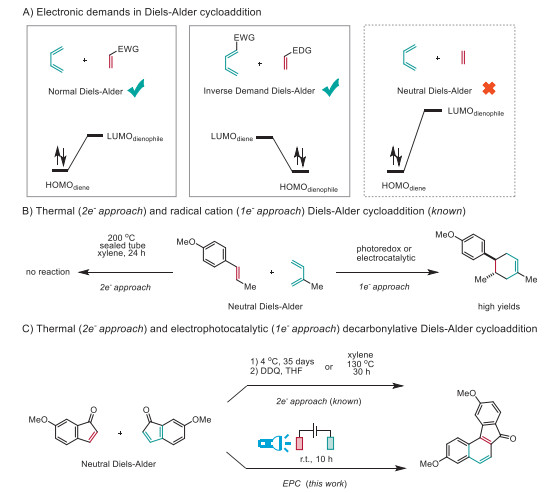
Scheme 1.
The Diels-Alder cycloaddition.
Electrophotocatalytic decarbonylative [4+2] cyclization of indenones
Guo-Cai Yuan , Ming-Gang Li , Sha Yang , Kanghui Song , Chen-Xu Gong , Shuang-Jun Zhu , Yuanming Li , Ke-Yin Ye

The Diels-Alder reaction [1] is one of the most powerful carbon-carbon bond-forming reactions that forges a diene and a dienophile into a new ring system with high synthetic fidelity [2-4]. Among many factors, the electronic demands of the reaction partners stand dominant. For instance, the normal Diels-Alder reaction entails an electron-rich diene and an electron-deficient dienophile because of a constructive overlap between the highest-occupied molecular orbital (HOMO) of the diene with the lowest-unoccupied molecular orbital (LUMO) of the dienophile (Scheme 1A) [5-7]. Conversely, the inverse Diels-Alder reaction operates with an electron-deficient diene and electron-rich dienophile [8-10]. In the absence of enough electronic bias, forcing conditions and significantly longer reaction time are necessary to compensate for the enlarged energy gap between HOMO and LUMO for a neutral Diels-Alder reaction to proceed (Scheme 1B) [11]. The sluggish reactivity of the thermal reaction (2e-approach) can be profoundly facilitated via photoredox [12,13] or electrocatalytic [14,15] enabled radical cation (1e-approach) Diels-Alder reaction [16-18] with several orders of magnitude enhanced reaction rates. Despite these advances, the generality and synthetic utility of the radical cation Diels-Alder reaction is still much less explored.
Among diverse extended fluorenones [19], benzo[c]fluorenones are promising photocatalysts [20,21] and active organic materials [22], but limited synthetic accessibility greatly impeded their further investigations. The neutral Diels-Alder cycloaddition of two indenones is a straightforward approach, but only occurs over 35 days when stored in a fridge [23]. The subsequent extrusion of one carbon monoxide followed by the chemical oxidation with superstoichiometric DDQ (2,3-dichloro-5,6-dicyano-1,4-benzoquinone) resulted in the formation of benzo[c]fluorenone (Scheme 1C). The reaction rate was largely accelerated upon heating at 130 ℃, but still needed 30 h for the reaction to complete [24]. Unfortunately, we found the generality and efficiency of this approach difficult to achieve in our hands (vide infra).
Electrophotocatalysis (EPC) [25-28] integrates electrochemical [29-32] and photochemical [33-35] energy to address challenging transformations that are otherwise difficult or impossible to achieve [36-38]. We envision that EPC may serve as a new solution for benzo[c]fluorenones through a consecutive photocatalytic decarbonylative Diels-Alder cycloaddition and electrochemical oxidative aromatization. Unlike the harsh thermal conditions, our reaction readily occurs at ambient temperature to afford various benzo[c]fluorenones that can be further elaborated as competent photocatalysts both in the oxidation of benzhydrol and also as an auto-photocatalyst [39-41] in the current electrophotocatalysis.
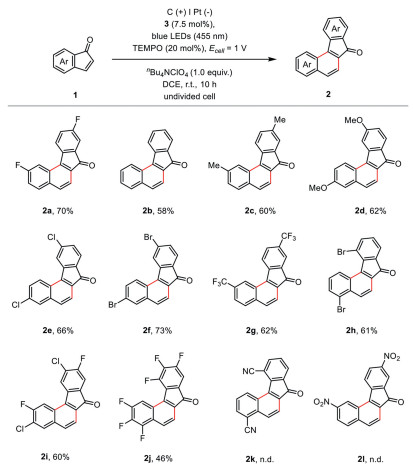
We initiated our study by optimizing the electrophotocatalytic conditions for the decarbonylative [4 + 2] cyclization of indenone (1a). Constant-cell potential electrolysis (Ecell = 1 V) was performed with 20 mol% of 2, 2, 6, 6-tetramethylpiperidinooxy (TEMPO) as the mediator in an undivided cell equipped with a carbon cloth anode and a platinum plate cathode. A 10 W blue LED (455 nm) was employed as the light source with 7.5 mol% of organic dye [Mes-Acr+]ClO4 (3) [42-44] as the catalyst in DCE at room temperature. Under these conditions, the desired benzo[c]fluorenones 2a was isolated in 70% yield (Table 1, entry 1). The formation of product 2a was not detected in the absence of light or photocatalyst 3 (entries 2 and 3). Screening of photocatalysts (entries 4–6) suggested the organic dye [Mes-Acr+] (3 and 4), outcompeted Ir(ppy)3 (5) and Ru(bpy)32+ (6). In the absence of electric current (entry 7) or TEMPO (entry 8), however, moderate yields were still observed, suggesting the inclusion of the TEMPO-mediated electrolysis accelerated this transformation. Other redox mediators, such as ferrocene (entry 9) and Ph3N (entry 10), were also examined but were less effective.
 DownLoad:
CSV
DownLoad:
CSV
 |
||
| Entry | Deviation from standard conditions | Yield (%)b |
| 1 | None | 56 (70)c |
| 2 | No light | n.d. |
| 3 | No [Mes-Acr+]ClO4- (3) | n.d. |
| 4 | [Mes-Acr+]BF4- instead of 3 | (67)c |
| 5 | Ir(ppy)3 instead of 3 | 25 |
| 6 | [Ru(bpy)32+]2PF6- instead of 3 | 10 |
| 7 | No electricity | 38 |
| 8 | No TEMPO | 30 |
| 9 | Ferrocene instead of TEMPO | 28 |
| 10 | Ph3N instead of TEMPO | 34 |
| a Reaction conditions: 1a (0.2 mmol, 2.0 equiv.), 3 (0.0075 mmol, 7.5 mol%), TEMPO (0.02 mmol, 20 mol%), nBu4NClO4 (0.1 mmol, 1.0 equiv.), a carbon cloth anode, a platinum plate cathode, Ecell = 1 V, a 10 W blue LED (455 nm), 10 h, r.t., under an N2 atmosphere. b Yields determined by 19F NMR analysis using PhOCF3 as the internal standard. c Isolated yield. | ||
The generality of this electrophotocatalytic decarbonylative [4 + 2] cyclization was then delineated (Scheme 2). Various indenones substituted with electron-donating Me (2c), MeO (2d), and electron-withdrawing Cl (2e), Br (2f, 2h), CF3 (2g), and fluorinated (2a, 2i, 2j) groups were all readily transformed into the corresponding benzo[c]fluorenones. However, indenones with cyano and nitro groups were not reactive in this reaction, probably due to their high oxidation potentials. We have also tried to use benzo[b]thiophene sulfoxide or benzo[b]thiophene sulfone as the substrate under standard conditions. However, no anticipated desulfonylative [4 + 2] cyclization products were generated. Attempts to achieve the electrophotocatalytic decarbonylative [4 + 2] cyclization between two different indenones were not successful because of the competing homo-dimerization reaction (see Supporting information for details).
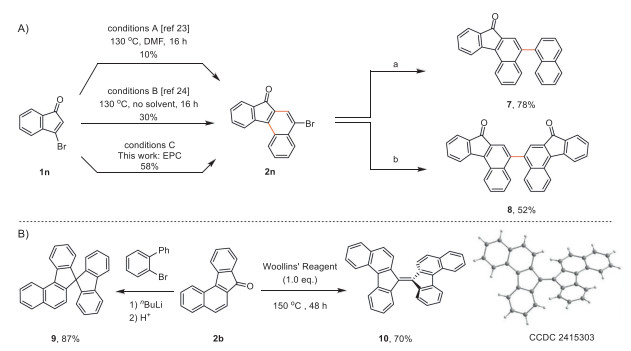
The survey of literature revealed that 3–bromo-indenone (1n) is a challenging substrate for the synthesis of the corresponding benzo[c]fluorenone using the existing methods (Scheme 3). For instance, the thermal approach required the reaction to be performed at 130 ℃ in DMF but only afforded benzo[c]fluorenone (2n) in 10% yield (conditions A) [23]. The solvent-free thermolysis increased the yield to 30% (conditions B) [24]. By contrast, 1n proceeded smoothly with the anticipated electrophotocatalytic decarbonylative [4 + 2] cyclization in 58% yield at ambient temperature (conditions C). The obtained 2n readily proceeded with Suzuki-Miyaura cross-coupling (7) and Ni-mediated dimerization (8). In addition, the carbonyl group in benzo[c]fluorenones can also be readily derivatized. For instance, 2–bromo-1,1′-biphenyl was first treated with nBuLi and then reacted with benzo[c]fluorenone (2b) to give the unsymmetric spirobifluorene derivative (9) as a potential optoelectronic material [22,45]. The carbonyl coupling reaction of 2b with Woollins' reagent gave the dimer 10 in 70% yield, which can be used as an electron acceptor and represents the planar fragmentation of C60.
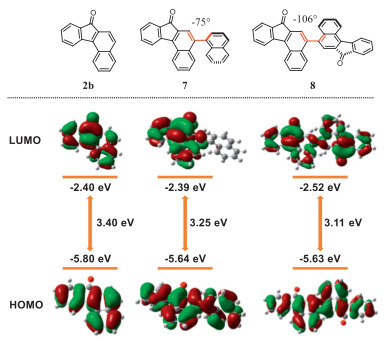
To explore benzo[c]fluorenones as potential carbonyl photocatalysts, the photophysical and redox properties of compounds 2b, 7, and 8 were further investigated (Table 2). The maximum absorption and maximum emission of the parent benzo[c]fluorenone 2b appeared at 426 and 554 nm, respectively. Compounds 7 and 8 displayed similar photophysical properties. The photochemistry of benzo[c]fluorenones is primarily dominated by the triplet excited state with a lifetime in the nanosecond range, i.e., 3.7 ns for 2b, 5.0 ns for 7, and 1.8 ns for 8, respectively. When photoexcited, the benzo[c]fluorenones 2b and 7 become competent oxidants (1.24 V for 2b and 1.42 V for 7, see Supporting information for details). Unfortunately, the low solubility of 8 in most common organic solvents inhibited the determination of its redox properties.
DownLoad:
CSV
| Compd. | λabs (nm) | λem (nm) | τT (ns) | Ered (V) | E*red (V) |
| 2b | 426 | 554 | 3.7 | −1.40 | 1.24 |
| 7 | 440 | 566 | 5.0 | −1.15 | 1.42 |
| 8 | 446 | 566 | 1.8 | ‒ | ‒ |
DFT calculations at the B3LYP/6–31G(d) level were performed to investigate the electronic properties of benzo[c]fluorenones (Fig. 1). The results indicate that the HOMO and LUMO of benzo[c]fluorenones are uniformly distributed across the entire molecular skeleton. The LUMO energy levels for all benzo[c]fluorenones are approximately −2.40 eV. Compared with the 2b, the HOMO energy level increases with the extra incorporation of naphthalene (−5.64 eV) and dimerization (−5.63 eV), respectively. These calculations agree with the experimental results of fluorescence spectrum and CV studies.
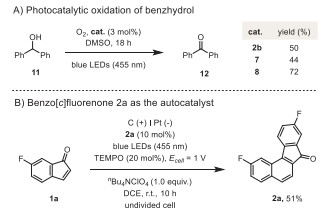
The performance of these benzo[c]fluorenones in the photocatalytic oxidation of benzhydrol (11) was investigated. While only moderate yields of benzophenone (12) were obtained with 2b and 7, using 10 mol% of photocatalyst 8 afforded the target product in 72% yield (Scheme 4A). These results further inspired us to probe the possibility of using benzo[c]fluorenone as the potential autocatalyst in this electrophotocatalytic decarbonylative [4 + 2] cyclization of indenones (Scheme 4B). Indeed, replacing photocatalyst Mes-Acr+ (3) by 2a under the otherwise same conditions still resulted in the formation of the desired product 2a in 51% yield, wherein the weight of the initially added 2a has been deduced. Therefore, the catalytic loading of 2a could be further reduced to 2 mol% while retaining a similar catalytic efficiency, indicative of a virtual autocatalysis scenario (see Supporting information for details).
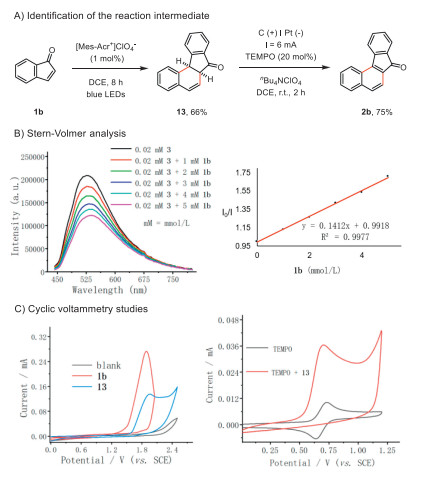
To shed light on the mechanism, several mechanistic experiments were performed (Fig. 2). It was found that the indanone (1b) proceeded with the Mes-Acr+ catalyzed photochemical decarbonylative [4 + 2] cycloaddition to afford the dihydro-benzo[c]fluorenone (13), which was readily oxidized under the electrochemical conditions (Fig. 2A). The Stern-Volmer analysis revealed that the indanone (1b) readily quenched the photo-excited state of Mes-Acr+ and thus is suggestive of a single-electron transfer process (Fig. 2B). Consistent with this hypothesis, cyclic voltammetry studies showed indanone (1b, Ep/2 = 1.65 V versus SCE) was prone to oxidation (Fig. 2B). Regarding the follow-up electrochemical oxidation of the dihydro-benzo[c]fluorenone (13), a catalytic current was observed when 13 was added to TEMPO (Fig. 2C). Therefore, TEMPO functioned as a redox mediator in the final dehydrogenation step.
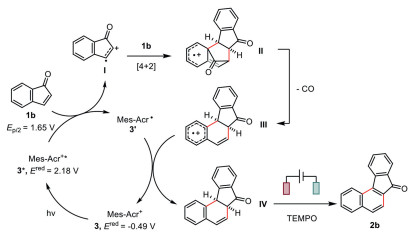
A possible mechanism for this electrophotocatalytic decarbonylative [4 + 2] cyclization of indenones was proposed (Scheme 5). Irradiation of the organic dye Mes-Acr+ (3) leads to its highly oxidizing excited state 3* [44], whose oxidation potential (Ered = 2.18 V vs. SCE in MeCN) is sufficiently positive to oxidize the indenone (1b). The resulting radical cation (Ⅰ) is then activated toward [4 + 2] cycloaddition to afford the radical cation Diels-Alder product (Ⅱ), which readily extrudes one carbon monoxide to release ring strain [23]. This new radical cation (Ⅲ) then abstracts an electron from the acridinyl radical Mes-Acr• (3′) to form the dihydro-benzo[c]fluorenone (Ⅳ) and meanwhile regenerates the ground-state catalyst (3). The final TEMPO-mediated electrochemical dehydrogenation affords the anticipated benzo[c]fluorenone (2b).
In summary, we have unraveled an electrophotocatalytic decarbonylative [4 + 2] cyclization of indenones for facile access toward various benzo[c]fluorenones. This approach integrates a photocatalytic radical cation Diels-Alder reaction and an electrochemical oxidative aromatization that overcomes limitations in a thermal approach, including high temperature, long reaction time, and low yields. The obtained benzo[c]fluorenones are promising photocatalysts in the photocatalytic oxidation of benzhydrol and can also function as an auto-photocatalyst in the current electrophotocatalysis. We anticipate that the electrophotocatalysis reported in this work should demonstrate much broader generality and synthetic utility beyond the radical cation Diels-Alder reaction.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Guo-Cai Yuan: Writing – original draft, Conceptualization. Ming-Gang Li: Data curation. Sha Yang: Data curation. Kanghui Song: Data curation. Chen-Xu Gong: Software, Data curation. Shuang-Jun Zhu: Data curation. Yuanming Li: Writing – review & editing, Conceptualization. Ke-Yin Ye: Writing – review & editing, Conceptualization.
Financial support from the National Natural Science Foundation of China (Nos. 22471038 and 22421002), and the Open Research Fund of School of Chemistry and Chemical Engineering, Henan Normal University (No. 2024Z08) is gratefully acknowledged.
Supplementary material associated with this article can be found, in the online version, at doi:
O. Diels, K. Adler, Ann. 460 (1928) 98–122. doi: 10.1002/jlac.19284600106
E.M. Stocking, R.M. Williams, Angew. Chem. Int. Ed. 42 (2003) 3078–3115. doi: 10.1002/anie.200200534
K.I. Takao, R. Munakata, K.I. Tadano, Chem. Rev. 105 (2005) 4779–4807. doi: 10.1021/cr040632u
Y. Sun, Y. Meng, J. Jiang, et al., Chin. Chem. Lett. 30 (2019) 966–968. doi: 10.1016/j.cclet.2019.02.015
R. Hoffmann, R.B. Woodward, J. Am. Chem. Soc. 87 (1965) 4388–4389. doi: 10.1021/ja00947a033
J.I. García, J.A. Mayoral, L. Salvatella, Acc. Chem. Res. 33 (2000) 658–664. doi: 10.1021/ar0000152
K. Chen, Z. He, W. Xiong, et al., Chin. Chem. Lett. 32 (2021) 1701–1704. doi: 10.1016/j.cclet.2020.12.047
Z.M. Png, H. Zeng, Q. Ye, et al., Chem. Asian J. 12 (2017) 2142–2159. doi: 10.1002/asia.201700442
M.M. Xu, L. Yang, K. Tan, et al., Nat. Catal. 4 (2021) 892–900. doi: 10.1038/s41929-021-00687-x
M.M. Xu, X.Y. You, Y.Z. Zhang, et al., J. Am. Chem. Soc. 143 (2021) 8993–9001. doi: 10.1021/jacs.1c04759
S. Lin, M.A. Ischay, C.G. Fry, et al., J. Am. Chem. Soc. 133 (2011) 19350–19353. doi: 10.1021/ja2093579
D. Wei, Y. Li, F. Liang, Adv. Synth. Catal. 358 (2016) 3887–3896. doi: 10.1002/adsc.201600587
L. Wang, F. Wu, J. Chen, et al., Angew. Chem. Int. Ed. 56 (2017) 6896–6900. doi: 10.1002/anie.201702940
Y. Okada, Y. Yamaguchi, A. Ozaki, et al., Chem. Sci. 7 (2016) 6387–6393. doi: 10.1039/C6SC02117D
X. Hu, L. Nie, G. Zhang, et al., Angew. Chem. Int. Ed. 59 (2020) 15238–15243. doi: 10.1002/anie.202003656
G. Zhang, Y. Lin, X. Luo, et al., Nat. Commun. 9 (2018) 1225–12231. doi: 10.1038/s41467-018-03534-z
Y. Sun, Z. Wang, S. Wu, et al., Green Synth. Catal. 3 (2022) 84–88. doi: 10.1117/12.2635681
S. Zhou, Y. Liu, Y. Hao, et al., Chin. Chem. Lett. 35 (2024) 108325–108333.
B. Large, D. Prim, Eur. J. Org. Chem. 2022 (2022) e202101032. doi: 10.1002/ejoc.202101032
J.R. Darwent, J. Chem. Soc., Chem. Commun. (1982) 798–799.
L. Capaldo, D. Ravelli, M. Fagnoni, Chem. Rev. 122 (2022) 1875–1924. doi: 10.1021/acs.chemrev.1c00263
C. Poriel, C. Quinton, F. Lucas, et al., Adv. Funct. Mater. 31 (2021) 2104980–2104997. doi: 10.1002/adfm.202104980
S. Zheng, H. Tan, X. Zhang, et al., Tetrahedron Lett. 55 (2014) 975–978. doi: 10.1016/j.tetlet.2013.11.081
A. Tutar, O. Cakmak, M. Balci, Tetrahedron. 57 (2001) 9759–9763. doi: 10.1016/S0040-4020(01)00978-4
H. Yan, Z.W. Hou, H.C. Xu, Angew. Chem. Int. Ed. 58 (2019) 4592–4595. doi: 10.1002/anie.201814488
Y. Yu, P. Guo, J.S. Zhong, et al., Org. Chem. Front. 7 (2020) 131–135. doi: 10.1039/c9qo01193e
D. Zhang, S. Yang, X. Fang, et al., Chin. Chem. Lett. 33 (2022) 4669–4674. doi: 10.1016/j.cclet.2022.02.001
H. Huang, K.A. Steiniger, T.H. Lambert, J. Am. Chem. Soc. 144 (2022) 12567–12583. doi: 10.1021/jacs.2c01914
M. Yan, Y. Kawamata, P.S. Baran, Chem. Rev. 117 (2017) 13230–13319. doi: 10.1021/acs.chemrev.7b00397
H. Wang, X. Gao, Z. Lv, et al., Chem. Rev. 119 (2019) 6769–6787. doi: 10.1021/acs.chemrev.9b00045
J. Liu, L. Lu, D. Wood, et al., ACS Cent. Sci. 6 (2020) 1317–1340. doi: 10.1021/acscentsci.0c00549
Y. Wang, S. Dana, H. Long, et al., Chem. Rev. 123 (2023) 11269–11335. doi: 10.1021/acs.chemrev.3c00158
J.M.R. Narayanam, C.R.J. Stephenson, Chem. Soc. Rev. 40 (2011) 102–113. doi: 10.1039/B913880N
C.K. Prier, D.A. Rankic, D.W.C. MacMillan, Chem. Rev. 113 (2013) 5322–5363. doi: 10.1021/cr300503r
N.A. Romero, D.A. Nicewicz, Chem. Rev. 116 (2016) 10075–10166. doi: 10.1021/acs.chemrev.6b00057
T. Shen, T.H. Lambert, Science 371 (2021) 620–626. doi: 10.1126/science.abf2798
C.Y. Cai, X.L. Lai, Y. Wang, et al., Nat. Catal. 5 (2022) 943–951. doi: 10.1038/s41929-022-00855-7
T. Shen, Y.L. Li, K.Y. Ye, et al., Nature 614 (2023) 275–280. doi: 10.1038/s41586-022-05608-x
J.M. Kollman, A. Merdes, L. Mourey, et al., Nat. Rev. Mol. Cell Biol. 12 (2011) 709–721. doi: 10.1038/nrm3209
R. Li, Y. Zhu, X. Gong, et al., J. Am. Chem. Soc. 145 (2023) 2999–3007. doi: 10.1021/jacs.2c11504
Y. Zhao, B. Li, X. Fu, et al., Angew. Chem. Int. Ed. (2024) e202415582.
S. Fukuzumi, H. Kotani, K. Ohkubo, et al., J. Am. Chem. Soc. 126 (2004) 1600–1601. doi: 10.1021/ja038656q
K. Ohkubo, K. Mizushima, R. Iwata, et al., Chem. Commun. 46 (2010) 601–603. doi: 10.1039/B920606J
N.A. Romero, D.A. Nicewicz, J. Am. Chem. Soc. 136 (2014) 17024–17035. doi: 10.1021/ja506228u
J. Ramakrishna, P. Venkatakrishnan, Chem. Asian J. 12 (2017) 181–189. doi: 10.1002/asia.201601359

Scheme 2 Substrate scope. Reaction conditions: 1 (0.2 mmol, 2.0 equiv.), 3 (0.0075 mmol, 7.5 mol%), TEMPO (0.02 mmol, 20 mol%), nBu4NClO4 (0.1 mmol, 1.0 equiv.), a carbon cloth anode, a platinum plate cathode, Ecell = 1 V, a 10 W blue LED (455 nm), 10 h, r.t., under an N2 atmosphere.

Scheme 3 Derivatizations of products. (a) 2n (0.2 mmol, 1.0 equiv.), naphthalen-1-ylboronic acid (0.21 mmol, 1.05 equiv.), Pd(PPh3)4 (0.01 mmol, 5 mol%), K2CO3 (1.2 mmol, 6.0 equiv.), H2O (0.3 mL) in THF at 70 ℃ for 24 h. (b) 2n (0.2 mmol, 2.0 equiv.), Ni(cod)2 (0.1 mmol, 1.0 equiv.), cycloocta-1,5-diene (0.1 mmol, 1.0 equiv.), bipyridine (0.1 mmol, 1.0 equiv.) in dioxane at 120 ℃ for 10 h under an N2 atmosphere.

Figure 1 Frontier molecular orbitals of compounds 2b, 7, and 8 calculated by DFT at the B3LYP/6–31G* level.
Table 1. Optimization of reaction conditions.a
|
||
| Entry | Deviation from standard conditions | Yield (%)b |
| 1 | None | 56 (70)c |
| 2 | No light | n.d. |
| 3 | No [Mes-Acr+]ClO4- (3) | n.d. |
| 4 | [Mes-Acr+]BF4- instead of 3 | (67)c |
| 5 | Ir(ppy)3 instead of 3 | 25 |
| 6 | [Ru(bpy)32+]2PF6- instead of 3 | 10 |
| 7 | No electricity | 38 |
| 8 | No TEMPO | 30 |
| 9 | Ferrocene instead of TEMPO | 28 |
| 10 | Ph3N instead of TEMPO | 34 |
| a Reaction conditions: 1a (0.2 mmol, 2.0 equiv.), 3 (0.0075 mmol, 7.5 mol%), TEMPO (0.02 mmol, 20 mol%), nBu4NClO4 (0.1 mmol, 1.0 equiv.), a carbon cloth anode, a platinum plate cathode, Ecell = 1 V, a 10 W blue LED (455 nm), 10 h, r.t., under an N2 atmosphere. b Yields determined by 19F NMR analysis using PhOCF3 as the internal standard. c Isolated yield. | ||
 下载: 导出CSV
下载: 导出CSV
Table 2. The photophysical and redox properties of compounds 2b, 7, and 8.
| Compd. | λabs (nm) | λem (nm) | τT (ns) | Ered (V) | E*red (V) |
| 2b | 426 | 554 | 3.7 | −1.40 | 1.24 |
| 7 | 440 | 566 | 5.0 | −1.15 | 1.42 |
| 8 | 446 | 566 | 1.8 | ‒ | ‒ |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们




 下载:
下载: