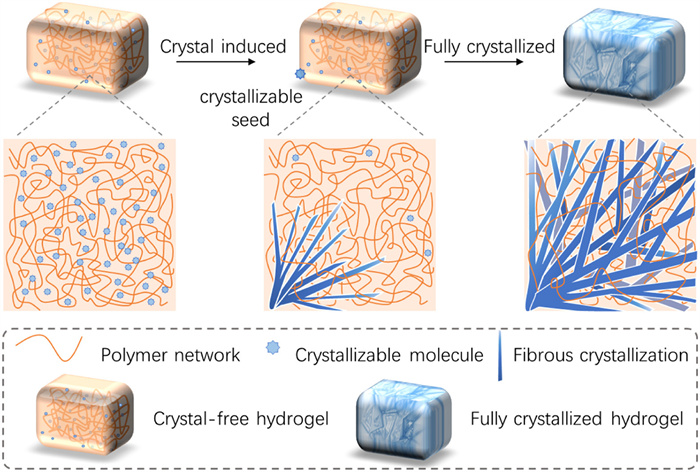
Figure 1.
Schematic illustration of crystal growth in the crystal hydrogels.
Crystal hydrogels: Strategies, properties, and applications
Qianwei Liu , Xinhong Xiong , Numan Ahmed , Peisong Tang , Jiaxi Cui

Hydrogels, soft materials made from hydrophilic crosslinked polymers and water, are attracting increasing attention due to their fascinating properties, such as excellent biocompatibility [1,2], tailorable mechanical properties [3,4], and great feasibility in integrating various functions [5-7]. These characteristics have led to a wide range of applications for hydrogels, including soft robotics [8,9], tissue engineering [10,11], sensors [12,13], optics [14,15]. Traditional hydrogels are typically static and fragile because of their irregular network topologies and the lack of an energy dissipation mechanism [16-19]. In the last two decades, many strategies have been developed to overcome these limitations, such as designing complex network topologies (e.g., double networks) [20,21], introducing reversible connecting interactions, and hybridizing functional fillers [22]. Among them, the hybridization method is versatile since the fillers can often strengthen and also functionalize materials [23,24]. Especially, many unique features are observed when substances that reversibly crystallize within networks are used as fillers to create novel materials named crystal hydrogels.
Crystal hydrogels are distinguished from other hydrogel composites because the rigid crystal fillers can rapidly grow in situ in a few minutes within the hydrogel matrices (Fig. 1). These crystals, typically exhibiting fibrous morphologies, spread extensively over the networks and frequently interconnect to construct robust frameworks, thereby imparting a high modulus. Such “double-network” crystal hydrogel structures mimic double-network hydrogels, combining flexible and rigid networks to form a spatial polymer network that integrates their complementary advantages [25,26]. Crystal hydrogels discussed here starkly contrast traditional crystal-containing hydrogels in which hydrogel matrices are used as viscous media to reach slow crystal growth [27,28]. This new method of modifying polymer networks by embedding crystals can be adapted to various complex environmental conditions. Additionally, neither photonic crystal hydrogels nor liquid crystal hydrogels are considered typical crystal hydrogels since they lack a representative process of crystal growth. Currently, an extensive body of sophisticated research has been undertaken on biomineralized hydrogels [29-32]. Based on these scientific investigations, we assume that biomineralized hydrogels do not conform to the classification of crystal hydrogels, primarily due to the inherently slow kinetics of the inorganic mineral salt deposition during the biomineralization process. Importantly, two primary conditions must be met to classify a hydrogel as a crystal hydrogel. Firstly, the crystallizable molecules must be evenly and stably dispersed within the hydrogel matrix. Secondly, upon exposure to specific external stimuli, these molecules must be capable of undergoing crystallization within the entire hydrogel matrix. The core feature of crystal hydrogels lies in the controllable crystallization behavior of crystallizable molecules within the matrix under external stimuli. Recently, various crystal hydrogels have emerged, showing many unique properties, including a highly rigid nature [33], self-transition in mechanical properties [34], stimuli-responsiveness [35], ionic conductance [36], self-adaptive surface morphologies [37], light emission [38], and shape self-recovering [39]. These materials also show excellent application potential in a wide range of fields, such as soft robotics [33,40], energy storage devices [36,41,42], hydrated lubrication [43], flexible sensors [44-46], recycled adhesive [38], and information encryption [47]. Therefore, an updated review to conclude the basic preparation methods and the features of these materials is favorable for providing a comprehensive understanding of this emerging field.
This review starts from the conceptual foundations of crystal hydrogels, summarizing the recent development. The main content is divided into three thematic sections: the initial section discusses the basic strategies of constructing crystal hydrogels, the subsequent section highlights the distinct properties of these materials, and the third section describes the potential and emerging applications of crystal hydrogels. Finally, we explore the opportunities and challenges in this emerging field.
Constructing a crystal network within a hydrogel is critical for preparing crystal hydrogels. This section categorizes crystallization strategies based on the methods to induce crystal nucleation and growth. Some typical examples show the primary feature of crystallization in the hydrogel but do not use the concept of crystal hydrogel in the literature, which is also concluded here for discussion. Predominant strategies for integrating polymer networks to construct crystal hydrogels include the crystallization of phase change materials, polymerization-induced crystallization, and redox-induced crystallization. We will discuss these crystalline manners of the products as follows.
Phase change materials (PCM) are functional compounds that reversibly store and release substantial latent heat during melting and freezing [48,49]. According to phase change forms, PCMs can be divided into solid-liquid, solid-solid, solid-gas, and liquid-gas. Solid-liquid PCMs exhibit the advantages of a wide phase change temperature range, large latent heat, high energy storage density, low toxicity, and low cost [50]. The solid-liquid PCMs, transitioning from liquid to crystalline phase, demonstrate significant potential as crystallizable fillers that can be seamlessly integrated into hydrogel matrices. The crystal hydrogels can be innovatively designed based on this transition. Two ground states correspond to the lowest energy state in the PCMs [51]. Similarly, crystal hydrogels in their crystal-free state, referred to as the supercooled state, exhibit metastability, which belongs to the first ground state [52]. Crystallization in a hydrogel initially requires the formation of sufficient nucleation sites. The strategies that come into contact with crystal seeds or pressure can be employed to achieve crystallization [53]. Upon the growth of crystallization throughout the polymer network within the crystal hydrogel, the hydrogel attains a fully crystallized state, representing the second ground state.
Inorganic hydrated salt PCMs, a typical class within solid-liquid PCMs, demonstrate promising performance as crystalline phases within crystal hydrogels. Recent advancements in these hydrated salts have been documented, encompassing sodium acetate trihydrate (NaAc·3H2O) [33-36], calcium chloride hexahydrate (CaCl2·6H2O) [45], and sodium thiosulfate pentahydrate (Na2S2O3·5H2O) [42]. Sodium acetate (NaAc) is particularly noted for its robust supercooling capabilities; despite a melting point of 58 ℃, it can remain as a supercooled liquid at room temperature [54,55]. Building on these properties, a crystal hydrogel has innovatively been prepared by integrating supersaturated NaAc into a polyacrylamide (PAAm) hydrogel [33]. Moreover, the original brittle PAAm network through crystallization has been significantly enhanced, but the mechanisms underlying the growth and evolution of crystallization within the crystal hydrogels remain largely unexplored. In a very recent report, we have elucidated the nucleation and growth processes of NaAc·3H2O crystals within the PAAm network, offering detailed insights into the crystal evolution dynamics. In our study, a system made from crosslinked PAAm and NaAc is used to demonstrate that metastable fibrous crystals of NaAc·3H2O preferably form in PAAm hydrogels via polymer-involving mismatch nucleation (Fig. 2a) [34]. The formation of fibrous crystals is mainly ascribed to the kinetic energy dissipation in the crystal tip direction, which is significantly stronger than in other directions [56]. Ubiquitous polymer networks in crystal hydrogels are known to hinder crystal growth [57]. Due to the low energy required to create steps on the crystal surface, NaAc molecules randomly settle on the parent fiber, forming mismatch nucleation-inducing daughter crystals. These branching structures are attributed to the formation of multiple nucleation sites, which induce the growth of daughter crystals on the parent fiber. Furthermore, the growth of NaAc·3H2O crystals within the polymer network is inhibited compared to their growth in a precursor solution containing numerous monomers. The crystal morphology varies across different regions (Fig. 2b) [35]. Notably, the growth rate of crystals in the polymerized zone is remarkably slower than in the non-polymerized zone. Increasing the monomer concentration significantly enhances the crystallization rate, while the content of crosslinkers has no effect.
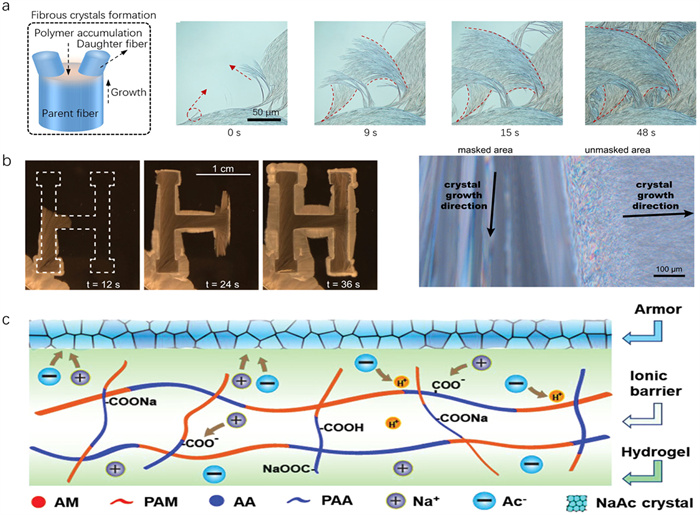
Varying polymer networks have different influences on crystallization growth in crystal hydrogels. Taking polyacrylic acid (PAA) crystal hydrogels as a typical example, the matrix of PAA restricts the crystallization of NaAc crystals. Specifically, the interaction between the carboxyl groups on the PAA chain and sodium ions confines the growth of NaAc crystals primarily to the hydrogel’s surface, resulting in hydrogels that resemble armored structures (Fig. 2c) [44]. Heating highly crystalline polymers above their melting point induces a liquid transformation [58,59]. Recognizing this property, a phase-change crystal hydrogel has been developed through an innovative one-pot strategy involving the in-situ formation of a crosslinked PAA network within a molten phase-changed polyethylene glycol (PEG) [60]. PAA and PEG have excellent compatibility, ensuring the stable presence of PEG in the PAA polymer network without leakage. PEG crystallizes upon cooling, causing the composite hydrogel to change from transparent to opaque. When heated to the melting point of PEG, the crystals dissolve, and the composite hydrogel becomes transparent again. However, due to the strong interaction between PEG and PAA chains, the crystallization of PEG molecules is significantly limited as the content of the PAA network increases. In summary, integrating phase-change-driven crystallization within a hydrogel network provides a direct and efficient method for designing crystal hydrogels. This technique not only endows crystal hydrogels with superior properties but also supports material recyclability due to the reversibility of the phase change process.
Many crystallizable molecules have greater solubility in the precursor solution than in the polymer solution. Therefore, an in-situ decrease in solubility is expected during a polymerization process (Fig. 3a). Here, the conditions that induce polymerization can be utilized as a switch to trigger crystallization for the crystal hydrogel. In contrast to the methods of inducing phase transitions through cooling, this strategy uses mixed molecules with variable solubility pre- and post-polymerization, offering a simpler alternative. For example, dibenzo-24-crown-8-ether (CE) is used as a crystallizable molecule, initially soluble in an acrylamide (AAm) solution [38]. Molecular interactions, including hydrogen bonds and C–H···π interactions between CE and AAm, facilitate the solvation of CE, enhancing its solubility in water. As AAm polymerizes, the solubility of CE diminishes, prompting the crystallization of large spherulites within the hydrogel. The addition of CE does not affect the polymerization and gelation process. The gradual formation of CE spherulites is attributed to both the high aspect ratio of CE crystals and the inhibition of nucleation caused by the network. These spherulites are large (millimeters to centimeters in diameter) with a characteristic void-eye-like structure. Notably, the crystal hydrogel with these large spherulites is stable, mechanically robust, and exhibits good stretchability.
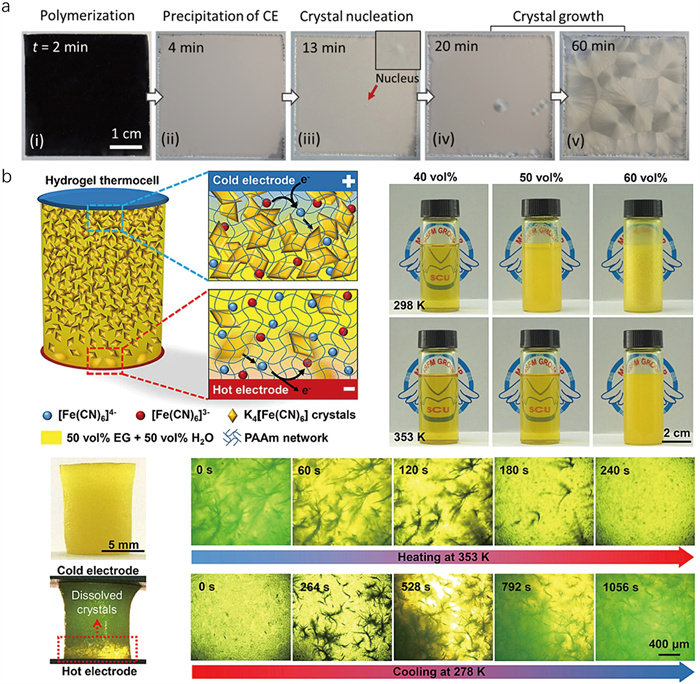
Many metal complexes display significantly variable solubility with the valence states of metal cores. By controllable redox, the solvation structure of redox ions can be rearranged, creating a concentration gradient that induces crystal formation [61]. This gradient causes the coupled redox ions to exhibit different solubility, leading to crystallization. The crystallization of [Fe(CN)6]4− is selectively induced in a hydrogel network by using ethylene glycol (EG) as a solvent for the redox coupled [Fe(CN)6]4− and [Fe(CN)6]3− (Fig. 3b) [46]. This redox couple [Fe(CN)6]4− and [Fe(CN)6]3−, widely recognized in electrochemistry and energy storage, is the focus of this method [62,63]. EG modifies the solvation shells of the redox couple, resulting in lower solubility for [Fe(CN)6]4− compared to [Fe(CN)6]3− in the EG-water mixture. The temperature gradient also significantly affects the solubility of the [Fe(CN)6]4− crystal. During this process, crystal dissolution and increased solubility provide a high local concentration of [Fe(CN)6]4− near the hot electrode, promoting its oxidation to [Fe(CN)6]3−. Conversely, crystallization and decreased solubility provide a low local concentration of [Fe(CN)6]4− near the cold electrode, promoting the reduction of [Fe(CN)6]3− to [Fe(CN)6]4−. This balance of dissolution and crystallization enables selective, limited micro-crystallization of [Fe(CN)6]4−. Within the mesh-like PAAm networks, the confined microcrystals exhibit branch-like microstructures. In contrast to the crystallization of phase change materials and polymerization-induced crystallization, the proposed strategy facilitates a more versatile modulation of crystal growth in crystal hydrogels by strategically restricting the concentration of coupled redox ions in distinct regions, thereby enabling localized crystallization. In addition, the [Fe(CN)6]4− crystal hydrogel can be reversibly used through heating and cooling, achieving reversible and thermo-sensitive confined micro-crystallization in the crystal hydrogel.
In summary, every preparation strategy aims to integrate crystals into the polymer matrix, ensuring global distribution throughout the material system. The reviewed strategies for the preparation of crystal hydrogels predominantly build upon the solid foundations of classical crystal growth theory [64-66], while simultaneously combining the principles of mismatch nucleation theory to improve the exploration of crystal growth [34]. Documented instances encompass a range of materials such as NaAc·3H2O, CaCl2·6H2O, Na2S2O3·5H2O, PEG, CE, and [Fe(CN)6]4−. The preparation methods for crystal hydrogels exhibit distinct advantages and limitations that determine their application suitability. While phase-change material doping offers simplicity and versatility, its concentration-dependent crystal nucleation poses challenges in growth control. In contrast, polymer-induced crystallization demonstrates superior spatial control through template effects, though restricted to specific polymer-crystal pairs (e.g., CE systems). Similarly, redox-mediated approaches enable remarkable temporal control but require specialized redox-active components (e.g., redox couple [Fe(CN)6]4− and [Fe(CN)6]3−. In principle, any material that can undergo similar crystallization processes should be a good candidate for designing a novel crystal hydrogel.
The proposed crystal hydrogels offer simpler and more convenient preparation methods and conditions, while also exhibiting various meaningful and intriguing properties. The crystal hydrogels can endow these materials with diverse and intriguing functionalities, making them applicable across various fields. Crystal hydrogels typically involve crystal growth, dissolution, and regeneration processes, enabling their properties to be switched repeatedly and making them reusable. Furthermore, introducing crystals into the hydrogel can significantly enhance the material’s mechanical properties, achieving variable stiffness and effectively toughening the hydrogel. This section explores typical demonstrations to illustrate their great potential for improving various materials’ performances and functions.
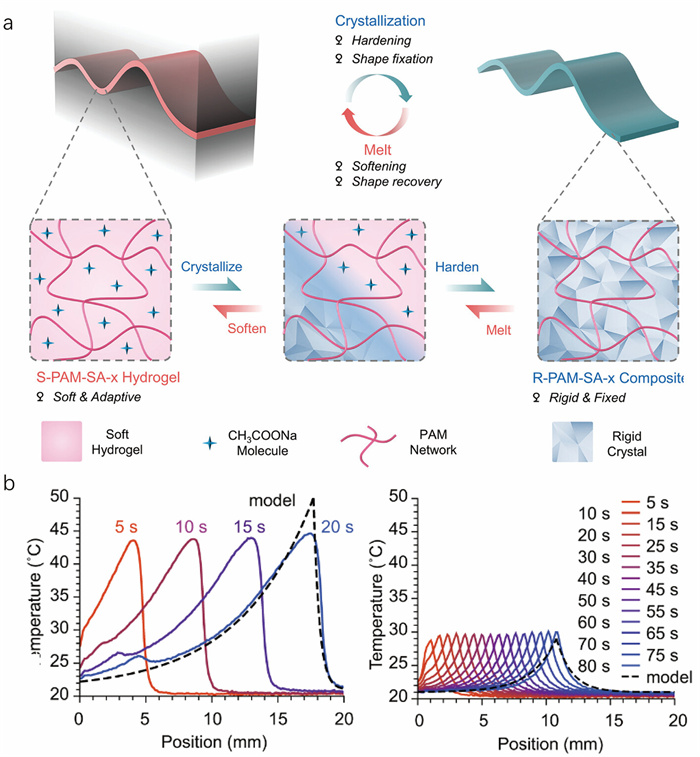
Traditional synthetic hydrogels comprise a single, irregular hydrophilic polymer network, and their limited mechanical properties greatly restrict their development [67]. Therefore, developing tough hydrogels is one of the most critical challenges in hydrogels. Soft and hard switching materials with significant stiffness variations are becoming increasingly important in applications such as soft robotics and adhesives [68]. The stiffness of polymer networks can be enhanced by incorporating solid-liquid phase change materials, including hydrated inorganic salts or crystalline polymers [60,69]. The transparent, uncrystallized hydrogel, known for its good elasticity, undergoes a dramatic transformation under transient stimulation from crystal nucleation. Specifically, the crystal hydrogel transitions from a transparent and soft state to an opaque and rigid state (Fig. 4a). Mechanically, upon complete crystallization, the stiffness of the crystal hydrogel increases by more than four orders of magnitude (from 15 kPa to 385 MPa) [33]. The mechanical properties of these crystal hydrogels can be modulated by varying the concentrations of monomers, crosslinkers, and NaAc [34]. Crystal hydrogels with higher crosslinker concentrations demonstrate better homogeneity than those with lower crosslinker concentrations. Remarkably, repeated crystallization cycles do not cause permanent damage to the polymer network [33]. The phase change crystallization of NaAc·3H2O induces significant stiffness changes. Besides, there are strong hydrogen bonds and electrostatic interactions between hydrated crystals and polymer chains to enhance the strength of the composite crystal hydrogel [36]. Similar results are also observed in the crystallization processes of other materials, such as CaCl2·6H2O [45], Na2S2O3·5H2O [42], PEG [60], and CE [38]. This cycle can be repeated by heating the hydrogel to dissolve the crystals, cooling to room temperature, and restarting crystallization. This phase transition process is crucial, driving the soft-hard transition and defining the most outstanding characteristics of crystal hydrogels. Furthermore, the dynamic transformation in mechanical properties lays the foundation for multiple applications of crystal hydrogels in technology and industry.
Thermo-responsive materials display temperature-dependent behaviors that transform heat into optical [70,71], mechanical [72-74], or chemical variations [75,76]. While most materials respond equally to temperature fluctuations, only a few can facilitate complex reaction processes through spatially defined response regions [77]. The occurrence of crystallization within the crystal hydrogel often involves a considerable amount of heat production. This heat generation can be effectively utilized to achieve thermo-responsive functionalities. Aizenberg et al. have pioneered a method involving a heat source that not only stores energy but also the patterns necessary to trigger intricate behaviors in thermally responsive materials upon demand [35]. This method takes advantage of the rapid growth and latent heat release of supersaturated NaAc·3H2O crystals in solution and hydrogel, initiating subsequent thermal response processes. Since the polymer network significantly inhibits crystal growth more than the monomer solution, the regional thermal response can be controlled by manipulating the polymerization conditions in the crystal growth region. Notably, the growth rate of crystals is markedly slower in polymerized zones compared to non-polymerized areas, resulting in a substantial temperature differential peak temperature in the polymerized region is 30 ℃, which is 15 ℃ lower than the 45 ℃ observed in the non-polymerized region (Fig. 4b). This regional temperature difference induced by polymerization can achieve a custom dynamic heat map, enabling regional temperature distribution to respond to downstream thermal activation effects. This photo-polymerization strategy effectively regulates the growth rate and direction of the crystals, allowing selective activation of thermal response characteristics in predetermined regions and offering innovative ideas for thermally responsive materials.
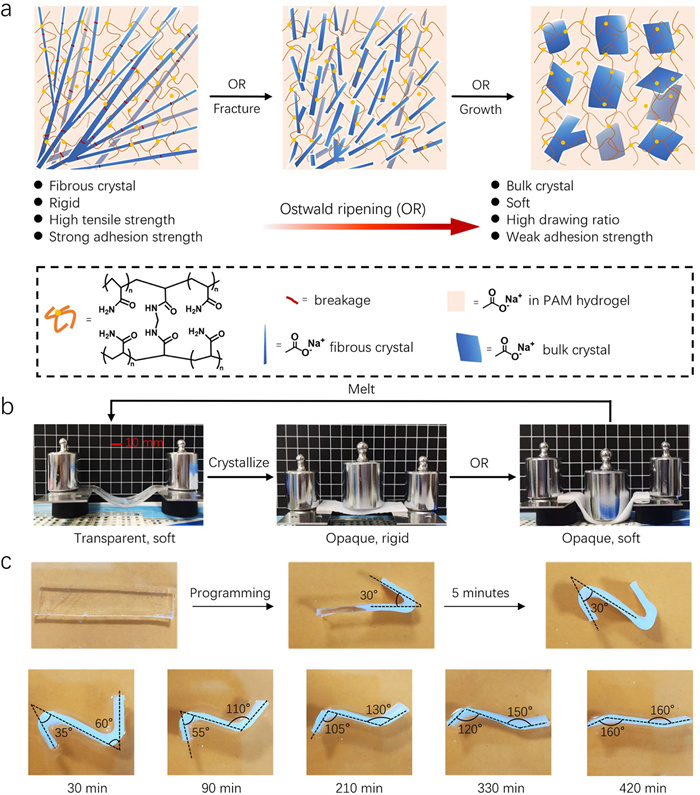
Ostwald ripening (OR) is a classic solution theory describing molecular transfer from a metastable state to a stable one [78]. Our study reveals that the initial fibrous NaAc·3H2O crystals formed within hydrogels are metastable. Due to mismatch nucleation, which often occurs at the fiber tip where the polymer accumulates, a series of cracks and branching points form. When these crystal hydrogels are stored at room temperature, OR will occur beginning at these points, causing fine fibrous branching crystals to dissolve gradually (Fig. 5a) [34]. In contrast, larger crystals grow in situ at the fracture boundaries. This OR-induced morphological change results in a significant decrease in the mechanical properties of hydrogels. Specifically, the complete cycle of NaAc-based crystal hydrogel stiffness transformation can be described as follows: The crystal growth process promotes the transition of the crystal hydrogel from a soft to a rigid state, while the OR-induced crystal structure transformation promotes the transition from a hard to a soft state (Fig. 5b). The OR-induced transformation also affects the adhesion and self-recovery properties of crystal hydrogels. The adhesion of crystal hydrogels declines with the change in crystal structure until adhesion fails. The failure time of mechanical and adhesion properties induced by OR can be controlled by adjusting the monomer and NaAc concentrations in crystal hydrogels. The shape self-recovery performance of crystal hydrogels is another profound demonstration of the OR process (Fig. 5c). Delayed shape recovery can be achieved by adjusting the sequence of crystallization. Therefore, introducing OR-induced crystal structure transformation into crystal hydrogels is an effective way to design advanced transient soft materials.
The application of traditional hydrogel electrolytes is often limited by their narrow operating voltage, weak mechanical strength, and susceptibility to solvent interference [79,80]. Introducing inorganic salt crystals into polymer hydrogels enhances their mechanical properties through hydrogen bonding, electrostatic interaction, and other synergies while inhibiting the electrochemical activity of water and expanding the operating voltage [81-83]. Thus, crystal hydrogel electrolytes represent a new class of non-liquid electrolytes with comprehensive properties. Inspired by the synergistic properties of composite materials and the beneficial characteristics of hydrated crystals, a crystal hydrogel electrolyte exhibiting superior comprehensive properties through the crystal transition of NaAc·3H2O within a hydrogel matrix (Fig. 6a) is innovatively synthesized [36]. These NaAc-based crystal hydrogels demonstrate a higher operating voltage of 2.0 V compared to traditional hydrogels. The hydrated crystals and hydrophilic polymer chains facilitate strong interactions with water molecules, while the presence of Zwitter ionic groups creates specialized ion migration channels, significantly boosting the supercapacitor’s energy density. Moreover, crystal hydrogel-based supercapacitors maintain functionality under extreme conditions, operating effectively at temperatures from 80 ℃ to −40 ℃ and briefly withstanding direct exposure to flames exceeding 200 ℃ and immersion in liquid nitrogen at −196 ℃. This exceptional temperature resilience is attributed mainly to the robustness of NaAc·3H2O crystals within the hydrogel structure. Similarly, integrating Na2S2O3·5H2O crystals into a hydrogel matrix composed of sodium polyacrylate (PAANa) results in a tri-phase skeleton structure [42]. This structure combines a high-strength crystalline hydrate phase, a polymer phase, and an intermediate water layer, endowing the crystal hydrogel electrolyte with high ionic conductivity. Na2S2O3-based crystal hydrogel supercapacitors use oxidation-reduced Na2S2O3·5H2O as a hydrated salt, enhancing the system’s capacitance and improving energy density. Therefore, crystal hydrogel electrolytes emerge as a highly safe and efficient option for advanced electrolytic applications.
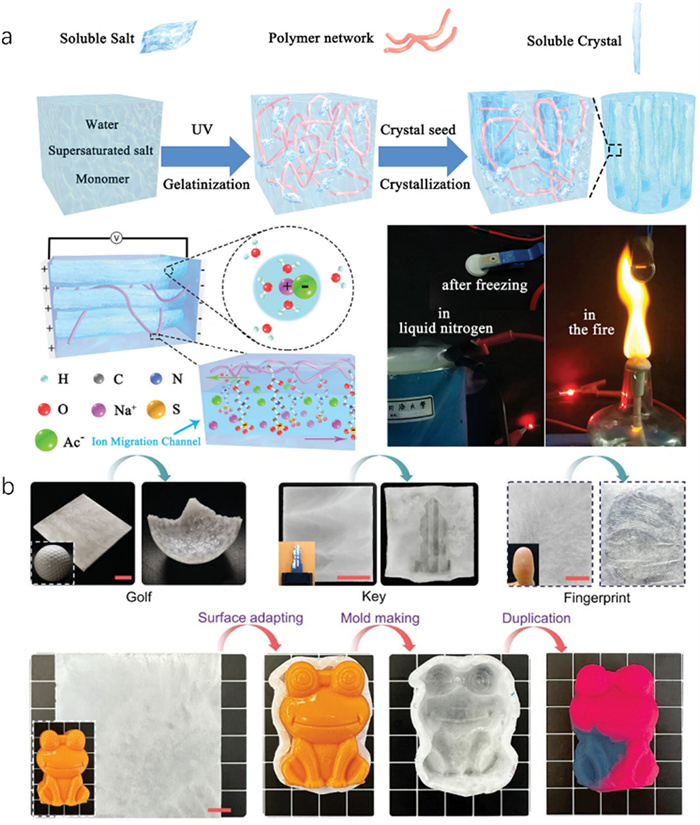
Crystal hydrogels in their non-crystallized state with ultra-low modulus exhibit exceptional adaptability to complex surfaces, thereby increasing the contact area with the substrate [84]. Upon stimulation-induced crystallization, these hydrogels harden, effectively interlocking with the substrate surface [40]. This transformation is primarily facilitated by the rigid support provided by the crystal phase transition within the hydrogel, which enhances the van der Waals adhesive forces and disperses the collapse force [85]. Leveraging this mechanism, we have incorporated supersaturated NaAc into a PAAm hydrogel to develop a surface-adaptive binder [37]. In its transparent state, this binder conforms to various complex surfaces, including wood, glass, and steel. With the growth of NaAc·3H2O crystallization, strong self-adaptivity occurs. Further explorations have revealed that this crystalline phase transition can replicate a range of surface textures, capturing intricate details such as those found on golf balls, keys, finger joints, and fingerprints (Fig. 6b) [39]. The self-adaptability and printability of crystal hydrogels will promote their functional applications.
Conventional PAAm hydrogels typically do not exhibit room-temperature phosphorescence (RTP) [86-88]. In PAAm, luminescence primarily arises from carbonyl clusters [89]. Based on this characteristic, a simple strategy has been devised to induce RTP in PAAm hydrogels by facilitating polymer-induced crystallization of the crystallizable molecule dibenzo-24-crown-8-ether (CE) (Fig. 7a) [38]. The crystallizable molecules CE exhibit excitation-independent phosphorescence emission [90]. The solubility differences between AAm and PAAm lead to CE crystallization. The resultant CE-PAAm hydrogels exhibit distinct green phosphorescence at room temperature when excited by 250−280 nm ultraviolet (UV) light, visible to the naked eye for several seconds post-illumination. These CE crystals compress and confine the surrounding PAAm matrix, diminishing the mobility of polymer segments and facilitating the aggregation of carbonyl groups. This confinement leads to the formation of clustered carbonyl groups with significantly reduced mobility, resulting in extraordinary phosphorescence while still maintaining good stretchability. This excitation-dependent emission underscores that carbonyl clusters act as the chromophore for the unique phosphorescence of the CE-PAAm gel. Such aggregation is pivotal for enhancing phosphorescence by limiting non-radiative decay pathways. The hydrated materials contrast with the dried PAAm film, where carbonyl clusters are sparse and flexible, leading to phosphorescence quenching due to low polymer density and high segment mobility. This method maintains mechanical integrity while offering considerable versatility and scalability, making it suitable for applications in chemical sensing, soft electronics, and information encryption. However, the formation of spherules compromises the optical clarity of hydrogels, posing challenges for applications requiring transparency.
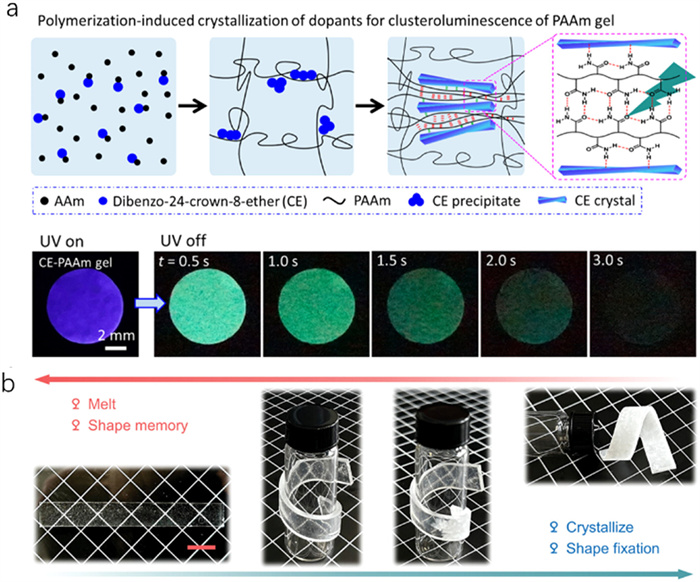
The function of shape memory, an ability to recover the original shape of a material, is desirable for many applications in soft robotics and medicine [91,92]. This property is usually realized in hydrogels by introducing reversible molecular bonds (e.g., hydrogen bonding, ionic assembly, dipole-dipole) [93,94]. The rigid crystal frames in crystal hydrogels provide a new approach to achieve this characteristic. We have demonstrated this idea using NaAc-based crystal hydrogels. Before the crystallization of NaAc, the materials are soft, transparent, and metastable. They can be molded into various temporary shapes. Subsequent crystallization, induced by either external force or crystal seed stimulation, fixes these shapes. The fully crystallized NaAc-based hydrogels become hard and exhibit a high modulus. Eventually, high-temperature heating melts the NaAc-based crystals, allowing the hydrogel to revert to its original shape. The shape memory effect is not confined to simple geometric transformations; it extends to complex manipulations such as loading, stretching, and compressing within the composite network of the hydrogels, demonstrating versatile shape memory capabilities (Fig. 7b) [39]. Furthermore, this effect is observable not only in hydrogels involving inorganic salts but also in systems where PEG and PAA undergo polymer phase change crystallization [60]. In conclusion, the shape memory effect within these hydrogels showcases that crystallization does not permanently alter the polymer network, which fully recovers upon reheating, reinstating the original structural integrity.
Crystals enhance hydrogels and also bring about various unique properties, which expand their applications. Note that transparent uncrystallized hydrogels initially possess a low modulus, providing excellent shape-editing capabilities for adaptation to various complex environments. Upon induced crystallization, these hydrogels transform into fully crystallized composites with a high modulus, which can be customized based on the type of crystal incorporated. The dynamic properties of crystal hydrogels make them exceptionally versatile, opening up a wide range of potential applications. The following sections will explore typical demonstrations to illustrate their capabilities and potential across diverse applications.
The crystal growth within the crystal hydrogel facilitates the material’s transition from a flexible, soft state to a remarkably stable, tough one, thereby elevating its potential for applications in soft robotics. Unlike their rigid counterparts, soft robots offer enhanced flexibility, superior adaptability, and simpler designs, making them ideal for applications in actuation, sensing, and biologically related fields [95,96].
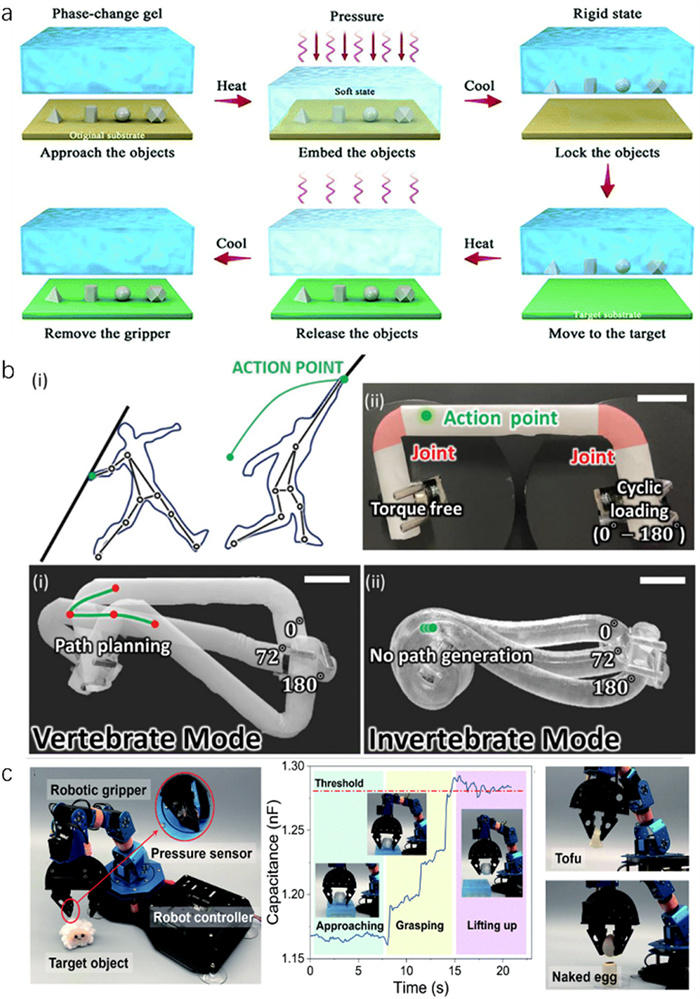
Hydrogels can be widely used in soft robots because of their high water content, excellent softness, and editable properties [97]. A strategy that utilizes mechanically dynamic crystal hydrogels to form PAA crosslinked networks through the phase transformation of PEG melt, thereby enabling shape memory effects for bionic grasping (Fig. 8a) [60]. This shape memory phenomenon in crystal hydrogels containing PEG melt allows for precise control over the material’s temporary and permanent shapes. The bionic gripper handles 3D objects of various shapes through a dual-state mechanical transformation, i.e., hard for contact and shape retention, and soft for adaptability and deformation. During a typical grasping cycle, the phase change gel softens, allowing the crystal hydrogel to mold around an object. The hardening process imparts a high modulus to the gel, which secures the temporary shape and provides the capability to grip objects. Upon reheating, the gel reverts to its permanent shape, releasing the object onto a designated substrate. This process benefits from adhesion, friction, and interlocking mechanisms, providing substantial gripping power. A spatial micro-water molecular manipulation strategy has been further employed to explore mechanical versatility. This involved integrating supersaturated NaAc·3H2O into a poly(acrylamide/2-acrylamido-2-methylpropane sulfonic acid) network (P(AAm/AMPS)), enabling the alternation between soft and hard states (Fig. 8b) [69]. This approach modeled the kinetic forces in a javelin throw using a rigid V-shaped crystal hydrogel as a four-bar linkage, demonstrating the material’s capacity for kinematic multi-modality under rigid conditions. Moreover, CaCl2·6H2O crystal hydrogels, with similar phase change capabilities, have also been fabricated into effective grippers (Fig. 8c) [45]. Integrating mechanically dynamic crystal hydrogels into soft robotics represents a significant advancement in material science inspired by biological adaptation mechanisms.
Hydrogel adhesives are emerging as a versatile class of multifunctional soft materials with broad applications in wound dressing [98,99], tissue engineering [71,100-102], implantable devices [103-105], and wearable electronics [106-108]. Typically, these adhesives exhibit weak adhesion strength due to their inherently soft mechanical properties [109,110]. Under stress, this often leads to binder failure from concentrated load points [111,112]. Introducing a stimulus-responsive hardening mechanism via phase change crystallization in crystal hydrogels enhances their adhesion capabilities, providing a critical improvement over traditional formulations. The NaAc-based crystal hydrogels exemplify this transformative property. In their uncrystallized state, these hydrogels are soft and amorphous, easily deforming to maximize contact area and adapt to complex surfaces. The hydrogel hardens significantly upon stimulation-induced crystallization, increasing van der Waals forces and providing robust support (Fig. 9a). This transformation allows for strong and immediate adhesion. Furthermore, these crystal hydrogel adhesives can undergo repeated cycles of heating, dissolution, and recrystallization without losing adhesion strength or fatigue resistance, underscoring their durability and reusability (Fig. 9b) [37]. The ability of these hydrogels to switch from a soft, amorphous state to a hard, crystalline state allows for dynamic control of adhesive properties, maximizing contact area and mechanical support on demand.
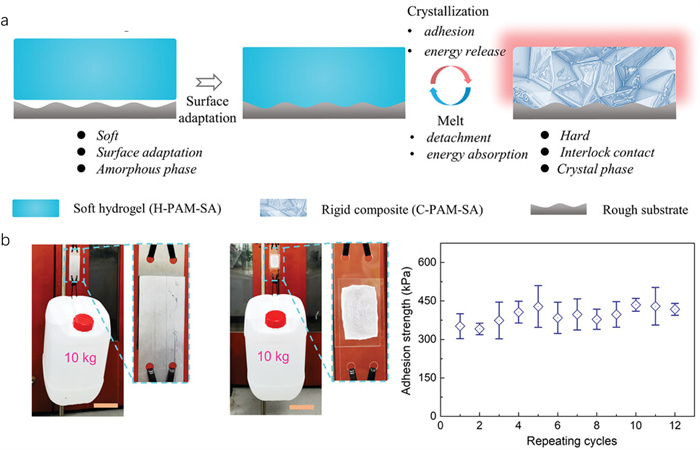
Additionally, expanding the application of this technology to integrate bioactive compounds could revolutionize medical adhesive applications, offering not only structural support but also therapeutic benefits. In summary, crystal hydrogel adhesives present a transformative approach in soft material technology, leveraging the unique properties of phase change crystallization to enhance adhesive strength and adaptability across various applications. Their dynamic switching capability between soft and hard states makes them ideal for applications requiring strong, durable, and reusable adhesives.
Hydrogel electrolytes represent a novel advancement in flexible energy storage devices [113-115]. Despite their potential, a narrow operating voltage range and insufficient mechanical strength often hinder practical applications. A promising solution to these challenges involves the integration of hydrated salt crystals into the hydrogel matrix [116,117]. Incorporating supersaturated NaAc·3H2O and Na2S2O3·5H2O into the hydrogels can suppress the electrochemical activity of water, thereby broadening the operating voltage [36,42]. This effect resembles the solvation sheath observed in “water-in-salt” electrolytes [118,119].
These crystal hydrogel electrolytes broaden the operating voltage and exhibit enhanced mechanical strength, improved electrochemical properties, and greater tolerance to extreme environmental conditions. Different inorganic salts variably impact the properties of salt composite hydrogels [120,121]. An innovative methodology incorporating the synergistic effects of co-nonsolvency [122] and salting-out utilizing potassium acetate (KAc) and zinc acetate dihydrate (ZnAc) has been introduced, resulting in the development of a hydrogel electrolyte exhibiting superior anti-freezing properties, robust mechanical stability, and enhanced mass transport capabilities [123]. This method effectively suppresses dendrite formation and side reactions (Fig. 10a). When fabricated into a soft pack battery, this hydrogel electrolyte demonstrates stable operation over 30,000 cycles at low temperatures and exhibits exceptional impact resistance. The development of crystal hydrogel-based electrolytes has successfully addressed fundamental limitations traditionally associated with hydrogel electrolytes in energy storage, such as narrow operating voltage and low mechanical strength. These electrolytes enhance electrochemical stability and mechanical properties by incorporating hydrated salt crystals, expanding their applicability under diverse environmental conditions. Crystal hydrogel electrolytes mark a significant advancement in the field of flexible energy storage devices. Their ability to combine broad operating voltage ranges, enhanced mechanical strength, and tolerance to extreme conditions makes them a superior alternative to traditional hydrogel electrolytes.
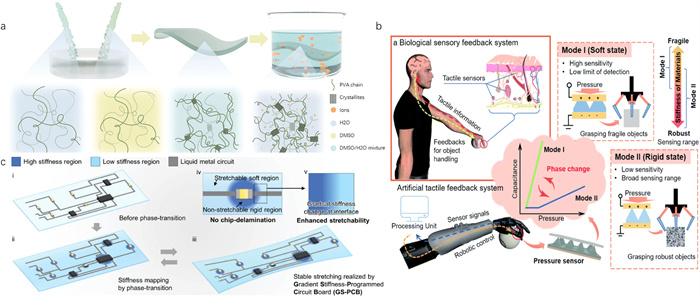
Sensing in the human body is a complex process predominantly managed by the nervous system and involves the coordinated operation of various organs [124,125]. Active sensing, particularly, is facilitated by the hands, with signals transmitted to the brain, which in turn coordinates the hand muscles for activities such as grasping [126,127]. In this sensory pathway, the skin, the largest and one of the most sensitive organs, plays a pivotal role due to its extensive sensing capabilities [128,129].
Recognizing the skin’s attributes, a pressure sensor utilizing the phase change crystallization of CaCl2-based crystal hydrogels has been developed (Fig. 10b) [45]. This sensor features a soft-hard conversion mechanism that allows for adjustable operational modes. The conical crystal hydrogel films employed in this sensor demonstrate varying load-bearing capacities, enhancing their sensitivity. Similarly, the stiffness variation during the phase transition of supersaturated NaAc-based crystal hydrogels has been exploited (Fig. 10c) [130]. A finger sensor exhibiting remarkable stability has been designed through the integration of hydrogels with laser-patterned liquid metal conductors, renowned for their superior metallic conductivity and exceptional stretchability. The distinct stiffness difference between the uncrystallized soft zones and the crystallized rigid zones in the substrate significantly enhances the sensor’s durability by preventing chip delamination. This ensures both excellent stability and a high stretch ratio. The innovative use of crystal hydrogels in the design of tactile sensors demonstrates their significant potential to replicate and enhance the human body’s sensory capabilities. By exploiting the inherent properties of phase change crystallization, these hydrogels offer adjustable mechanical properties crucial for developing sensitive, adaptable, and durable sensors. Their ability to switch between soft and hard states, combined with their high sensitivity and durability, makes them ideal for developing innovative tactile sensors.
Throughout the evolution of organisms, various hydration lubrication systems have developed, including the cardiovascular system, tendon tissue [131], digestive tract [132], oral mucosa [133], and articular cartilage [134,135]. Hydrogels, with their extensive three-dimensional hydrophilic networks, are ideally suited for simulating these systems due to their ability to reduce drag through hydration [136]. In the current research on lubricating hydrogels, the following strategies are usually adopted to achieve hydrated lubrication: Gel-sol transition of supramolecular hydrogels [137,138], construction of double/multi-layer hydrogel coatings [139,140], and hybridization of lipid boundary layer hydrogels [141,142]
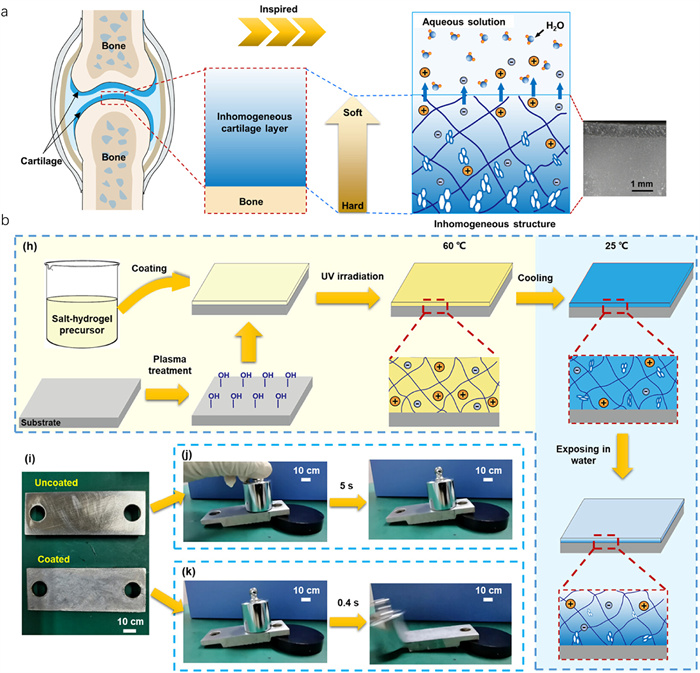
A novel strategy employing crystal hydrogels for the optimization of hydrated lubrication has been introduced [43]. The influence of water exposure on a composite hydrogel composed of NaAc-based crystals has been examined, revealing variations in the solubility of NaAc molecules across distinct regions within the hydrogel. This heterogeneous solubility distribution consequently led to the formation of an inhomogeneous hydrogel structure. This variability resulted in a hydrogel with an inhomogeneous structure (Fig. 11a). Through meticulous measurements of the elastic moduli at three distinct locations across the cross-sectional plane of the hydrogel, a gradual reduction in the moduli values from the interior to the exterior of the material was observed. This observation confirms the inhomogeneous distribution of stiffness within the hydrogel matrix, indicating variations in its mechanical properties. Specifically, the topmost polymer network, where NaAc molecules dissolve more rapidly, forms a hydration layer that provides effective lubrication (Fig. 11b). Conversely, in the bottom polymer network, where NaAc dissolves more slowly, the hydrogel exhibits a higher elastic modulus, enhancing its load-bearing capacity and durability. The innovative approach of incorporating NaAc·3H2O crystals to create inhomogeneous structural distributions within hydrogels enhances both their lubrication effectiveness and mechanical robustness. This heterogeneous structure thus combines flexibility with toughness, showcasing the hydrogel’s dual functionality. Therefore, the inhomogeneous crystal hydrogel represents a promising material for hydration lubrication applications, offering tailored properties that mimic natural lubrication systems effectively. These advancements can significantly improve the performance and application range of hydration lubrication systems in medical and engineering fields, fostering the development of more efficient and durable biomimetic materials.
Crystal hydrogel materials, capable of undergoing repeated melting and recrystallization processes through external stimulation, present significant opportunities in information encryption communication due to associated changes in optical transparency. These optical transitions, while potentially limiting in specific environments, are complemented by photoluminescence, providing a robust and versatile method for information encryption across diverse settings.
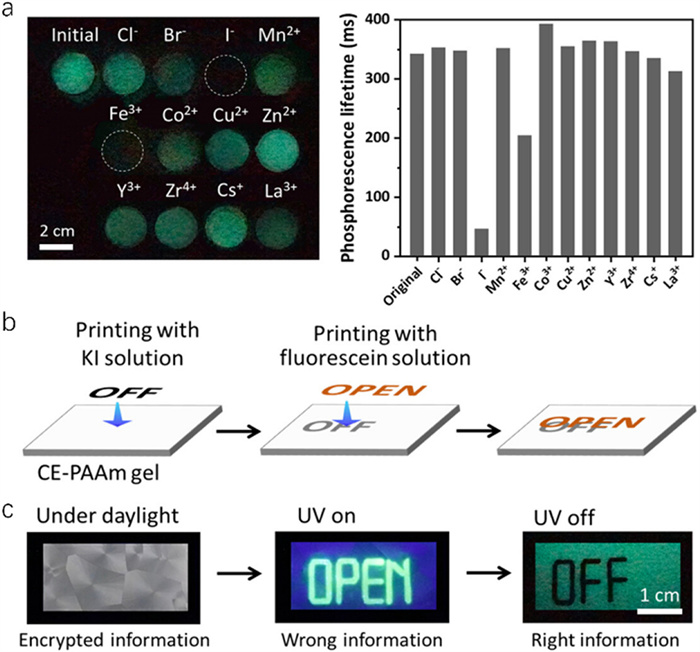
The innovative approach induces the crystallization of CE within hydrogels, which produces room-temperature phosphorescence properties under ultraviolet irradiation [38]. The quenching effect of diverse ions on phosphorescence exhibits notable variations, with iodine and iron ions exhibiting particularly significant impacts on the phosphorescent properties (Fig. 12a). An efficient information encryption and display system can be developed by quenching the phosphorescence and re-exciting it (Fig. 12b). This approach enhances the security features of crystal hydrogels, making them suitable for use in complex environmental conditions. The targeted phosphorescence quenching by specific ions improves security, ensuring that the encrypted information can only be revealed under controlled conditions (Fig. 12c). Crystal hydrogels, with their unique optical properties and dynamic response to external stimuli, hold significant promise for advancing the field of information encryption. They offer innovative solutions for enhancing information security, making them ideal for applications requiring robust and versatile encryption methods. In conclusion, crystal hydrogels represent an excellent material for information encoding with great potential for development. Their ability to combine photoluminescence with dynamic structural transformations provides a foundation for future advancements in secure communication technologies.
Recently, crystal hydrogels have emerged as a popular topic, showcasing many unique properties. Their synthetic methods have been summed up in this review, primarily based on inducing fast crystallization within polymer matrices. It predominantly relies on the various methods for inducing crystal nucleation and growth, including the crystallization of phase change materials at supersaturated states, polymerization-induced crystallization, and redox-involved crystallization of crystals. The obtained crystal networks would substantially enhance the hydrogels, resulting in distinctive characteristics as opposed to the hydrogels in crystal-free states. The sharpest difference is material’s rigidity, for example, the 25,000-fold enhancement in stiffness observed in NaAc-based crystal hydrogels. Besides mechanical properties, the crystal fillers within the hydrogel matrix significantly enhance the material’s exceptional characteristics, encompassing thermo-responsiveness, Ostwald ripening, superior electrochemical properties, self-adaptability, photoluminescence, shape memory effects, etc. Owing to these noteworthy attributes, crystal hydrogels have garnered significant attention in diverse research domains, including applications in soft robotics, adhesives, energy storage devices, sensors, hydrated lubrication systems, and information encryption. These unique features and interesting applications have promoted crystal hydrogels to be a new field that possesses promising developmental prospects.
Despite notable advancements in the field of crystal hydrogels, the technology remains in its nascent stage, leaving ample room for further research. In principle, various methods of inserting crystals into a hydrogel matrix can be used to design crystal hydrogels, but few systems are involved at present. Since suitable crystal systems for use as fillers are abundant, there is a large space to be explored by designing innovative crystal hydrogels. In the reported studies, crystals in hydrogels are mainly employed for structural reinforcement. The functionalities of the crystal molecules themselves are rarely utilized. Hence, it could be a direction for forthcoming studies to prioritize the integration of functional crystals with hydrogels, aiming to explore and elaborate on the unique functionalities and applications of crystal hydrogels. Of course, there are also many challenges. For example, the intricate interaction between polymeric networks and crystals is crucial for the progression of crystallization, yet the process is rapid and complex due to the concurrent variations in numerous parameters (polymer chain conformations, temperatures, crystallization velocity, etc.). Therefore, a sustained and rigorous exploration of crystal growth mechanisms is imperative to fully realize the potential of crystal hydrogels in the fields of smart materials and biotechnology.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Qianwei Liu: Writing – review & editing, Writing – original draft. Xinhong Xiong: Writing – review & editing, Supervision. Numan Ahmed: Writing – review & editing. Peisong Tang: Writing – review & editing. Jiaxi Cui: Writing – review & editing, Funding acquisition, Conceptualization.
This work was financially supported by the National Natural Science Foundation of China (Nos. 52203135 and 51973023), Huzhou Science and Technology Program Projects (No. 2023GZ18), Zhejiang Postdoctoral Research Project (No. ZJ2023133), and Science and Technology Cooperation Fund Program of Chengdu–Chinese Academy of Sciences (2023-2025).
J. Wang, X. Ge, Y. Xiang, et al., Chin. Chem. Lett. 36 (2025) 109819. doi: 10.1016/j.cclet.2024.109819
X. Liang, C. Huang, H. Liu, et al., Chin. Chem. Lett. 35 (2024) 109442. doi: 10.1016/j.cclet.2023.109442
M.A. Ghanem, A. Basu, R. Behrou, N. Boechler, et al., Nat. Rev. Mater. 6 (2021) 84–98.
Y. Kamiyama, R. Tamate, T. Hiroi, et al., Sci. Adv. 8 (2022) eadd0226. doi: 10.1126/sciadv.add0226
X. Xiong, H. Wang, L. Xue, J. Cui, Angew. Chem. Int. Ed. 62 (2023) e202306565. doi: 10.1002/anie.202306565
N. Gao, Y. Zhang, Z. Yang, et al., Chin. Chem. Lett. 35 (2024) 108820. doi: 10.1016/j.cclet.2023.108820
T. Zhang, X. Liang, L. Wang, et al., Chin. Chem. Lett. 36 (2025) 109889. doi: 10.1016/j.cclet.2024.109889
L. Chen, C. Zhao, J. Huang, J. Zhou, M. Liu, Nat. Commun. 13 (2022) 6821. doi: 10.1038/s41467-022-34677-9
Y. Fang, C. Liang, V. Liljestrom, et al., Adv. Mater. 36 (2024) 2402282. doi: 10.1002/adma.202402282
V.G. Muir, J.A. Burdick, Chem. Rev. 121 (2021) 10908–10949. doi: 10.1021/acs.chemrev.0c00923
R. Song, H. Xie, G. Liu, Chin. Chem. Lett. 36 (2025) 110442. doi: 10.1016/j.cclet.2024.110442
M. Wang, Y. Luo, T. Wang, et al., Adv. Mater. 33 (2020) 2003014.
Q. Zhang, S. Niu, L. Wang, et al., Adv. Mater. 30 (2018) 1801435. doi: 10.1002/adma.201801435
T. Ku, J. Swaney, J.Y. Park, et al., Nat. Biotechnol. 34 (2016) 973–981. doi: 10.1038/nbt.3641
Z. Hu, H. Zhang, Z. Li, et al., Chin. Chem. Lett. 35 (2024) 109527. doi: 10.1016/j.cclet.2024.109527
X. Li, J.P. Gong, Nat. Rev. Mater. 9 (2024) 380–398. doi: 10.1038/s41578-024-00672-3
Y.S. Zhang, A. Khademhosseini, Science 356 (2017) eaaf3627. doi: 10.1126/science.aaf3627
J.Y. Sun, X. Zhao, W.R. Lleperuma, et al., Nature 489 (2012) 133–136. doi: 10.1038/nature11409
X. Liu, J. Liu, S. Lin, X. Zhao, Mater. Today 36 (2020) 102–124. doi: 10.1016/j.mattod.2019.12.026
L. Hu, Y. Wang, Q. Liu, et al., Chin. Chem. Lett. 34 (2023) 108262. doi: 10.1016/j.cclet.2023.108262
X. Li, X. Ding, J. Zhou, et al., Chin. Chem. Lett. 35 (2024) 109158. doi: 10.1016/j.cclet.2023.109158
X. Zhang, B. Wu, S. Sun, P. Wu, Adv. Funct. Mater. 30 (2020) 1910425. doi: 10.1002/adfm.201910425
S. Choi, Y. Choi, J. Kim, Adv. Funct. Mater. 29 (2019) 1904342. doi: 10.1002/adfm.201904342
X. Cao, L. Sun, D. Xu, et al., Adv. Sci. 10 (2023) 2300902. doi: 10.1002/advs.202300902
J. Gong, Y. Katsuyama, T. Kurokawa, Y. Osada, Adv. Mater. 15 (2003) 1155–1158. doi: 10.1002/adma.200304907
J. Gong, Soft Matter 6 (2010) 2583–2590. doi: 10.1039/b924290b
S. Sugiyama, M. Maruyama, G. Sazaki, et al., J. Am. Chem. Soc. 134 (2012) 5786–5789. doi: 10.1021/ja301584y
R. Contreras-Montoya, M. Arredondo-Amador, G. Escolano-Casado, et al., ACS Appl. Mater. Interfaces 13 (2021) 11672–11682. doi: 10.1021/acsami.1c00639
L. Mao, H. Gao, H. Yao, et al., Science 354 (2016) 107–110. doi: 10.1126/science.aaf8991
Y. Ju, Y. Zhao, Q. Guan, et al., Angew. Chem. Int. Ed. 61 (2022) e202211254. doi: 10.1002/anie.202211254
H. Wei, B. Zhang, M. Lei, et al., ACS Nano 16 (2022) 4734–4745. doi: 10.1021/acsnano.1c11589
N. Kim, H. Lee, G. Han, et al., Adv. Sci. 10 (2023) 2300816. doi: 10.1002/advs.202300816
F.K. Yang, A. Cholewinski, L. Yu, G. Rivers, B. Zhao, Nat. Mater. 18 (2019) 874–882. doi: 10.1038/s41563-019-0434-0
Q. Liu, Y. Fang, X. Xiong, W. Xu, J. Cui, Angew. Chem. Int. Ed. 63 (2024) e202320095. doi: 10.1002/anie.202320095
T.B.H. Schroeder, J. Aizenberg, Nat. Commun. 13 (2022) 259.
J. Wei, G. Wei, Y. Shang, et al., Adv. Mater. 31 (2019) 1900248. doi: 10.1002/adma.201900248
Y. Fang, X. Xiong, L. Yang, et al., Adv. Funct. Mater. 33 (2023) 2301505. doi: 10.1002/adfm.202301505
H. Ju, H. Zhang, L. Hou, et al., J. Am. Chem. Soc. 145 (2023) 3763–3773. doi: 10.1021/jacs.2c13264
Y. Fang, Z. Bai, L. Yang, et al., Adv. Funct. Mater. 34 (2024) 2314353. doi: 10.1002/adfm.202314353
J. Wei, R. Li, L. Li, W. Wang, T. Chen, Nano-Micro Lett. 14 (2022) 182. doi: 10.1007/s40820-022-00931-4
J. Wei, C. Yin, H. Wang, Q. Wang, J. Mater. Chem. A 6 (2018) 58–64. doi: 10.1039/C7TA09616J
Z. Tai, J. Wei, J. Zhou, et al., Nat. Commun. 11 (2020) 1843. doi: 10.1038/s41467-020-15415-5
Y. Yan, Y. Shi, C. Liu, et al., ACS Appl. Mater. Interfaces 15 (2023) 48632–48644. doi: 10.1021/acsami.3c10271
Y. Zhao, Y. Chen, X. Song, et al., Adv. Funct. Mater. 33 (2023) 2304439. doi: 10.1002/adfm.202304439
H. Jing, L. Xu, X. Wang, Y. Liu, J. Hao, J. Mater. Chem. A 9 (2021) 19914–19921. doi: 10.1039/d1ta02791c
P. Li, T. Hu, T. Luo, et al., Chem. Eng. J. 478 (2023) 147380. doi: 10.1016/j.cej.2023.147380
S. Deng, L. Huang, J. Wu, et al., Adv. Mater. 33 (2021) 2008119. doi: 10.1002/adma.202008119
K. Matuszek, M. Kar, J.M. Pringle, D.R. MacFarlane, Chem. Rev. 123 (2023) 491–514. doi: 10.1021/acs.chemrev.2c00407
M.A. Gerkman, G.G.D. Han, Joule 4 (2020) 1621–1625. doi: 10.1016/j.joule.2020.07.011
G. Wang, Z. Tang, Y. Gao, et al., Chem. Rev. 123 (2023) 6953–7024. doi: 10.1021/acs.chemrev.2c00572
G.C. Sosso, J. Chen, S.J. Cox, et al., Chem. Rev. 116 (2016) 7078–7116. doi: 10.1021/acs.chemrev.5b00744
Y. Zhao, X. Zhang, X. Xu, S. Zhang, J. Energy Storage 27 (2020) 101156. doi: 10.1016/j.est.2019.101156
I. Shamseddine, F. Pennec, P. Biwole, F. Fardoun, Renew. Sust. Energ. Rev. 158 (2022) 112172. doi: 10.1016/j.rser.2022.112172
L. Wei, K. Ohsasa, ISIJ Int. 50 (2010) 1265–1269. doi: 10.2355/isijinternational.50.1265
M. Dannemand, J.M. Schultz, J.B. Johansen, S. Furbo, Appl. Therm. Eng. 91 (2015) 671–678. doi: 10.1016/j.applthermaleng.2015.08.055
T. Vetter, M. Iggland, D.R. Ochsenbein, F.S. Hänseler, M. Mazzotti, Cryst. Growth Des. 13 (2013) 4890–4905. doi: 10.1021/cg4010714
X. Liu, P.D. Sawant, W. Tan, et al., J. Am. Chem. Soc. 124 (2002) 15055–15063. doi: 10.1021/ja0206137
J. Yang, G. Qi, Y. Liu, et al., Carbon 100 (2016) 693–702. doi: 10.1016/j.carbon.2016.01.063
J. Yang, G. Qi, L. Tang, et al., J. Mater. Chem. A 4 (2016) 9625–9634. doi: 10.1039/C6TA03733J
X. Zhao, L. Peng, Y. Chen, et al., Mater. Horiz. 8 (2021) 1230–1241. doi: 10.1039/D0MH02069A
T. Kim, J.S. Lee, G. Lee, et al., Nano Energy 31 (2017) 160–167. doi: 10.1016/j.nanoen.2016.11.014
C. Han, X. Qian, Q. Li, et al., Science 368 (2020) 1091–1098. doi: 10.1126/science.aaz5045
J. Duan, G. Feng, B. Yu, et al., Nat. Commun. 9 (2018) 5146. doi: 10.1038/s41467-018-07625-9
J.A. Dirksen, T.A. Ring, Chem. Eng. Sci. 46 (1991) 2389–2427. doi: 10.1016/0009-2509(91)80035-W
D. Kashchiev, J. Chem. Phys. 118 (2003) 1837–1851. doi: 10.1063/1.1531614
J.J. De Yoreo, P.G. Vekilov, Rev. Mineral. Geochem. 54 (2003) 57–93. doi: 10.2113/0540057
R. Long, C. Hui, Soft Matter 12 (2016) 8069–8086. doi: 10.1039/C6SM01694D
J.R. Capadona, K. Shanmuganathan, D.J. Tyler, S.J. Rowan, C. Weder, Science 319 (2008) 1370–1374. doi: 10.1126/science.1153307
I. Ha, M. Kim, K.K. Kim, et al., Adv. Sci. 8 (2021) 2102536. doi: 10.1002/advs.202102536
M. Sun, Y. Zhong, J. Yao, Angew. Chem. Int. Ed. 57 (2018) 7820–7825. doi: 10.1002/anie.201803546
L. Mei, S. An, K. Hu, et al., Angew. Chem. Int. Ed. 59 (2020) 16061–16068. doi: 10.1002/anie.202003808
C. Sun, J. Luo, S. Yan, et al., Adv. Funct. Mater. 33 (2022) 2211035.
W. Zhang, T. Aida, Science 337 (2012) 1462–1463. doi: 10.1126/science.1228178
Y. Cui, Y. Wang, Z. Shao, et al., Adv. Mater. 32 (2020) 1908249. doi: 10.1002/adma.201908249
B. Li, J.J. Whalen, M.S. Humayun, M.E. Thompson, Adv. Funct. Mater. 30 (2020) 1907478. doi: 10.1002/adfm.201907478
M. Li, Y. Shi, H. Gao, Z. Chen, Adv. Funct. Mater. 30 (2020) 1910328. doi: 10.1002/adfm.201910328
A. Kotikian, C. McMahan, E.C. Davidson, et al., Sci. Robot. 4 (2019) eaax7044. doi: 10.1126/scirobotics.aax7044
W. Ostwald, Z. Phys. Chem. 22 (1897) 289–330. doi: 10.1515/zpch-1897-2233
B. Yao, S. Wu, R. Wang, et al., Adv. Funct. Mater. 32 (2022) 2109506. doi: 10.1002/adfm.202109506
K. Leng, G. Li, J. Guo, et al., Adv. Funct. Mater. 30 (2020) 2001317. doi: 10.1002/adfm.202001317
G. Yang, Z. Hao, C. Fang, et al., Chin. Chem. Lett. 36 (2025) 111185. doi: 10.1016/j.cclet.2025.111185
N. Rauner, M. Meuris, M. Zoric, J.C. Tiller, Nature 543 (2017) 407–410. doi: 10.1038/nature21392
M. Yu, Y. Lu, H. Zheng, X. Lu, Chem. Eur. J. 24 (2018) 3639–3649. doi: 10.1002/chem.201704420
H. Cho, G. Wu, J.C. Jolly, et al., Proc. Natl. Acad. Sci. U. S. A. 116 (2019) 13774–13779. doi: 10.1073/pnas.1818534116
J. Cui, D. Drotlef, I. Larraza, et al., Adv. Mater. 24 (2012) 4601–4604. doi: 10.1002/adma.201200895
X. Ma, W. Zhang, Z. Liu, et al., Adv. Mater. 33 (2021) 2007476. doi: 10.1002/adma.202007476
J. Yang, X. Zhen, B. Wang, et al., Nat. Commun. 9 (2018) 840. doi: 10.1071/mf17375
B. Ding, L. Ma, Z. Huang, X. Ma, H. Tian, Sci. Adv. 7 (2021) eabf9668. doi: 10.1126/sciadv.abf9668
Q. Zhou, Z. Wang, X. Dou, et al., Mater. Chem. Front. 3 (2019) 257–264. doi: 10.1039/c8qm00528a
P. Wei, X. Zhang, J. Liu, et al., Angew. Chem. Int. Ed. 59 (2020) 9293–9298. doi: 10.1002/anie.201912155
Y. Xia, Y. He, F. Zhang, Y. Liu, J. Leng, Adv. Mater. 33 (2021) 2000713. doi: 10.1002/adma.202000713
Q.J. Ze, X. Kuang, S. Wu, et al., Adv. Mater. 32 (2020) 1906657. doi: 10.1002/adma.201906657
J. Wu, Z. Zhang, Z. Wu, et al., Adv. Funct. Mater. 33 (2023) 2210395. doi: 10.1002/adfm.202210395
Y. Cui, D. Li, C. Gong, C. Chang, ACS Nano 15 (2021) 13712–13720. doi: 10.1021/acsnano.1c05019
J. Shintake, V. Cacucciolo, D. Floreano, H. Shea, Adv. Mater. 30 (2018) 1707035. doi: 10.1002/adma.201707035
G.M. Whitesides, Angew. Chem. Int. Ed. 57 (2018) 4258–4273. doi: 10.1002/anie.201800907
C.S. Park, Y. Kang, H. Na, J. Sun, Prog. Polym. Sci. 150 (2024) 101791. doi: 10.1016/j.progpolymsci.2024.101791
C. Ghobril, M.W. Grinstaff, Chem. Soc. Rev. 44 (2015) 1820–1835. doi: 10.1039/C4CS00332B
T. Chen, Y. Chen, H.U. Rehman, et al., ACS Appl. Mater. Interfaces 10 (2018) 33523–33531. doi: 10.1021/acsami.8b10064
Y. Hong, F. Zhou, Y. Hua, et al., Nat. Commun. 10 (2019) 2060. doi: 10.1038/s41467-019-10004-7
L. Zhou, C. Dai, L. Fan, et al., Adv. Funct. Mater. 31 (2021) 2007457. doi: 10.1002/adfm.202007457
J. Shin, J.S. Lee, C.Y. Lee, et al., Adv. Funct. Mater. 25 (2015) 3814–3824. doi: 10.1002/adfm.201500006
Z. Jia, X. Lv, Y. Hou, et al., Bioact. Mater. 6 (2021) 2676–2687.
H. Yuk, C.E. Varela, C.S. Nabzdyk, et al., Nature 575 (2019) 169–174. doi: 10.1038/s41586-019-1710-5
M. Singh, D.L. Teodorescu, M. Rowlett, et al., Adv. Mater. 36 (2024) 2307288. doi: 10.1002/adma.202307288
L. Wang, Y. Wang, X. Bo, et al., Adv. Funct. Mater. 32 (2022) 2204304. doi: 10.1002/adfm.202204304
Z. Zhang, C. Qin, H. Feng, et al., Nat. Commun. 13 (2022) 6964. doi: 10.1038/s41467-022-34816-2
Y. Xue, J. Zhang, X. Chen, et al., Adv. Funct. Mater. 31 (2021) 2106446. doi: 10.1002/adfm.202106446
J. Yang, R. Bai, B. Chen, Z. Suo, Adv. Funct. Mater. 30 (2020) 1901693. doi: 10.1002/adfm.201901693
S. Wu, C. Cai, F. Li, Z. Tan, S. Dong, Angew. Chem. Int. Ed. 59 (2020) 11871–11875. doi: 10.1002/anie.202004104
X. Li, Z. Wang, W. Li, J. Sun, ACS Mater. Lett. 3 (2021) 875–882. doi: 10.1021/acsmaterialslett.1c00167
Y. Chen, J. Meng, Z. Gu, et al., Adv. Funct. Mater. 30 (2020) 1905287. doi: 10.1002/adfm.201905287
Z. Wang, H. Li, Z. Tang, et al., Adv. Funct. Mater. 28 (2018) 1804560. doi: 10.1002/adfm.201804560
Q. Liu, Z. Yu, Q. Zhuang, et al., Adv. Mater. 35 (2023) 2300498. doi: 10.1002/adma.202300498
Y. Wang, K. Ding, X. Gong, et al., Chin. Chem. Lett. (2025), doi: 10.1016/j.cclet.2025.111078.
S. Chen, P. Sun, B. Sun, et al., Energy Storage Mater. 37 (2021) 598–608. doi: 10.1016/j.ensm.2021.02.038
S. Chen, P. Sun, J. Humphreys, et al., Energy Storage Mater. 42 (2021) 240–251. doi: 10.1016/j.ensm.2021.07.033
L. Suo, O. Borodin, Y. Wang, et al., Adv. Energy Mater. 7 (2017) 1701189. doi: 10.1002/aenm.201701189
L. Suo, F. Han, X. Fan, et al., J. Mater. Chem. A 4 (2016) 6639–6644. doi: 10.1039/C6TA00451B
D. Bao, Z. Wen, J. Shi, et al., J. Mater. Chem. A 8 (2020) 13787–13794. doi: 10.1039/d0ta03215h
S. Subramanian, H.Y. Wu, T. Constant, J. Xavier, F. Vollmer, Adv. Mater. 30 (2018) 1801246. doi: 10.1002/adma.201801246
R. Khodambashi, Y. Alsaid, R. Rico, et al., Adv. Mater. 33 (2021) 2005906. doi: 10.1002/adma.202005906
Y. Yan, S. Duan, B. Liu, et al., Adv. Mater. 35 (2023) 2211673. doi: 10.1002/adma.202211673
S. Wang, J.Y. Oh, J. Xu, H. Tran, Z. Bao, Acc. Chem. Res. 51 (2018) 1033–1045. doi: 10.1021/acs.accounts.8b00015
X. Qiao, Z. Xu, Z. Wei, et al., Chin. Chem. Lett. 36 (2025) 110884. doi: 10.1016/j.cclet.2025.110884
Y. Chang, L. Wang, R. Li, et al., Adv. Mater. 33 (2021) 2003464. doi: 10.1002/adma.202003464
S.R.A. Ruth, V.R. Feig, H. Tran, Z. Bao, Adv. Funct. Mater. 30 (2020) 2003491. doi: 10.1002/adfm.202003491
B. Zhu, H. Wang, Y. Liu, et al., Adv. Mater. 28 (2016) 1559–1566. doi: 10.1002/adma.201504754
Z.H. Yang, J. Yin, L. Xin, et al., Chin. Chem. Lett. 35 (2024) 109558. doi: 10.1016/j.cclet.2024.109558
M. Kim, S. Hong, J.J. Park, et al., Adv. Mater. 36 (2024) 2313344. doi: 10.1002/adma.202313344
L. Xiang, J. Liang, Z. Wang, et al., Sci. Adv. 9 (2023) eadc9375. doi: 10.1126/sciadv.adc9375
I. Sensoy, Curr. Res. Food Sci. 4 (2021) 308–319. doi: 10.1016/j.crfs.2021.04.004
A. Sarkar, E. Andablo-Reyes, M. Bryant, D. Dowson, A. Neville, Curr. Opin. Colloid Interface Sci. 39 (2019) 61–75. doi: 10.1016/j.cocis.2019.01.008
Z. Zhang, C. Shen, P. Zhang, et al., Adv. Colloid Interface Sci. 327 (2024) 103145. doi: 10.1016/j.cis.2024.103145
W. Lin, J. Klein, Acc. Mater. Res. 3 (2022) 213–223. doi: 10.1021/accountsmr.1c00219
T. Shoaib, R.M. Espinosa-Marzal, Colloids Interfaces 4 (2020) 54. doi: 10.3390/colloids4040054
X. Zhang, J. Wang, H. Jin, S. Wang, W. Song, J. Am. Chem. Soc. 140 (2018) 3186–3189. doi: 10.1021/jacs.7b12886
J. Kang, X. Zhang, X. Yang, et al., Adv. Mater. 35 (2023) 2307705. doi: 10.1002/adma.202307705
J. Huang, Y. Tang, P. Wang, et al., Adv. Mater. 36 (2024) 2309141. doi: 10.1002/adma.202309141
M.H. Bai, B. Zhao, Z.Y.T. Liu, et al., Adv. Mater. 34 (2022) 2108848. doi: 10.1002/adma.202108848
T.A. Schmidt, Science 370 (2020) 288–289. doi: 10.1126/science.abd3831
Y. Lei, X. Wang, J. Liao, et al., Bioact. Mater. 16 (2022) 472–484.

Figure 2 Phase-change-induced crystallization in crystal hydrogel. (a) Strategies for crystal growth induced by mismatch nucleation in polymer network matrix. Polarizing micrographs showing the crystallization process of the crystal hydrogel. Copied with permission [34]. Copyright 2024, Wiley-VCH. (b) Crystal morphology images of polymerized and unpolymerized regions in NaAc-based crystal hydrogels. Copied with permission [35]. Copyright 2022, Springer Nature. (c) Mechanism diagram of armor formation by restricting the growth of NaAc crystals on the surface of the PAA matrix. Copied with permission [44]. Copyright 2023, Wiley-VCH.

Figure 3 Polymerization-induced CE crystallization in crystal hydrogel. (a) Photos showing the polymerization-induced precipitation and crystallization of CE during the synthesis of the hydrogel at 70 ℃. Copied with permission [38]. Copyright 2023, American Chemical Society. (b) Schematic of the redox-induced [Fe(CN)6]4− crystallization. Photos of EG/H2O solutions with [Fe(CN)6]4−/3− (0.125 mol/L) at different EG contents at room temperature (upper) and 80 ℃ (lower). Optical micrographs show heating-induced dissolution and cooling-induced recrystallization of [Fe(CN)6]4− in the crystal hydrogel. Copied with permission [46]. Copyright 2023, Elsevier B.V.

Figure 4 Mechanical properties and thermo-responsiveness of the NaAc-based crystal hydrogel. (a) The NaAc-based crystal hydrogel shows the transition from a soft and adaptive state to a rigid and fixed state. Copied with permission [39]. Copyright 2024, Wiley-VCH. (b) Spatial temperature distribution of NaAc·3H2O crystal growth in the unpolymerized (left) and polymerized regions (right), recorded every 5 seconds. Copied with permission [35]. Copyright 2022, Springer Nature.

Figure 5 Design of NaAc-based crystal hydrogels utilizing the OR process. (a) Schematic of OR progression in NaAc·3H2O fibrous crystal within the NaAc-based crystal hydrogels, illustrating changes in material properties. (b) Transformation of the crystal hydrogel from a transparent, crystal-free soft state to an opaque, rigid state, and then to an opaque, soft state. (c) Preprogrammed shape recovery behavior of the crystal hydrogel. Copied with permission [34]. Copyright 2024, Wiley-VCH.

Figure 6 Electrochemical and self-adaptivity properties of the NaAc-based crystal hydrogel. (a) Design of the NaAc·3H2O crystal processable ultra-tough hydrogel electrolyte (up). Schematic illustration of the ion migration channel built by zwitterionic groups under an external electric field in NaAc·3H2O crystal hydrogel (bottom, left). Extreme temperature tolerance of NaAc-based crystal hydrogel supercapacitors (bottom, right). Copied with permission [36]. Copyright 2019, Wiley-VCH. (b) Complex surface morphologies and self-adaptivity of the NaAc-based crystal hydrogel. Copied with permission [39]. Copyright 2024, Wiley-VCH.

Figure 7 Photoluminescence and shape memory properties of crystal hydrogels. (a) Polymerization-induced crystallization of CE results in crystals embedded and constrained in the polymeric matrix, acting as cluster phosphors with potential for enhanced optical properties. The luminescent photographs of the CE-based hydrogel were taken under 280 nm UV irradiation and at different time intervals after turning off the UV lamp. Copied with permission [38]. Copyright 2023, American Chemical Society. (b) Graphs of NaAc-based crystal hydrogels for shape fixation (crystallization) and shape recovery (melting). Copied with permission [39]. Copyright 2024, Wiley-VCH.

Figure 8 (a) Schematic illustration of the gripping and manipulation process of the crystal hydrogel gripper. Digital photographs of the gripping, transportation, and release process of different materials using the phase change gel gripper. Copied with permission [60]. Copyright 2021, Royal Society of Chemistry. (b) Kinematics and expandability of NaAc·3H2O crystal hydrogel. Copied with permission [69]. Copyright 2021, Wiley-VCH. (c) The artificial tactile feedback system for adaptive grasping of the CaCl2·6H2O crystal hydrogel. Copied with permission [45] Copyright 2021, Royal Society of Chemistry.

Figure 9 (a) Surface adaptation of the NaAc·3H2O soft crystal hydrogel (increase contact area) and the crystal solidification (lock the interface). (b) Demonstration of the instant and robust self-adhesion of the NaAc·3H2O crystal hydrogel. Copied with permission [37]. Copyright 2023, Wiley-VCH.

Figure 10 (a) Strong and anti-freezing crystal hydrogel electrolyte through the synergy of co-nonsolvency and salting-out. Copied with permission [123]. Copyright 2023, Wiley-VCH. (b) Schematic illustration of a biological sensory feedback system and an artificial tactile feedback system. Copied with permission [45]. Copyright 2021, Royal Society of Chemistry. (c) Gradient stiffness-programmed circuit board of crystal hydrogel. Copied with permission [130]. Copyright 2024, Wiley-VCH.

Figure 11 (a) The mechanism of inhomogeneous structure crystal hydrogel for hydration lubrication. (b) Scheme of fabricating NaAc-based crystal hydrogel coating by heat-UV irradiation and the cooling method. Visual demonstration of the hydrated lubrication performance of the inhomogeneous NaAc-based crystal hydrogel coating iron substrate (uncoated and coated). Copied with permission [43]. Copyright 2023, American Chemical Society.

Figure 12 (a) Information encryption phosphorescence photos of CE-based crystal hydrogel treated by different ions and the corresponding phosphorescence lifetimes. (b) Schematic for the encoding of digital information by printing of KI solution and fluorescein solution on the CE-based crystal hydrogel. (c) Photos of the patterned CE-based crystal hydrogel used for information encryption under daylight and UV light, as well as just after turning off the UV lamp. Copied with permission [38]. Copyright 2023, American Chemical Society.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:
 下载:
下载: