through increased surface sites enabling favorable CH4 interactions. To explore the specific adsorption capacity of Na species for CH4, temporal variations in the C 1s NAP-XPS spectra were analyzed under light-protected conditions. As shown in Fig. 4c, a small amount of residual carbon impurities (284.8 eV) exists on the surface of Na1-ZnO. However, these impurities do not compromise the assessment of CH4 adsorption capacity, as their characteristic peaks remain unchanged upon exposure to 0.45 mbar CH4 [38,39]. Furthermore, the time parameters show a significant increase in characteristic peak signals linked to adsorbed CH4 molecules, demonstrating stable adsorption and accumulation on Na1-ZnO. Consistent with in-situ DRIFTS results, NAP-XPS spectra show substantially stronger CH4 adsorption on Na1-ZnO than on pure ZnO, further confirming the specific adsorption capability of Na species (Fig. S9 in Supporting information). The in-situ DRIFTS and NAP-XPS spectra of CH4 adsorbed on Co5/Na1-ZnO show characteristic peaks intensities similar to those observed for Na1-ZnO alone, revealing that the presence of CoO nanoparticles has a negligible effect on CH4 adsorption.
Density functional theory (DFT) calculations elaborate the distinct CH4 adsorption and activation behavior on ZnO and Na1-ZnO surface. The structural configurations of ZnO and Na1-ZnO are established and optimized (Figs. S10 and S11 in Supporting information). The Na1-ZnO exhibits a stronger CH4 adsorption energy (−37.7 eV) than that of pristine ZnO (−29.7 eV), as listed in Table S5 (Supporting information). This aligns with the observed reduction in the distance between CH4 and the Na1-ZnO surface (3.316 Å) compared to pristine ZnO (3.627 Å), indicating enhanced chemisorption stability on Na1-ZnO. Na functions as a structural promoter, elevating the concentration of surface oxygen vacancies and basic sites, which is beneficial for CH4 adsorption. As depicted in Fig. S12 (Supporting information), Differential charge density analysis for pristine ZnO unveils negligible charge transfer between CH4 and the surface, maintaining CH4 in gas molecular state. In contrast, Na1-ZnO surface displays significant charge accumulation between the C atom of CH4 and the Na dopant, forming a covalent σ bond (Fig. 4d), indicating Na-mediated electron redistribution. Moreover, the C—H bond length in CH4 adsorbed on Na1-ZnO elongates from 1.100 Å (free CH4) to 1.201 Å, whereas it remains nearly unchanged (1.110 Å) on pristine ZnO (Table S5 in Supporting information). The elongation of chemical bonds suggests that Na doping, owing to its relatively lower electronegativity compared to Zn, generates electron-enriched sites within the ZnO crystal lattice. This modification effectively increases the surface electron concentration while enhancing interfacial interactions between the catalytic material and CH4 molecules. Consequently, these combined effects substantially weaken the C—H bond strength and lower the activation energy barrier for CH4 activation.
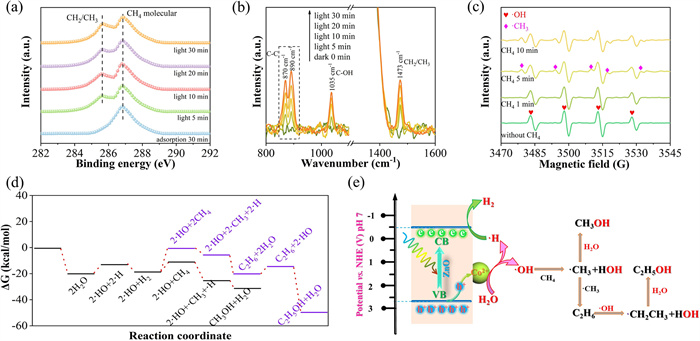
Ulteriorly, C 1s NAP-XPS and in-situ DRIFTS spectra were utilized to investigate photocatalytic CH4 oxidation mechanism over Co5/Na1-ZnO. C 1s NAP-XPS reveals the electronic states of carbon species, while DRIFTS tracks dynamic intermediates. As depicted in Figs. 5a and b, after CH4 adsorption and light exposure, both the C 1s NAP-XPS and in-situ DRIFTS spectra detected characteristic peaks corresponding to hydroxyl- activated methyl/methylene radicals [40–42]. DRIFTS further identifies the existing intermediates, including C—C bonds from methyl radical coupling (potential ethane/ethanol precursors) and C—OH bonds from methyl-hydroxyl interactions (methanol/ethanol sources). All peaks intensified progressively with reaction time, confirming their origin as reaction intermediates rather than impurities. This combined evidence supports a radical-mediated pathway involving sequential C—C coupling and hydroxylation.
Figure 5
Figure 5.
(a) C 1s near ambient pressure XPS spectra over Co5/Na1-ZnO in Ar purging and upon exposure to 0.45 mbar of CH4 under light irradiation. (b) In-situ DRIFTS spectra at room temperature with CH4 oxidation reaction under light irradiation for different time Co5/Na1-ZnO. (c) In-situ EPR spectra of Co5/Na1-ZnO in water with CH4 dissolved under light irradiation. (d) Calculated potential energy diagrams for CH4 oxidation to CH3OH and C2H5OH on Co5/Na1-ZnO hybrid photocatalyst. (e) Schematic diagram illustrates the pathway for photocatalytic coupling conversion of CH4 to CH3OH and C2H5OH on the designed Co5/Na1-ZnO catalyst.
To clarify the role of water in CH4 activation, in-situ EPR with DMPO was conducted (Fig. 5c). In the absence of CH4, only the signal peaks of ·OH with the intensity ratio of 1:2:2:1 were observed. Upon introducing CH4 under light irradiation, a distinct EPR signal with the intensity ratio of 1:1:1:1:1 emerged and intensified, while ·OH signals weakened proportionally. This inverse correlation conclusively demonstrates that ·CH3 generation is entirely dependent on ·OH consumption, proving that water-derived hydroxyl radicals directly mediate CH4 activation in photocatalytic oxidation.
Based on the in-situ experiments and theoretical calculations, the reaction mechanisms on the Co5/Na1-ZnO catalyst were systematically elucidated. The structural configurations of Co5/Na1-ZnO are established and optimized (Fig. S13 in Supporting information). The entire methane oxidation conversion pathway is presented in Figs. 5d and e. CoO nanoparticles capture photogenerated holes under light irradiation, and oxidize H2O to ·OH. In the subsequent reaction process, the ·OH radicals in the liquid phase oxidize CH4 molecules adsorbed on Na sites. The strong oxidative capability of ·OH attacks C—H bond cleavage in CH4, producing ·CH3 radicals and ·H atoms. These generated reactive intermediates will rapidly combine with available ·OH radicals, including ·CH3 reacts to form CH3OH, while ·H recombination regenerates H2O, thus completing the primary methanol synthesis pathway. It is worth noting that there are also some secondary reactions in the entire system. A fraction of ·CH3 radicals will still undergo self-coupling to form C2H6. This unstable state C2H6 intermediate further reacts with ·OH radicals by C—H bond cleavage to produce ·C2H5 radicals. Subsequent ·OH combination converts these radicals into minor amounts of C2H5OH. While a relatively small proportion in the entire reaction pathway, these side reactions reveal the system's chemical complexity and product diversity, providing critical insights for optimizing conditions and improving the selectivity toward the target product.
In summary, we successfully constructed CoO nanoparticle-modified Na-doped ZnO nanosheets (Co5/Na1-ZnO) through a solvent heat treatment and calcination process. In the photocatalytic oxidation of CH4 with H2O as the oxidant, compared with pure ZnO, the co-introduction of Na species and CoO nanoparticles, significantly enhanced the yield and selectivity of low alcohol products (mainly CH3OH and C2H5OH). The yields of CH3OH and C2H5OH reached 91.2 µmol g-1 h-1 and 38.8 µmol g-1 h-1 respectively, with a selectivity accounting for 96.44 %. Moreover, this reaction was completely photo-driven and independent of thermal effects, showing a significant advantage over other similar materials. In-situ tests and theoretical calculations demonstrated that Co5/Na1-ZnO catalyst significantly improved the adsorption capacity of CH4 through Na doping. CoO nanoparticles could capture photogenerated holes and promote the activation of H2O to generate hydroxyl radicals. In-situ EPR experiments confirmed that H2O (in the form of hydroxyl radicals) was the primary reactive species for CH4 activation. The Co5/Na1-ZnO catalyst exhibited excellent performance in the photocatalytic oxidation of CH4 to methanol through its unique heterostructure and efficient separation of photogenerated charges, providing important guidance for the design and optimization of more efficient photocatalytic systems.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The authors acknowledge support from the Zhejiang Provincial Natural Science Foundation of China (No. LQ24B030011), the Ningbo Natural Science Foundation (No. 2023J181), the Open Research Fund of Key Laboratory of Functional Inorganic Materials Chemistry of the Ministry of Education (Heilongjiang University) and the Start-up Funding offered by Ningbo University of Technology to J.D. Li. National Natural Science Foundation of China (No. U24A2071), Postdoctoral Research Start-up Fund (No. 2111224002), and Harbin Normal University Talent Plan (No. 1305124213) to Y.D. Liu.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.111741
.
[1]
C.Q. Han, Y.H. Cao, W. Yu, et al., J. Am. Chem. Soc. 145 (2023) 8609–8620. doi: 10.1021/jacs.3c01317
[2]
J. Duan, S.Y. Fan, X.Y. Li, et al., Chem. Eng. J. 485 (2024) 149904.
[3]
Z.S. Yang, Q.Q. Zhang, H. Song, et al., Chin. Chem. Lett. 35 (2024) 108418.
[4]
Y.H. Jiang, Y.Y. Fan, X.L. Liu, et al., J. Am. Chem. Soc. 146 (2024) 16039–16051. doi: 10.1021/jacs.4c03083
[5]
L.Q. Li, X.W. Shi, L.Y. Liu, et al., Small 21 (2025) 2500835.
[6]
R.X. Zhang, J.L. Shi, L. Fu, et al., ACS Nano. 18 (2024) 12994–13005. doi: 10.1021/acsnano.4c01318
[7]
Z.Y. Huang, C. Guo, Q.X. Zheng, et al., Chin. Chem. Lett. 35 (2024) 109580.
[8]
Y.H. Zhi, C. Gu, H.C. Ji, et al., Chin. Chem. Lett. 36 (2025) 110234.
[9]
S.Y. Nie, L. Wu, X. Wang. J. Am. Chem. Soc. 145 (2023) 23681–23690. doi: 10.1021/jacs.3c07984
[10]
J. Ding, Z.Y. Teng, X.Z. Su, et al., Chem 9 (2023) 1017–1035.
[11]
Q. Zhang, C. Yang, Y.S. Chen, et al., Angew. Chem. Int. Ed. 64 (2025) e202419282.
[12]
T. Luo, Y. Peng, L. Chen, et al., Environ. Sci. Technol. 54 (2020) 10261–10269. doi: 10.1021/acs.est.9b07078
[13]
Y.D. Liu, Y.H. Chen, W.B. Jiang, et al., Research 2022 (2022) 9831340. doi: 10.34133/2022/9831340
Figure 1
(a) Schematic diagram of Co5/Na1-ZnO sample preparation process. (b) TEM image, (c) HRTEM image, (d) HAADF-STEM images and (e) The corresponding EDX elemental mapping images of Co5/Na1-ZnO. (f) Na 1s XPS spectra and (g) Co 2p XPS spectra of Na1-ZnO, Co5/ZnO and Co5/Na1-ZnO.
Figure 2
(a) Photocatalytic activity of CH4 conversion over different samples. (b) Time-dependent photocatalytic activity over Co5/Na1-ZnO under light. (c) Production yields in the cyclic tests of Co5/Na1-ZnO. (d) GC–MS spectra of CH318OH and 13CH3OH produced over Co5/Na1-ZnO using an equivalent amount of H218O and 13CH4 as the reactant.
Figure 3
(a) TR-SPV responses of ZnO, Na1-ZnO and Co5/ZnO. (b) In situ NAP-XPS spectra of Co 2p over Co5/ZnO under light irradiation. (c) EPR spectra and (d) fluorescence spectra related to the amounts of hydroxyl radical formed after irradiation for 1 h of ZnO, Na1-ZnO and Co5/ZnO.
Figure 4
(a) CH4-TPD spectra of Na1-ZnO. (b) In-situ DRIFTS spectra at room temperature with CH4 adsorption in dark for different time Na1-ZnO. (c) C 1s near ambient pressure XPS spectra over Na1-ZnO in Ar purging and upon exposure to 0.45 mbar of CH4 in dark. (d) Different dimensional plots of electron density differences for CH4 adsorbed on Na1-ZnO.
Figure 5
(a) C 1s near ambient pressure XPS spectra over Co5/Na1-ZnO in Ar purging and upon exposure to 0.45 mbar of CH4 under light irradiation. (b) In-situ DRIFTS spectra at room temperature with CH4 oxidation reaction under light irradiation for different time Co5/Na1-ZnO. (c) In-situ EPR spectra of Co5/Na1-ZnO in water with CH4 dissolved under light irradiation. (d) Calculated potential energy diagrams for CH4 oxidation to CH3OH and C2H5OH on Co5/Na1-ZnO hybrid photocatalyst. (e) Schematic diagram illustrates the pathway for photocatalytic coupling conversion of CH4 to CH3OH and C2H5OH on the designed Co5/Na1-ZnO catalyst.

 DownLoad:
DownLoad:

 下载:
下载:






 下载:
下载:
