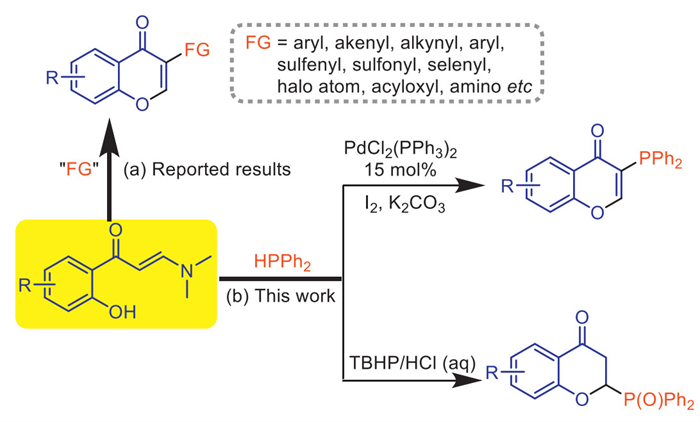
Scheme 1.
Known and the current works for chromone and chromanone synthesis.

Organophosphine compounds, especially tertiary organophosphines, are indispensable chemicals in modern organic synthesis by acting as ligands in numerous catalytic reactions owing to their extraordinary excellent ability of coordination with different metal species and other electron deficient structures [1-5]. In addition, the organic molecules containing phosphorus also have found attractive application in pharmaceuticals, materials and food industry, etc. [6-11]. However, comparing with the plenty of methods for the synthesis of P(Ⅴ) containing molecules such as phosphorylated compounds and phosphates, the synthesis of organophosphines remains as challenge because of the sensitivity of P(Ⅲ) toward oxidation. Therefore, many reactions starting from the P(Ⅲ) reagents result in P(Ⅴ) centered products [12-15]. Currently, the synthesis of organophosphines relies mainly on the reduction of P(Ⅴ) precursors such as phosphine oxides [16-18]. The nucleophilic substitution [19-21], the cross coupling of the P-H phosphines [22-26], and reactions with heteroatom functionalized phosphines [27-31] could also be employed for the synthesis of tertiary phosphines, but require usually either pre-functionalized & sensitive substrates, multi-step operation or harsh reaction conditions. In this context, developing more facile synthetic methods such as direct phosphination of C—H bonds with easily accessible starting materials is urgent for organophosphines.
On the other hand, since chromone is a typical heterocyclic moiety in many natural products and pharmaceuticals [32-35], the synthesis of chromones by employing the cascade vinyl C—H functionalization and chromone annulation of o-hydroxyaryl enaminones has received splendid advances [36,37]. By using proper reaction partners, numerous chromones with functional structure in the C3-site have been practically accessed via the formation of C—C [38-43], C-halogen [44,45], C—N [46,47], C-S/Se [48-52], C—O [53] and C-P [54] bonds as a typical transformation (Scheme 1a). Extensive efforts have also been made to realize the synthesis of chromones with other functionalization modes by such chromone annulation [55,56]. However, like the challenge in most known methods for tertiary phosphine synthesis, the synthesis of 3-phosphinylated chromones via such cascade reactions involving C—H bond functionalization and annulation remains inapplicable. During our longstanding efforts in devising step-economical coupling reactions [57-60], we have proven that the in situ halogenation of a raw C—H bond could be practical for a series of challenging coupling reaction via much simpler operation without relying on the application of prior prepared C-halogen substrates [61,62]. Under the inspiration of the previous works as well as the notable application of enaminone C—H activation or functionalization [63-67], we envision that the in situ halogenation tactic may also be applicable for the C-P(Ⅲ) bond formation via cross coupling process based on the in situ generated C-halogen bonds. Herein, we report the first method on the synthesis of 3-phosphinyl chromones via the reactions of o-hydroxyaryl enaminones and diaryl phosphine wherein the in situ C—H iodination plays the key role in mediating the subsequent C-P coupling. In addition, by modifying reaction conditions, the reaction pathway could be completely switched for the synthesis of novel 2-phosphoryl chromanones without using any metal reagent, enriching the enaminone-based synthetic diversity of chromones and their derivatives (Scheme 1b).
To start the work, the reaction of o-hydroxyphenyl enaminone 1a and diphenyl phosphine 2a was selected to probe applicable conditions. After systematic optimization on reaction parameters, including reaction medium, palladium catalyst species, base species, the loading of reagents, the reaction temperature, as well as reaction time, it was observed that running the reaction in MeCN in the presence of PdCl2(PPh3)2, molecular iodine, K2CO3 at 100 ℃ gave the target product 3-diphenylphosphinyl chromone 3a with optimal yield of 71% (Table S1 in the Supporting Information).
After optimization, the application scope of this method in the synthesis of 3-phosphinyl chromones was investigated by employing different o-hydroxyaryl enaminones to react with diphenyl phosphine under the optimal conditions. As outlined in Scheme 2, the current method displayed good tolerance to the synthesis of products 3 by tolerating a series of different functional groups in the enaminone component. For example, the alkyl (3b, 3c, 3i and 3j), alkoxy (3d and 3e), halo atoms (3f-3h) substituted enaminones reacted with 2 to give diphenyl phosphinylated products with moderate to good yields. In addition, the reactions employing enaminone bearing two different groups in the phenyl ring as well as the naphthyl derived enaminone were also practical substrates in the titled synthesis (3k and 3l). Some strong electron withdrawing group functionalized phenyl enaminones, such as nitro- and Cl-, Br-disubstituted phenyl enaminones were found inapplicable. Subsequently, the reactions using diadamantanyl phospine and phenyl phosphine were also performed, respectively. However, no target chromone products was observed (Scheme 2). Primary attempt by subjecting the reaction residue under oxygen (1 atm) after the completion of 3a formation did not provide 3-phosphoryl chromone. Moreover, no visible new product was observed by running the reaction of diphenyl phosphine oxide with enaminone 1a under the current conditions.

After the synthesis of 3-phosphinyl chromones 3, further efforts in switching the reaction pathways with identical substrates were continuously executed. Interestingly, after extensive attempts in probing reaction conditions (Table S2 in Supporting information), we successfully achieved the selective synthesis of 2-phosphoryl chromanones by running the reaction at room temperature in the presence hydrochloric acid and TBHP. Under the mild and simple conditions, the chromonone products resulting from the novel selective transformations were acquired with generally good to excellent yields (Scheme 3). Besides the reaction with enaminone 1a, typical functional groups in the form of single (4b-4k, 4q) and dual substitution (4l-4p) in the phenyl ring of enaminone substrates were proven generally applicable for the synthesis. Further, the naphthyl derived o-hydroxyaryl enaminone reacted with 2 to provide product 4r with moderated yield (Scheme 3). It should be noted that the synthesis of such phosphoryl chromanones has not be previously accessed by the o-hydroxyaryl enaminone annulation reactions. Afterwards, the product 4a was employed with stoichiometric zinc powder in MeCN at room temperature, but no 2-phosphinyl chromone was observed. Further, the reaction using enaminone without hydroxyl group in the phenyl ring, the (E)-3-(dimethylamino)-1-phenylprop-2-en-1-one with 2 was performed under the standard conditions in Schemes 2 and 3, respectively. Neither the C—H phosphinylation, nor the phosphorylation of the enaminone was observed.
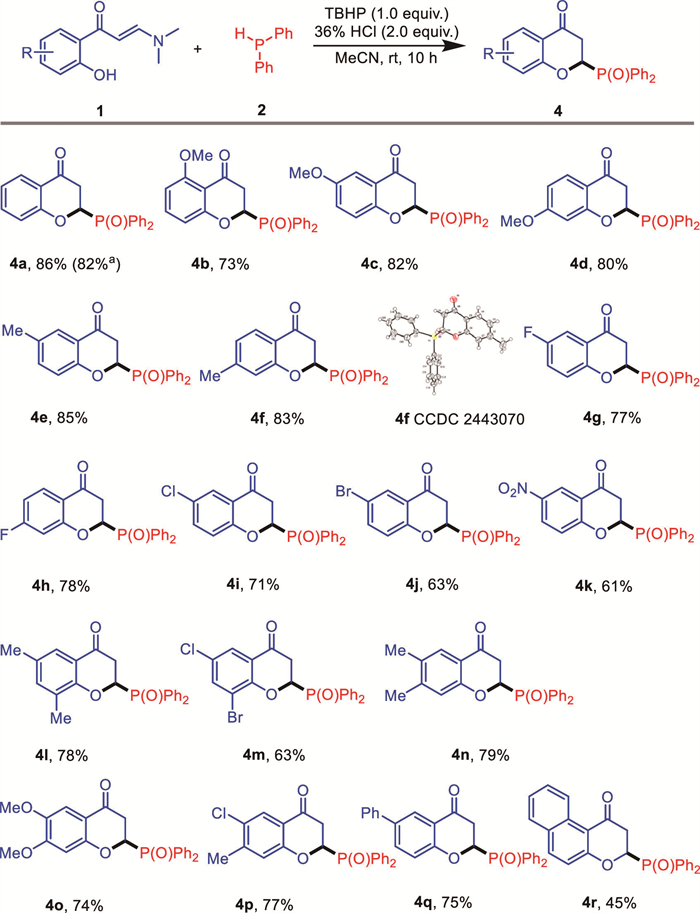
For the sake of investigating reaction mechanisms, a plethora of control experiments were designed and performed. For the reaction providing 3-phosphinyl chromone, employing the chromone 5 and diphenyl phosphine 2 to the standard reaction conditions did not provide 3a (Scheme 4a), confirming that the C—H bond in chromone was inactive for the phosphination. Analyzing the model reaction mixture at the stage of 2 h confirmed the formation of 3-iodochromone (see Supporting information for details). In addition, the reaction using 3-iodochromone 6 and 2 gave the product 3a with high yield (Scheme 4b), suggesting that 3-iodochromone was a key intermediate in the formation of 3. Further explore by subjecting chromone 5 alone to the standard conditions did not provide 6 (Scheme 4c), indicating that chromone 5 could not be the intermediate in the formation of 6. In addition, running the reaction in the presence of free radical scavenger (FRS), including TEMPO, BHT and DEP led to no evident inhibition to the synthesis of 3a (Scheme 4d). On the other hand, for the mechanism toward the selective synthesis of 2-phosphoryl chromanones 4, the reaction of 5 with 2 gave product 4a with excellent yield (Scheme 4e), and analogous results was observed by using diphenyl phosphine oxide 7 as alternative reaction partner (Scheme 4f), implying that the phosphine 2 might first undergo oxidation to generate phosphine oxide 7 to participate the 4a production. Indeed, the reaction of enaminone 1a with 7 could provide 4a with 88% yield in the absence of TBHP (Scheme 4g), further supporting this hypothesis. Employing diphenyl phosphine alone to the conditions led to the formation of diphenyl phosphine oxide 7 with 75% yield (Scheme 4h), and running the model reaction under air allowed the synthesis of product 4a with 67% yield (Scheme 4i). The results suggested that the diphenyl phosphine could be oxidized into corresponding phosphine oxide both by TBHP and air. Finally, the reaction under FRS was also run wherein no inhibition to the formation of 4a was observed, either (Scheme 4j).
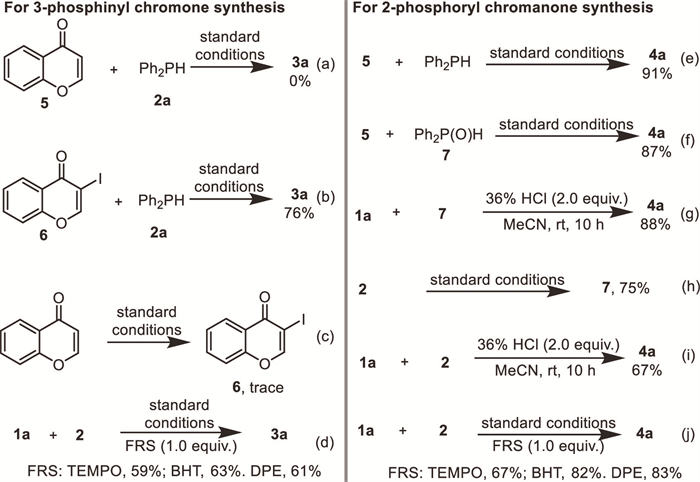
Following these information, the mechanisms for the two selective reactions are proposed (Scheme 5). For the formation of 3-phosphinyl chromones, enaminone 1a couples molecular iodine to provide 3-iodochromone 6 via chromone annulation and electrophilic iodination. In the presence of palladium catalyst, the in situ generated C(sp2)-I bond then undergoes oxidative addition to afford Pd(Ⅱ) inserted intermediated A. The substitution of diphenyl phosphine to the iodine anion leads to intermediate B. After that, a typical reductive elimination of B gave target product 3a and releases Pd(Ⅱ) for continuous catalysis. On the other hand, under the conditions with only hydrochloric acid and TBHP, the enaminone 1a proceeds to chromone 5 via typical acid promoted chromone annulation. Meanwhile, the oxidation of diphenyl phosphine 2 by TBHP or air gives diphenyl phosphine oxide 7. Subsequently, the typical nucleophilic addition of Ph2P(O)H to the polar enone fragment in chromone affords 2-phosphoryl chromonone 4a.
In conclusion, by making use of the in situ halogenation as the key tactic, we have designed and successfully achieved the first synthesis of 3-phosphinyl chromones involving the challenging C-P(Ⅲ) bond formation. In addition, to expande the diversity of functionalized chromone derivatives, we also have realized the selective synthesis of 2-phosphoryl chromanones by modifying the reaction conditions. The step economy of the C-P(Ⅲ) cross coupling without prior preparation of halogenated intermediate for the synthesis of phosphinyl chromones, as well as the metal-free selective synthesis of 2-phosphorylated chromanones provide practical accesses to divergent phosphorus functionalized chromones and chromanones.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Yongli Zhao: Methodology, Investigation. Dingsheng Cao: Methodology. Jie-Ping Wan: Writing – review & editing, Supervision, Funding acquisition, Conceptualization. Yunyun Liu: Writing – original draft, Supervision, Methodology.
The work is financially supported by the National Natural Science Foundation of China (No. 22261026).
Supplementary material associated with this article can be found, in the online version, at doi:
T. Hayashi, Acc. Chem. Res. 33 (2000) 354–362. doi: 10.1021/ar990080f
N.G. Andersen, B.A. Keay, Chem. Rev. 101 (2001) 997–1030. doi: 10.1021/cr000024o
L.C. Liang, Coord. Chem. Rev. 250 (2006) 1152–1177. doi: 10.1016/j.ccr.2006.01.001
X. Pan, W. Cai, Y. Huang, Chin. Chem. Lett. 36 (2025) 110628. doi: 10.1016/j.cclet.2024.110628
J. Feng, L. Nicchio, L. Liu, et al., Org. Chem. Front. 12 (2025) 1695–1707. doi: 10.1039/d4qo02079k
H. Liu, Y. Du, Y. Deng, P.D. Ye, Chem. Soc. Rev. 44 (2015) 2732–2743. doi: 10.1039/C4CS00257A
I. van der Veen, J. de Boer, Chemosphere 88 (2012) 1119–1153. doi: 10.1016/j.chemosphere.2012.03.067
Z.W. Liu, F. Peng, H.J. Wang, et al., Angew. Chem. Int. Ed. 50 (2011) 3257–3261. doi: 10.1002/anie.201006768
M.A. Ali, M.A. Maalouf, D. Feng, et al., Bioorg. Med. Chem. 122 (2025) 118140. doi: 10.1016/j.bmc.2025.118140
T.I. Rokitskaya, R.S. Kirsanov, L.S. Khailova, et al., ChemBioChem 25 (2024) e202300848. doi: 10.1002/cbic.202300848
V. Cecchini, A. Sabatino, B. Contzen, C.M. Avesani, Eur. J. Clin. Nutr. (2025), doi: 10.1038/s41430-025-01600-6.
J.C. Hoefler, D. Jackson, J. Blümel, ACS Catal. 63 (2024) 9275–9287. doi: 10.1021/acs.inorgchem.4c01027
T.A. Rusmore, C. Lander, K.M. Nicholas, Eur. J. Inorg. Chem. 27 (2024) e202300506. doi: 10.1002/ejic.202300506
Y. Tanabe, K. Nakajima, Y. Nishibayashi, Chem. Eur. J. 24 (2018) 18618–18622. doi: 10.1002/chem.201805129
S.J. Tereniak, C.R. Landis, S.S. Stahl, ACS Catal. 8 (2018) 3708–3714. doi: 10.1021/acscatal.8b01009
M. You, Z. Zhang, C. Chen, et al., Green Chem. 27 (2025) 3743–3750. doi: 10.1039/d5gc00477b
K. Yin, M. Wei, Z. Wang, W. Luo, L. Li, Org. Lett. 25 (2023) 5236–5241. doi: 10.1021/acs.orglett.3c01690
C. Laye, J. Lusseau, F. Robert, Y. Landais, Adv. Synth. Catal. 363 (2021) 3035–3043. doi: 10.1002/adsc.202100189
J.M. Brown, J.C.P. Laing, J. Organometal. Chem. 529 (1997) 435–444. doi: 10.1016/S0022-328X(96)06660-0
S. Singh, K.M. Nicholas, Chem. Commun. 1998 (1998) 149–150.
V.C. Chan, M. Chiu, R.G. Bergman, F.D. Toste, J. Am. Chem. Soc. 131 (2009) 6021–6032. doi: 10.1021/ja9014887
Y. Xiao, X. Yang, H. Li, et al., Org. Lett. 26 (2024) 10564–10569. doi: 10.1021/acs.orglett.4c03951
Y. Chai, Y.L. Tian, J.H. Jia, X.C. Wang, Z.J. Quan, Chem. Commun. 61 (2025) 6138–6141. doi: 10.1039/D5CC00210A
J. Sun, H. Tong, Y. Yan, et al., Org. Lett. 26 (2024) 7414–7418. doi: 10.1021/acs.orglett.4c02747
J. Sun, Y. Yan, X. Chen, Z. Huang, Y. Huang, Chem. Sci. 15 (2024) 6943–6948. doi: 10.1039/d4sc00446a
W. Liu, H. Hou, H. Jing, et al., Org. Lett. 25 (2023) 8350–8355. doi: 10.1021/acs.orglett.3c03473
C. Wang, M. Tang, Y. Wang, S. Huang, L.G. Xie, Org. Lett. 26 (2024) 8154–8158. doi: 10.1021/acs.orglett.4c03049
L. Dong, B. Zhong, Y.S. Zhang, J.D. Yang, J.P. Cheng, Chem. Commun. 60 (2024) 12549–12552. doi: 10.1039/d4cc04750h
J. Ma, L. Wang, Z. Duan, Org. Lett. 24 (2022) 1550–1555. doi: 10.1021/acs.orglett.2c00220
H. Sun, J. Wang, Z. Du, et al., Org. Lett. 26 (2024) 1618–1622. doi: 10.1021/acs.orglett.4c00001
G.D. Xia, Z.K. Liu, Y.L. Zhao, F.C. Jia, X.Q. Hu, Org. Lett. 25 (2023) 5279–5284. doi: 10.1021/acs.orglett.3c01824
V.M. Patil, N. Masand, S. Verma, V. Masand, Chem. Biol. Drug Des. 98 (2021) 943–953. doi: 10.1111/cbdd.13951
C.F.M. Silva, D.C.G.A. Pinto, A.M.S. Silva, ChemMedChem 11 (2016) 2252–2260. doi: 10.1002/cmdc.201600359
R.S. Keri, S. Budagumpi, R.K. Pai, R.G. Balakrishna, Eur. J. Med. Chem. 78 (2014) 340–374. doi: 10.1016/j.ejmech.2014.03.047
A. Gaspar, E.M.P.J. Garrido, F. Borges, J.M.P.J. Garrido, ACS Omega 9 (2024) 21706–21726. doi: 10.1021/acsomega.4c00771
L.L. Mao, Y. Liu, J.P. Wan, Chin. Chem. Lett. 36 (2025) 110784. doi: 10.1016/j.cclet.2024.110784
L. Fu, J.P. Wan, Asian J. Org. Chem. 8 (2019) 767–776. doi: 10.1002/ajoc.201900196
Z.Q. Zhu, J.Y. Hu, Z.B. Xie, J. Tang, Z.G. Le, Adv. Synth. Catal. 364 (2022) 2169–2173. doi: 10.1002/adsc.202200304
S. Han, C. Fang, Y. Zhang, et al., Eur. J. Org. Chem. 27 (2024) e202400401.
M. Zhang, M. Liu, Y. Qiu, et al., Adv. Synth. Catal. 366 (2024) 2363–2369. doi: 10.1002/adsc.202400093
F. Ahmad, P.K. Ranga, S. Fatma, R.V. Anand, J. Org. Chem. 89 (2024) 12104–12117. doi: 10.1021/acs.joc.4c01006
P.N. Bagle, M.V. Mane, S.P. Sancheti, et al., Org. Lett. 21 (2019) 335–339. doi: 10.1021/acs.orglett.8b03989
S. Mkrtchyan, V.O. Iaroshenko, J. Org. Chem. 86 (2021) 4896–4916. doi: 10.1021/acs.joc.0c02294
J.S.S. Neto, F.T. Coelho, C.V. Doerner, et al., Chem. Asian J. 19 (2024) e202300852. doi: 10.1002/asia.202300852
Y. Lin, J. Jin, C. Wang, J.P. Wan, Y. Liu, J. Org. Chem. 86 (2021) 12378–12385. doi: 10.1021/acs.joc.1c01347
W. Hu, X. Diao, J. Yuan, et al., J. Org. Chem. 89 (2024) 644–655. doi: 10.1021/acs.joc.3c02399
Z.W. Wang, Y. Zhang, Y.E. Qian, et al., J. Org. Chem. 87 (2022) 1477–1484. doi: 10.1021/acs.joc.1c02796
T. Zhou, J. Zhou, Y. Liu, J.P. Wan, F.E. Chen, Chin. Chem. Lett. 35 (2024) 109683. doi: 10.1016/j.cclet.2024.109683
T. Guo, C. Gao, Z. Li, et al., Chem. Commun. 60 (2024) 14866–14869. doi: 10.1039/d4cc04609a
K. Chen, Y. Liu, J. Yu, et al., J. Org. Chem. 89 (2024) 8632–8640. doi: 10.1021/acs.joc.4c00567
J. Du, S.Y. Zhao, Z.C. Hu, Z.B. Dong, Asian J. Org. Chem. 12 (2023) e202300499. doi: 10.1002/ajoc.202300499
T. Zhang, W. Yao, J.P. Wan, Y. Liu, Adv. Synth. Catal. 363 (2021) 4811–4816. doi: 10.1002/adsc.202100617
Y. Guo, Y. Xiang, L. Wei, J.P. Wan, Org. Lett. 20 (2018) 3971–3974. doi: 10.1021/acs.orglett.8b01536
D. Cao, J.P. Cao, Y. Liu, Adv. Synth. Catal. 366 (2024) 3670–3675. doi: 10.1002/adsc.202400587
S.G. Lei, Y. Zhou, L.S. Wang, et al., Org. Chem. Front. 10 (2023) 4843–4847. doi: 10.1039/d3qo01149f
Y. Lin, J.P. Wan, Y. Liu, J. Org. Chem. 88 (2023) 4017–4023. doi: 10.1021/acs.joc.3c00206
C. Wang, Y. Liu, J.P. Wan, Org. Lett. 27 (2025) 3983–3987. doi: 10.1021/acs.orglett.5c00946
Z. Wang, L. Gan, Z. Song, Y. Liu, J.P. Wan, Chin. J. Chem. 42 (2024) 3041–3046. doi: 10.1002/cjoc.202400664
X. Li, Y. Liu, J.P. Wan, J. Org. Chem. 89 (2024) 16049–16054. doi: 10.1021/acs.joc.4c02127
D. Chen, J.P. Wan, Y. Liu, Org. Lett. 27 (2025) 2371–2376. doi: 10.1021/acs.orglett.5c00213
L. Fu, Z. Xu, J.P. Wan, Y. Liu, Org. Lett. 22 (2020) 9518–9523. doi: 10.1021/acs.orglett.0c03548
Y. Zhao, D. Cao, J.P. Wan, Y. Liu, Asian J. Org. Chem. 14 (2025) e202400599. doi: 10.1002/ajoc.202400599
L. Gao, M. Wang, H. Ren, et al., J. Org. Chem. 90 (2025) 116–123. doi: 10.1021/acs.joc.4c01956
C. Yang, B. Li, P. Shi, et al., Org. Lett. 27 (2025) 2964–2969. doi: 10.1021/acs.orglett.5c00556
C. Yang, X. Zhang, X. Fan, Org. Lett. 26 (2024) 6602–6607. doi: 10.1021/acs.orglett.4c02197
M. Liu, Z. Zhang, Y. Xu, et al., Adv. Synth. Catal. 367 (2025) e202500172. doi: 10.1002/adsc.202500172
H. Guo, Y. Liu, J.P. Wan, Green Synth. Catal. 6 (2025) 206–210.

Scheme 2 Scope on the synthesis of 3-phosphinyl chromones. a Yield from 1 mmol-scale reaction.

Scheme 3 Scope on the synthesis of 2-phosphoryl chromanones. a Yield from 1 mmol-scale reaction.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:


 下载:
下载: