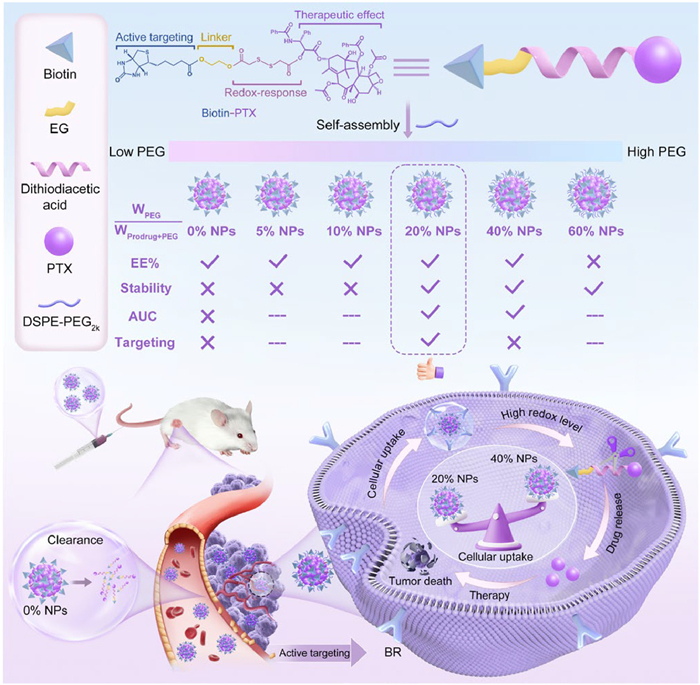
Scheme 1.
The content of DSPE-PEG2k influenced the stability and targeting efficiency of Biotin-PTX NPs.
Fine-tuning PEGylation: Leveraging the self-assembly stability and targeting efficiency of biotinylated-paclitaxel prodrug nanoassemblies
Lurong Zhang , Xin Wang , Xuan Li , Minglong Huang , Bowen Zhang , Shengyao Xu , Jialin Xing , Yafan Xiao , Yi Zheng , Zhenzhen Zhao , Jin Sun , Zhonggui He , Lingxiao Li , Bingjun Sun

Cancer represents a major public health concern in the world, with the number of new cancer cases worldwide up to 20 million in 2022 [1]. Although some advancements have been made in cancer treatment so far, chemotherapy remains the irreplaceable mainstream regimen to treat cancer [2]. However, most chemotherapy drugs suffer from the poor solubility, limited tumor specificity and serious off-target toxicity, leading to limited clinical applications [3,4]. For example, paclitaxel (PTX) is used as a first-line chemotherapeutic agent in the treatment of many cancers, including breast cancer, ovarian cancer and non-small cell lung cancer [5,6]. However, due to the poor water solubility of PTX, its commercial injection, named as Taxol, has to use polyoxyethylene castor oil and anhydrous ethanol as the solubilizer, which could cause severe allergic reactions and tend to precipitate after dilution [7]. These drawbacks bring security risks and greatly limit the clinical application of Taxol. Therefore, it is important to design suitable delivery systems to settle these deficiencies.
Nano-delivery system offers an alternative to address the shortcomings of chemotherapy drugs [8,9]. For instance, Abraxane employs natural serum albumin to deliver PTX molecules, thereby eliminating the need for solubilizers and addressing the issue of allergies. However, Abraxane loads PTX through physical encapsulation, leading to complex preparation process, limited drug loading, and poor pharmaceutical behavior [10,11]. To address the above problems, a novel carrier-free prodrug self-assembled nano-delivery system has emerged [12]. It combines the advantages of prodrug strategy and nano-delivery system, exhibiting high drug loading, facile preparation, superior tumor selectivity and pharmaceutical behavior. Prodrugs typically consist of drug molecules linked by functional modules and tumor microenvironment sensitive modules [13,14]. As a lot of work reported, disulfide bond was widely utilized in the design of prodrug nanoassemblies (NPs) to achieve intelligent drug release in the tumor redox microenvironment [15,16]. Despite its good tumor selectivity, most existing prodrug nano-delivery systems can only accumulate at the tumor site through the enhanced permeability and retention effect (EPR), exhibiting limited tumor-targeting efficiency [17,18]. Therefore, the rational design of functional modules with tumor-targeting ability is crucial for the development of advanced NPs.
Biotin, also known as vitamin H, is an essential micronutrient that maintains normal physiological functions in humans. Compared with normal cells, cancer cells require greater quantity of biotin to maintain rapid growth and proliferation, leading to the overexpression of biotin receptors (BRs, proteins encoded by the SLC5A6 gene) in many cancer cells, including breast cancer cells, colon cancer cells, kidney cells and lung cancer cells [19–21]. There are numerous studies to attach biotin to carriers or drugs for tumor-targeting diagnosis and treatment [22–24]. However, studies on the integration of biotin as a functional module into NPs have not yet been reported. Notably, pure NPs are susceptible to clearance in vivo, leading to poor pharmacokinetic behavior and consequently limited tumor accumulation [25–27]. Polyethylene glycol (PEG) surface modification strategy is widely used to improve the stability and circulation time of nanomaterials. Especially, distearoyl phosphatidylethanolamine-polyethylene glycol2000 (DSPE-PEG2k) is the most representative derivatives of PEG, which is widely used in the marketed formulations, such as Doxil and AmBisome [28–30]. However, DSPE-PEG2k may mask the biotin target head, thereby affecting their tumor-targeting efficiency [31–33]. Therefore, it is crucial to optimize the content of DSPE-PEG2k to achieve a balance between the self-assembly stability, pharmacokinetic behavior, and tumor-targeting efficiency of NPs.
Based on the above considerations, we synthesized the targeting prodrug using PTX as the drug module, disulfide bond as the sensitive module, and biotin as the targeting functional module, named as Biotin-PTX (Scheme 1). Then, we investigated the self-assembly performance and targeting efficiency of Biotin-PTX with different DSPE-PEG2k ratios (0%, 5%, 10%, 20%, 40% and 60%, WPEG/Wprodrug+PEG) to form Biotin-PTX NPs. In details, PTX as the hydrophobic component tended to aggregate together, biotin as the hydrophilic component was exposed to the outside, and DSPE-PEG2k modified on the surface of nanoparticles. We found that the Biotin-PTX NPs exhibited poor stability at lower DSPE-PEG2k content (0%, 5%, 10%) and weak targeting efficiency at higher DSPE-PEG2k content (40%). When the content of DSPE-PEG2k was too high (60%), the encapsulation efficiency (EE%) was unsatisfactory. Interestingly, Biotin-PTX NPs containing 20% DSPE-PEG2k (20% NPs) exhibited significantly improved colloidal stability and tumor-targeting efficiency. As a result, 20% NPs exhibited good antitumor efficacy and safety compared with Taxol, Abraxane and Prodrug Sol. In summary, this work emphasized the key role of moderate PEGylation in regulating the self-assembly stability and tumor-targeting efficiency of NPs, providing a new way of thinking for tumor-targeting treatment.
The tumor-targeting prodrug was rationally designed and synthesized by conjugating PTX to biotin via a disulfide linker (Fig. S1 in Supporting information), and the resulting compound was denoted as Biotin-PTX. Given the well-documented targeting ability and specificity of biotin toward certain tumor cells, a control prodrug lacking biotin was not included in this study. The structure of Biotin-PTX was successfully identified by 1H nuclear magnetic resonance (1H NMR) and high-resolution mass spectrometry (HRMS) (Fig. S2 in Supporting information). In addition, the purity of Biotin-PTX exceeded 99%, meeting the requirements of the subsequent studies.
Due to the high hydrophobicity of Biotin-PTX, the Biotin-PTX NPs may be unstable and susceptible to clearance in the body, leading to poor pharmacokinetic behavior. To improve the blood circulation of Biotin-PTX NPs, we selected DSPE-PEG2k to modify their surface. However, it was still a problem to determine the optimal amount of DSPE-PEG2k. This was because too little DSPE-PEG2k may not be sufficient to enhance their stability, and too much may affect their tumor-targeting efficiency. Therefore, we modified Biotin-PTX NPs using different DSPE-PEG2k ratios (0%, 5%, 10%, 20%, 40% and 60%, WPEG/Wprodrug+PEG) and considered the colloidal stability and targeting efficiency to censor the optimal amount. The obtained formulations were denoted as 0% NPs, 5% NPs, 10% NPs, 20% NPs, 40% NPs and 60% NPs, respectively (Scheme 1). The preparation process of nanoassemblies was shown in Fig. 1A. The particle sizes were between 118 nm and 163 nm, which increased subsequently with the increase of DSPE-PEG2k ratios (Fig. 1B and Table S1 in Supporting information). This was because DSPE-PEG2k formed a hydration layer on the out layer of nanoparticles, thereby increasing their particle size. The more amount of DSPE-PEG2k, the larger hydration layer formed. The results from transmission electron microscopy (TEM) revealed that they were all homogeneous and spherical in structures. Interestingly, the EE% was around 88% when the DSPE-PEG2k ratio was 60%, which was significantly lower than other nanoparticle groups. This may be attributed to that an excessive amount of DSPE-PEG2k can act as a solubilizing agent, resulting in the partial dissolution of prodrugs, which cannot participate in the self-assembly process. Hence, the usage of DSPE-PEG2k should not be too much.
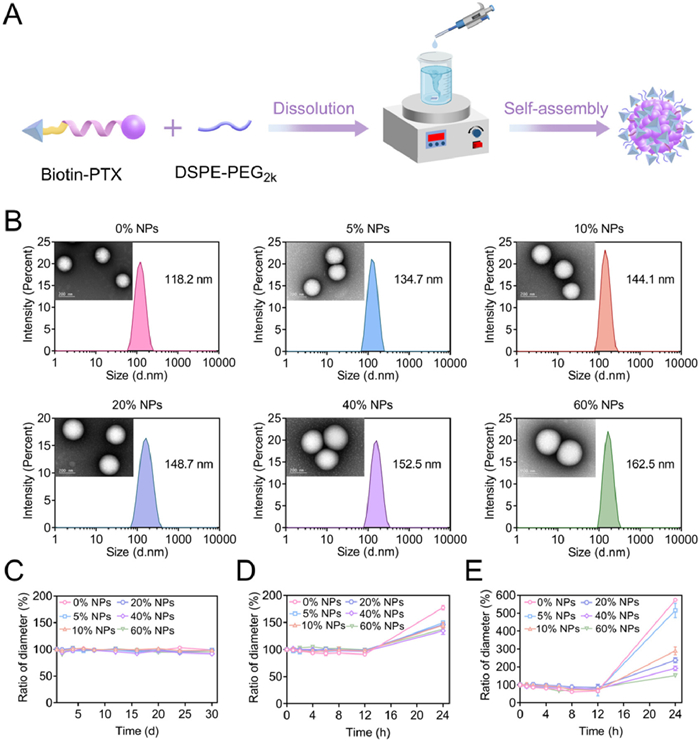
Then, the stability of Biotin-PTX NPs was determined at 4 ℃ using changes in particle size as an evaluation index. As shown in Fig. 1C, the particle size remained unchanged within 30 days, suggesting their excellent storage stability. Subsequently, the Biotin-PTX NPs were diluted with phosphate buffer saline (PBS, pH 7.4) containing 10% (v/v) fetal bovine serum (FBS). As shown in Fig. 1D, the particle size increased within 24 h, particularly the most obvious increase in 0% NPs. To further magnify the differences between Biotin-PTX NPs, we replaced 10% with 50% (v/v) FBS. Similar outcomes were displayed in Fig. 1E, the particle size was increased obviously in 0% NPs and 5% NPs within 24 h, while the particle size of 10% NPs, 20% NPs, 40% NPs and 60% NPs only slightly increased. Notably, as the content of DSPE-PEG2k increased, the particle size exhibited less change. These results illustrated that a sufficient DSPE-PEG2k content demonstrated relatively good assembly stability.
To investigate the self-assembly mechanism of Biotin-PTX, a molecular docking simulation was conducted. As shown in Fig. S3A (Supporting information), the self-assembly process of Biotin-PTX involved three types of intermolecular interactions, which were hydrophobic interaction, hydrogen bond and π-π stacking (a type of electrostatic interaction force). Moreover, the docking process between Biotin-PTX and DSPE-PEG2k involved two types of intermolecular interactions: hydrophobic interaction and hydrogen bond (Fig. S3B in Supporting information). Notably, no π-π stacking was detected after the addition of DSPE-PEG2k. This may be due to that the hydrophobic part of DSPE-PEG2k, when assembled with the precursor, it hinders the direct interaction between benzene ring and benzene ring (the main source of π-π stacking effect) to a certain extent, which making the π-π stacking weak and difficult to be detected by the software. To further validate the interactions within Biotin-PTX NPs, they were exposed to a series of concentrations of sodium chloride (NaCl, electrostatic interaction competitor), urea (hydrogen bond blocker), and sodium dodecyl sulfate (SDS, hydrophobic interaction competitor). As shown in Fig. S4 (Supporting information), after incubating Biotin-PTX NPs with urea or NaCl for 24 h, the particle size in each group increased. Meanwhile, as the DSPE-PEG2k ratio decreased, the particle size continued to expand, indicating that DSPE-PEG2k could improve the stability of nanoparticles to some extent, consistent with the results of molecular docking simulation. Especially, when Biotin-PTX NPs encountered SDS, the particle size and polydispersity index (PDI) in each group experienced significant expansion immediately, indicating the critical role of hydrophobic interactions in stabilizing the nanoassemblies. These findings revealed that the presence of intermolecular interactions within Biotin-PTX NPs, which facilitated their self-assembly process.
Considering the EE% and the stability of Biotin-PTX NPs, 20% NPs and 40% NPs were chosen for subsequent experiments, with 0% NPs as a control. Firstly, we analyzed the elemental distribution map and composition of these three nanoassemblies, which were characterized using scanning electron microscopy (SEM) and energy dispersive X-ray spectrometry (EDS). As shown in Fig. S5A (Supporting information), these three nanoassemblies exhibited near-homogeneous distributions of elements, further confirming the successful self-assembly of Biotin-PTX. Interestingly, the quantitative elemental analysis indicated that as the DSPE-PEG2k ratio increased, the content of phosphorus (P) increased, whereas the content of sulfur (S) decreased (Fig. S5B in Supporting information). This was because the increased DSPE-PEG2k content could lead to an increase in P content, which in turn masked the exposure of the S element in Biotin-PTX, indicating that excessive DSPE-PEG2k could mitigate the targeting efficiency of Biotin-PTX NPs.
Effective release of disulfide-bridged prodrugs under redox conditions and less release under normal conditions are relevant to their antitumor efficacy and safety. Therefore, the drug release profiles of 0% NPs, 20% NPs and 40% NPs were investigated respectively using glutathione (GSH) and hydrogen peroxide (H2O2) as reducing and oxidizing agents, with blank PBS to simulate normal conditions. As depicted in Fig. S6 (Supporting information), three nanoassemblies only released a small part of PTX in blank media, which may minimize premature drug leakage in the blood and reduce the systemic toxicity associated with PTX. In contrast, three nanoassemblies exhibited redox-sensitive drug release profiles, and the drug release trend was similar in the same media (Figs. 2A–D), suggesting that the DSPE-PEG2k coating on the Biotin-PTX NPs did not affect the drug release trend. Furthermore, even at smaller concentrations of GSH (0.1 mmol/L) and H2O2 (1 mmol/L), they released more PTX compared with the normal condition. These findings revealed that Biotin-PTX NPs could respond to the redox microenvironment to effectively release drugs, thereby facilitating their efficient antitumor efficacy. The drug release mechanism of Biotin-PTX was clarified in Fig. S7 (Supporting information), as reported in previous literatures [34–36]. When incubated with GSH, the disulfide bond underwent exchange reactions with GSH, forming hydrophilic intermediates that continued to hydrolyze and released PTX. The mass spectrometry confirmed the presence of PTX-SH intermediate and released PTX (Fig. S8 in Supporting information). When incubated with H2O2, the disulfide bond was oxidized to sulfoxide or sulfone, which increased the hydrophilicity of Biotin-PTX and subsequently facilitated the hydrolysis of the ester bond, leading to the release of PTX. Further mass spectrometric analysis of the release medium confirmed the presence of Biotin-PTX oxides and released PTX (Fig. S9 in Supporting information).
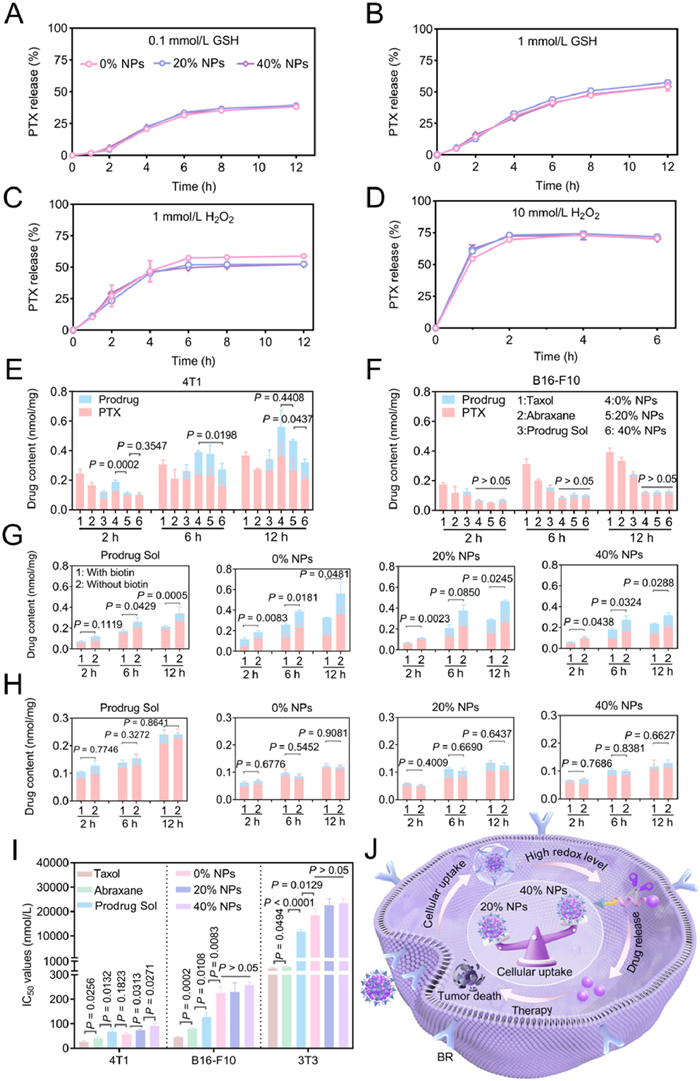
Biotin can enhance drug delivery by actively targeting biotin receptors on tumor cells. However, it was unknown whether the DSPE-PEG2k coating on the Biotin-PTX NPs affects cellular uptake. We therefore accurately examined the cellular uptake of Biotin-PTX NPs using ultra performance liquid chromatography-tandem mass spectrometry (UPLC-MS-MS). Prior experiments, we conducted a polymerase chain reaction (PCR) study on 4T1 cells, B16-F10 cells and 3T3 cells to detect the expression of biotin receptors (Fig. S10 in Supporting information). The results indicated that the expression of biotin receptors in 4T1 cells were dramatically higher than those in B16-F10 cells and 3T3 cells. Then, 4T1 cells (biotin receptor-positive) and B16-F10 cells (biotin receptor-negative) were selected to assess the cellular uptake of 0% NPs, 20% NPs and 40% NPs, with Taxol, Abraxane and Prodrug Sol as controls. As shown in Figs. 2E and F, three nanoassemblies can be effectively internalized by tumor cells and activated to release free PTX, and the cellular uptake increased with time. Specifically, the cellular uptake of three nanoassemblies by 4T1 cells was significantly higher than that by B16-F10 cells, which was in line with the PCR study. In 4T1 cells, the cellular uptake of three nanoassemblies followed the order of 0% NPs > 20% NPs > 40% NPs, suggesting that excessive DSPE-PEG2k coating may mask biotin on the surface of nanoparticles, resulting in low cellular uptake. Notably, 0% NPs and 20% NPs exhibited greater cellular uptake at 12 h compared with Taxol, Abraxane and Prodrug Sol, which may be attributed to biotin binding to biotin receptors. While in B16-F10 cells, the cellular uptake of three nanoassemblies was similar, yet lower than that of Taxol, Abraxane, and Prodrug Sol. This was because that the very low expression of biotin receptors in B16-F10 cells made the targeting efficiency of biotin negligible.
To further demonstrate biotin-mediated targeting, free biotin was added to bind to biotin receptors before incubating Prodrug Sol, 0% NPs, 20% NPs and 40% NPs with tumor cells. As depicted in Figs. 2G and H, the cellular uptake of Prodrug Sol and three nanoassemblies markedly decreased after 4T1 cells were pretreated with free biotin. In addition, because of fewer biotin receptors, the cellular uptake efficiency of B16-F10 cells was independent of biotin incorporation and lower than that of 4T1 cells. These findings revealed that biotin could mediate the access of Biotin-PTX NPs to cells that highly express biotin receptors.
Next, the cytotoxicity of 0% NPs, 20% NPs and 40% NPs was assessed against 4T1 cells, B16-F10 cells, and 3T3 cells, with Taxol, Abraxane and Prodrug Sol as controls. As depicted in Fig. 2I and Fig. S11 (Supporting information), three nanoassemblies and Prodrug Sol showed weaker cytotoxicity than Taxol and Abraxane because of the delayed drug release. In 4T1 cells, the median inhibitory concentration (IC50) values of three nanoassemblies followed the order of 0% NPs < 20% NPs < 40% NPs. In addition, they displayed lower cytotoxicity in B16-F10 cells, and the IC50 values were similar (Table S2 in Supporting information). These outcomes were attributed to that the Biotin-PTX NPs exerted cytotoxicity involving two steps: the uptake by the cell and the release of the prodrug inside the cell (Fig. 2J). In the experiments described above, we demonstrated that the content of DSPE-PEG2k had no effect on the release profiles (Figs. 2A–D). The lower amount of DSPE-PEG2k resulted in greater biotin exposure and cellular uptake. As a result, the stronger cytotoxicity was observed. Notably, Taxol and Abraxane exhibited a lower tumor selectivity index (TSI) compared with Prodrug Sol and three nanoassemblies. Among three nanoassemblies, 20% NPs showed the highest TSI, indicating its superior safety (Table S3 in Supporting information).
To further prove the existence of biotin-biotin receptor interactions, competitive cytotoxicity assays of Prodrug Sol, 0% NPs, 20% NPs and 40% NPs were conducted on 4T1 cells and B16-F10 cells in the presence of free biotin (Fig. S12 in Supporting information). With the increasing concentration of free biotin, the IC50 values of Prodrug Sol, 0% NPs, 20% NPs and 40% NPs increased sequentially in 4T1 cells, indicating that the cytotoxicity decreases sequentially. However, their IC50 values against B16-F10 cells remained similar at all biotin concentrations, with no significant differences. These results supported that the specific binding of Biotin-PTX NPs to biotin receptors is crucial for the tumor-targeting efficiency.
Before the in vivo experiments, we conducted a hemolysis assay on the 0% NPs, 20% NPs and 40% NPs. As shown in Fig. S13 (Supporting information), three nanoassemblies did not induce hemolysis, with the hemolysis percentage (HP%) being much less than 5%, meeting the requirement for intravenous injection. Given the significant impact of in vivo drug delivery on the therapeutic outcome of nanoassemblies, the pharmacokinetic behaviors of DiR Sol and DiR-labeled nanoassemblies (0% NPs, 20% NPs and 40% NPs) were therefore investigated using a fluorescence detection method (Fig. 3A). As illustrated in Fig. 3B, DiR Sol quickly disappeared in the blood, indicating poor pharmacokinetic performance of the free drug. Moreover, although the pharmacokinetic behavior of 0% NPs showed slight improvement compared with DiR Sol, the decline in blood concentration remained rapid. It was noteworthy that 20% NPs and 40% NPs demonstrated an extension in blood circulation, which may be favorable for their therapeutic efficiency. The area under the curve (AUC0-24 h) of 20% NPs and 40% NPs was 69.75 and 112.55 times greater than that of DiR Sol, which should be attributed to the excellent stability of PEGylated nanoassemblies (Table S4 in Supporting information). These results suggested that the DSPE-PEG2k coating on the Biotin-PTX NPs contributed to improving the pharmacokinetic behavior, laying the foundation for tumor accumulation.
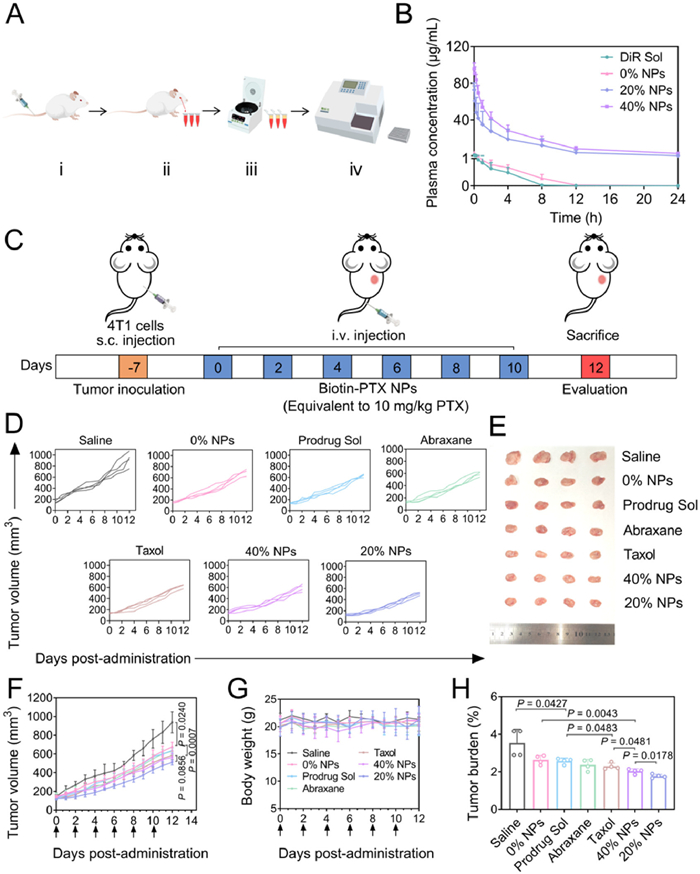
The accumulation of Biotin-PTX NPs in tumors was the key to achieve their therapeutic effect. Therefore, the fluorescence imaging was applied to investigate the tumor accumulation of DiR Sol and DiR-labeled nanoassemblies (0% NPs, 20% NPs and 40% NPs). As shown in Fig. S14 (Supporting information), DiR Sol and 0% NPs displayed negligible fluorescence signals in tumors, but high fluorescence signals were detected in spleens and livers, indicating their poor tumor accumulation and rapid metabolism in the body. In comparison, 20% NPs and 40% NPs exhibited higher tumor accumulation, and the fluorescence signals in tumors significantly increased from 1 h to 12 h. Notably, although the pharmacokinetic behavior of 20% NPs was not as favorable as that of 40% NPs, their tumor accumulation at 12 h was almost the same. This was likely due to that 20% NPs exposed more biotin, which enhanced their cellular uptake.
The therapeutic efficacy of 0% NPs, 20% NPs and 40% NPs was evaluated in mice bearing 4T1 tumors (Fig. 3C). The results were depicted in Figs. 3D–H, compared with the Saline group, all treatment groups exhibited antitumor efficacy to some extent. Among them, 0% NPs exhibited the largest tumor volume and tumor burden, followed by Prodrug Sol, Abraxane, Taxol, 40% NPs and 20% NPs. Although 40% NPs exhibited good tumor accumulation and optimal blood circulation, the surface PEGylated modification restricted their interaction with biotin receptors on tumor cells. In addition, 20% NPs exhibited the lowest tumor burden and tumor volume, suggesting the highest therapeutic efficiency. This result was attributed to a combination of good colloidal stability and excellent tumor-targeting efficiency.
In terms of safety, all treatment groups demonstrated good safety with no significant changes in body weight. Furthermore, liver and kidney function tests coupled with routine blood tests were conducted for each group (Figs. S15 and S16 in Supporting information). Compared with the Saline group, three nanoassemblies exhibited negligible systemic toxicity, whereas certain indicators for Taxol, Abraxane and Prodrug Sol were abnormal. These findings indicated that Biotin-PTX NPs alleviated the nonspecific toxicity of PTX, emphasizing their good safety. The animal assays conducted in this study followed the Guidelines for the Management and Use of Laboratory Animals. The study received ethical support from the Institutional Animal Ethical Care Committee (IAEC) of Shenyang Pharmaceutical University.
In this work, we selected PTX as the parent drug to link biotin via a disulfide bond to develop tumor-targeting prodrug, Biotin-PTX. We introduced DSPE-PEG2k into Biotin-PTX NPs and investigated the effect of DSPE-PEG2k ratios (0%, 5%, 10%, 20%, 40% and 60%, WPEG/Wprodrug+PEG) on their performance. We found that surface PEGylation affected the stability of Biotin-PTX NPs, and a sufficient DSPE-PEG2k content exhibited relatively good assembly stability, significantly extending the blood circulation of Biotin-PTX NPs. However, an excessive amount of DSPE-PEG2k could obscure the surface biotin density, thereby affecting the biotin-biotin receptor interactions, as evidenced by the decreased cytotoxicity of 4T1 cells. In general, 20% NPs balanced pharmacokinetics behavior and tumor-targeting efficiency, exhibiting good antitumor efficacy and safety compared with Taxol, Abraxane and Prodrug Sol.
Active targeting offers the potential to improve cellular uptake, reduce off-target effects, and enhance therapeutic efficacy compared with passive targeting via the EPR effect. In our study, biotin was employed as a targeting ligand to exploit the overexpression of biotin receptors in certain tumors, thereby enhancing the specificity and efficacy of Biotin-PTX NPs. However, active targeting strategies also face several limitations. For example, the heterogeneous expression of target receptors across different tumors and even within the same tumor may compromise the targeting efficiency. Surface ligands may also alter the protein corona of nanoparticles in the bloodstream, potentially affecting their in vivo fate, biodistribution, and ultimately reducing their targeting ability. These limitations contribute to the current challenges in the clinical translation of actively targeted nanomedicines. Therefore, rational design and careful optimization of ligand density, surface properties, and pharmacokinetics are essential to maximize therapeutic benefit.
In conclusion, our research developed tumor-targeting Biotin-PTX NPs, further revealed the effect of surface PEGylation on the stability, cytotoxicity, pharmacokinetics, and pharmacodynamics. This work provided new strategies for the development of tumor-targeting drug delivery systems.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Lurong Zhang: Writing – review & editing, Writing – original draft, Validation, Investigation, Formal analysis. Xin Wang: Writing – review & editing, Validation. Xuan Li: Visualization. Minglong Huang: Visualization. Bowen Zhang: Visualization. Shengyao Xu: Supervision. Jialin Xing: Supervision. Yafan Xiao: Methodology. Yi Zheng: Methodology. Zhenzhen Zhao: Supervision, Project administration. Jin Sun: Resources, Project administration. Zhonggui He: Resources, Project administration. Lingxiao Li: Supervision, Investigation. Bingjun Sun: Supervision, Resources, Funding acquisition, Conceptualization.
The research presented in this work received financial support from National Key R&D Program of China (No. 2022YFE0111600), Youth Innovation Team Project of Liaoning Provincial Education Department (No. LJ222410163049), Liaoning Revitalization Talents Program (No. XLYC2203083), Key Research and Development Program of Liaoning Province (No. 2024JH2/102500061), National Natural Science Foundation of China (No. 82302276), and the General Program of Department of Education of Liaoning Province (No. LJ212510163012).
Supplementary material associated with this article can be found, in the online version, at doi:
Z. Wu, F. Xia, R. Lin, J. Hematol. Oncol. 17 (2024) 119. doi: 10.1186/s13045-024-01640-8
R. Pirker, Curr. Opin. Oncol. 32 (2020) 63–67. doi: 10.1097/cco.0000000000000592
J. Zhu, Y. Xiong, X. Bai, et al., Chin. Chem. Lett. 36 (2025) 110799. doi: 10.1016/j.cclet.2024.110799
S. Hossen, M.K. Hossain, M.K. Basher, et al., J. Adv. Res. 15 (2019) 1–18.
H. Cai, Y. Xiang, Y. Zeng, et al., Acta Pharm. Sin. B 11 (2021) 544–559. doi: 10.1016/j.apsb.2020.07.023
X. Wu, X. Chen, X. Wang, et al., Chin. Chem. Lett. 35 (2024) 108756. doi: 10.1016/j.cclet.2023.108756
E.K. Rowinsky, R.C. Donehower, N. Engl. J. Med. 332 (1995) 1004–1014. doi: 10.1056/NEJM199504133321507
X. Sun, P. Zhao, J. Lin, K. Chen, J. Shen, Cancer Drug Resist. 6 (2023) 390–415. doi: 10.20517/cdr.2023.16
T. Liu, H. Zou, J. Mu, et al., Chin. Chem. Lett. 34 (2023) 108135. doi: 10.1016/j.cclet.2023.108135
M. Norouzi, M. Amerian, M. Amerian, F. Atyabi, Drug Discov. Today 25 (2020) 107–125. doi: 10.1016/j.drudis.2019.09.017
M. Zhao, H. Li, Y. Ma, et al., Int. J. Nanomed. 12 (2017) 1685–1697. doi: 10.2147/IJN.S129976
C. Guo, L. Lin, Y. Wang, et al., Theranostics. 15 (2025) 5440–5480. doi: 10.7150/thno.112475
F. Fang, X. Chen, ACS. Nano 18 (2024) 23827–23841. doi: 10.1021/acsnano.4c09027
G. Li, B. Sun, Y. Li, et al., Small. 17 (2021) e2101460. doi: 10.1002/smll.202101460
H. Shi, Y. Luo, S. Zhang, et al., Chin. Chem. Lett. 36 (2025) 110775. doi: 10.1016/j.cclet.2024.110775
Y. Sun, S. Wang, Y. Li, et al., Acta Biomater. 157 (2023) 417–427. doi: 10.1016/j.actbio.2022.12.005
D. Zeng, Z. Ma, X. Zan, et al., Chin. Chem. Lett. 35 (2024) 108433. doi: 10.1016/j.cclet.2023.108433
J. Peng, Y. Xiao, Q. Yang, et al., Acta Pharm. Sin. B 11 (2021) 1069–1082. doi: 10.1016/j.apsb.2020.06.013
H. Hou, S. Wei, Y. Shao, et al., Chin. Chem. Lett. 36 (2025) 110315. doi: 10.1016/j.cclet.2024.110315
S. Maiti, P. Paira, Eur. J. Med. Chem. 145 (2018) 206–223. doi: 10.1016/j.ejmech.2018.01.001
F. Amiri, S. Mahmazi, H. Danafar, J. Polym. Environ. 28 (2020) 2939–2946. doi: 10.1007/s10924-020-01831-7
Q. Li, Z. Zheng, Y. Chen, et al., Anal. Chem. 97 (2025) 1627–1634. doi: 10.1021/acs.analchem.4c04513
L. Lin, Y. Zheng, C. Huang, et al., Eur. Polym. J. 222 (2025) 113592. doi: 10.1016/j.eurpolymj.2024.113592
H.R. Jia, Y.X. Zhu, Y. Liu, et al., Exploration 2 (2022) 20220010. doi: 10.1002/EXP.20220010
Y. Li, X. Shen, H. Ding, et al., Acta Pharm. Sin. B 14 (2024) 3680–3696.
Y. Liu, X. Wang, Z. Wang, et al., Pharmaceutics. 15 (2023) 262. doi: 10.3390/pharmaceutics15010262
J.W. Shreffler, J.E. Pullan, K.M. Dailey, S. Mallik, A.E. Brooks, Int. J. Mol. Sci. 20 (2019) 6056. doi: 10.3390/ijms20236056
H. He, L. Wang, Y. Ma, et al., J. Control. Release 327 (2020) 725–736.
M. Zhou, H. Huang, D. Wang, et al., Nano Lett. 19 (2019) 3671–3675. doi: 10.1021/acs.nanolett.9b00737
Y. Xia, S. Fu, Q. Ma, Y. Liu, N. Zhang, Nano-micro Lett. 15 (2023) 145.
Y. Li, Y. Wu, Z. Fang, et al., Adv. Mater. 36 (2024) e2307263.
B. Zhang, L. Li, M. Huang, et al., Nano Lett. 24 (2024) 3759–3767. doi: 10.1021/acs.nanolett.4c00300
Z. Geng, L. Wang, K. Liu, J. Liu, W. Tan, Angew. Chem. Int. Ed. 60 (2021) 15459–15465. doi: 10.1002/anie.202102631
B. Sun, C. Luo, H. Yu, et al., Nano Lett. 18 (2018) 3643–3650. doi: 10.1021/acs.nanolett.8b00737
X. Xiang, X. Feng, S. Lu, et al., Exploration 2 (2022) 20220008.
L. Li, T. Liu, S. Zuo, et al., Adv. Mater. 36 (2024) e2310633.

Scheme 1 The content of DSPE-PEG2k influenced the stability and targeting efficiency of Biotin-PTX NPs.

Figure 1 Preparation and characterization of Biotin-PTX NPs. (A) Schematic illustration of preparation. (B) Particle size distribution and TEM images. Scale bar: 200 nm. The changes in particle size of Biotin-PTX NPs after being stored at (C) 4 ℃ for 30 days, (D) PBS (pH 7.4) containing 10% (v/v) FBS for 24 h and (E) PBS (pH 7.4) containing 50% (v/v) FBS for 24 h. Data were presented as mean ± SD (n = 3).

Figure 2 Redox dual-sensitive release and cytological evaluation. The PTX release process under conditions of (A) 0.1 mmol/L GSH, (B) 1 mmol/L GSH, (C) 1 mmol/L H2O2 and (D) 10 mmol/L H2O2. The cellular uptake and activation of Taxol, Abraxane, Prodrug Sol, 0% NPs, 20% NPs and 40% NPs against (E) 4T1 cells and (F) B16-F10 cells. In the absence of biotin or with 0.1 mmol/L biotin, the cellular uptake and activation of Prodrug Sol, 0% NPs, 20% NPs and 40% NPs against (G) 4T1 cells and (H) B16-F10 cells. (I) The IC50 values of Taxol, Abraxane, Prodrug Sol, 0% NPs, 20% NPs and 40% NPs against 4T1 cells, B16-F10 cells and 3T3 cells. (J) The diagram of Biotin-PTX NPs functioning in tumor cells. Data were presented as mean ± SD (n = 3). Statistical differences were analyzed with a two-tailed student’s t-test.

Figure 3 Pharmacokinetics and antitumor efficacy. (A) The process of pharmacokinetic experiment in Sprague-Dawley rats. ⅰ means intravenous injection, ⅱ means orbital blood collection, ⅲ means centrifuge to get supernatant and ⅳ means fluorescence measurement. (B) Plasma concentration-time curves following intravenous injection with an equivalent DiR dose of 1 mg/kg. Data were presented as mean ± SD (n = 5). (C) The protocol of antitumor efficacy study. (D–H) The antitumor effects of 0% NPs, 20% NPs and 40% NPs versus Prodrug Sol, Taxol and Abraxane. (D) Individual tumor volume. (E) Tumor photos. (F) Tumor volume. (G) Body weight. (H) Tumor burden. Data were presented as mean ± SD (n = 4). Statistical differences were analyzed with a two-tailed student’s t-test.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
 下载:
下载: