Citation:
Shao-Qi Guan, Chen-Hui Liu, Ya-Ping Wang, Xiao-Dong Zhang, Zhong-Min Cao, Mei Pan. Cancer cell membrane-biomimetic metallacycles for tumor microenvironment-responsive ferroptosis/apoptosis therapy with low systemic toxicity[J]. Chinese Chemical Letters,
2026, 37(7): 111692.
doi:
10.1016/j.cclet.2025.111692
Cancer cell membrane-biomimetic metallacycles for tumor microenvironment-responsive ferroptosis/apoptosis therapy with low systemic toxicity
English
Cancer cell membrane-biomimetic metallacycles for tumor microenvironment-responsive ferroptosis/apoptosis therapy with low systemic toxicity
MOE Laboratory of Bioinorganic and Synthetic Chemistry, Lehn Institute of Functional Materials, IGCME, GBRCE for Functional Molecular Engineering, School of Chemistry, Sun Yat-sen University, Guangzhou 510275, China
b.
Department of Radiology, Guangdong Provincial Key Laboratory of Malignant Tumor Epigenetics and Gene Regulation, Medical Research Center, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou 510030, China
Received Date:
25 April 2025 Accepted Date:
08 August 2025 Revised Date:
03 August 2025 Available Online:
15 July 2026
Abstract:
Ferroptosis, a form of programmed cell death driven by iron-dependent lipid peroxidation (LPO), has emerged as a promising therapeutic strategy for cancer. However, challenges such as uncontrolled iron delivery, insufficient ferroptosis induction efficiency, and off-target drug leakage limit its applications. To address these limitations, we developed a biomimetic nanoplatform (Fe-DTX@M) integrating cancer cell membrane-camouflaged Fe-based metallacycles with the chemotherapeutic drug docetaxel (DTX), which synergistically amplifies ferroptosis and apoptosis for precise and effective cervical cancer therapy. The membrane camouflage enabled highly efficient tumor-specific accumulation, achieving a 5.4-fold increase compared to non-targeted controls, and reduced systemic toxicity. Within tumors, Fe2+/Fe3+ cycles attributed to Fe-based metallacycle drove Fenton reactions to convert H2O2 into •OH, inducing LPO, while Fe3+-mediated glutathione depletion inhibited GPX4, amplifying ferroptosis with 2.4-fold malondialdehyde (MDA) increase. Acid-triggered DTX release of nanoplatform further promoted apoptosis, thereby enhancing therapeutic efficacy. Furthermore, Fe-DTX@M exhibited excellent long-term biocompatibility and safety in normal mice over 30 days post-intravenous injection. This combination of biomimetic metallacycles-induced ferroptosis and chemotherapy-induced apoptosis may provide a new paradigm for achieving effective cancer therapy.
Cervical cancer is a significant global public health concern and is one of the most common malignant tumors posing a severe threat to women’s health worldwide [1]. Current treatments, including radiotherapy, chemotherapy, and surgery, frequently prove inadequate in fully and effectively curing cervical cancer due to its poorly understood pathogenesis, resistance to platinum-based therapy, and late metastasis and recurrence [2]. Therefore, there is an urgent need to develop novel therapeutic strategies to address these limitations. Distinct from traditional forms of cell death such as apoptosis, autophagic, and necrosis, ferroptosis is a new iron-dependent and non-apoptotic form of cell death caused by lipid peroxidation (LPO) dysregulation [3,4]. Recent studies have demonstrated the effectiveness of ferroptosis in enhancing tumor immunotherapy outcomes across various cancer types, including lung [5,6], breast [7,8], and pancreatic cancer [9,10]. Despite growing interest in the therapeutic potential of targeting iron homeostasis, downregulating glutathione peroxidase 4 (GPX4), and inhibiting the SLC7A11/system xc− for cervical cancer treatment, the current research landscape remains underexplored [11–13]. Furthermore, the mechanistic insights into these approaches are still incomplete, warranting further in-depth investigation to validate their therapeutic efficacy and explore their translational potential in clinical applications [14,15].
Ferroptosis is distinguished by elevated levels of lipid peroxides caused by redox-active iron (Fe2+) and reactive oxygen species (ROS) [16–18]. However, the upregulation of LPO is inhibited by the low levels of labile iron pool (~1 µmol/L Fe2+ labile iron pool) and overexpressed glutathione (GSH) in the tumor microenvironment (TME), while GSH depletion can inactivate GPX4 and induce irreversible LPO [19]. Therefore, strategies to increase Fe content and deplete GSH have emerged as promising approaches to enhance the efficacy of existing treatments. Currently, various iron-based materials have been developed for ferroptosis-based cancer treatment, such as Ferumoxytol [20], amorphous iron [21,22], and Fe3O4 [23], which are often used as ferroptosis inducers, but these materials typically require high doses of iron (such as 75 mg/kg for amorphous iron and 10 mg/kg for ferumoxytol and Fe3O4) to induce ferroptosis and often pose long-term toxicity concerns. To address these issues, iron-based organic complexes supramolecular iron delivery system (SIDS) (10 mg/kg) [24] and PAB@MIL-53 (5 mg/kg) [25] have exhibited competitive ferroptosis induction with good biocompatibility and therapeutic efficacy (Table S1 in Supporting information). These studies collectively highlight that iron-based nanomaterials typically require high doses of iron to induce ferroptosis, which can accumulate in the body, posing potential risks. There is a pressing need for safer, more effective and lower-dose iron sources to induce ferroptosis. Compared with the reported Fe-based nanoparticles, metal-organic complexes composed of metal ions and organic ligands, including a series of intriguing metallacycles, have recently garnered attention as anticancer agents due to their flexibility and designability in molecular structure, photophysical properties, and multi-functionality [26,27]. Notably, recent studies have shown that macromolecular drugs (i.e., metal macrocycles) exhibit superior cellular uptake and longer tumor retention in cancer cells compared with small molecule drugs [28]. However, the application of metallacycles with high iron content in ferroptosis therapy for cervical cancer remains unexplored. Moreover, the nonspecific targeting of iron-based nanomaterials leads to leakage into normal tissues, causing toxicity and limited activation of ferroptosis. Therefore, selective delivery of iron to the target site and prevention of iron leakage are crucial to avoid potential toxicity and enhance treatment efficacy.
Although the current nano drug delivery systems can improve the pharmacokinetics and biodistribution of drugs to a certain extent through the enhanced permeability and retention effect (EPR) in solid tumors, they are easily recognized and cleared by the immune system due to their exogenous nature, resulting in poor internalization effects within tumor cells [29]. While conventional ligand-modified nanoparticles including peptide, antibody, or nucleic acid conjugated systems demonstrate tumor-targeting capabilities [30], their further applications are generally hindered by high production costs, complex characterization requirements, and unpredictable nanocarrier modification outcomes [31]. To address these limitations, cancer cell membrane-coated nanoparticle technology offers a new modification and camouflage strategy for developing highly targeted and low immunogenic nanoparticles [32,33]. These cancer cell membrane biomimetic nanoparticles retain the antigen and structural features of cell membrane, conferring them with immune evasion capabilities, prolonged circulation time, and strong target recognition abilities [34]. Furthermore, cell membrane biomimetics offers an effective top-down strategy for nanoparticle functionalization, thus simplifying the extensive development of nanocarrier platforms [35]. Therefore, the integration of cancer cell membrane camouflage technology is of great significance for nano-delivery platforms to target cancer cells, reduce toxic side effects, and improve effective drug delivery for enhanced therapeutic efficacy.
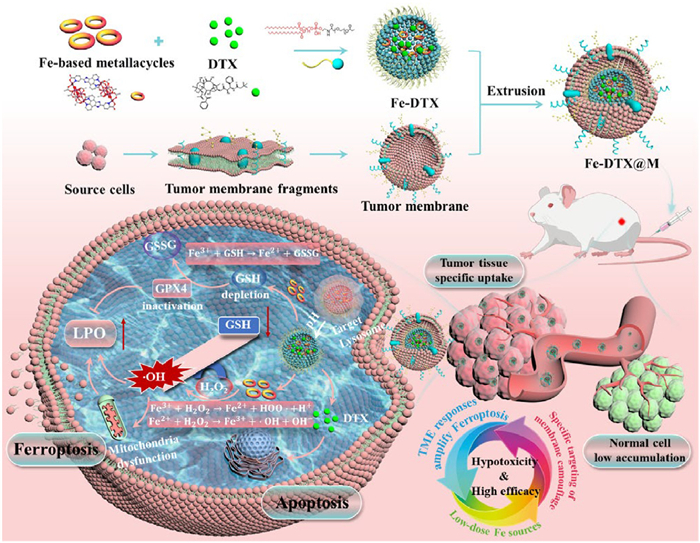
Herein, we developed a biomimetic drug delivery strategy by coating nanoparticles with cancer cell membranes to improve drug safety and efficacy of tumor treatment. As illustrated in Scheme 1, we initially prepared a Fe-based metallacycle as a ferroptosis inducer, which was then assembled with a biocompatible amphiphilic phospholipid-polymer 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-(methoxy(polyethylene glycol)-2000) (DSPE-PEG2000) through hydrophobic interaction to form nanopolymers with excellent physiological stability and biocompatibility. Simultaneously, docetaxel (DTX), a first-line chemotherapy drug for advanced cervical cancer [36,37], was loaded in the nanopolymers to prevent it from leaking prematurely and causing severe systemic side effects. Finally, the physical extrusion method was employed to camouflage the cancer cell membrane on the surface, obtaining Fe-DTX@M, which achieves extended circulation time and homologous targeting of tumor cells. Fe-DTX@M specifically targeted tumor cells, were internalized via endocytosis, and subsequently localized in lysosomes. The Fe-based metallacycle can react with overexpressed H2O2 in the TME to generate •OH, accompanied by GSH depletion, thereby accelerating the release of Fe2+ content, causing mitochondrial dysfunction and the blockage of GPX4 reduction of LPO. This process achieves a double amplification of ferroptosis. In vivo studies have demonstrated that a small injection dose can effectively induce a synergistic antitumor response by combining ferroptosis and chemotherapy, achieving both safety and efficacy in cervical cancer mice. Therefore, this biomimetic Fe-based metallacycle delivery system enables precise tumor-targeting combination therapy with TME response, holding considerable potential for clinical translation.
Scheme 1
Scheme 1.
Schematic illustration of the formation of Fe-DTX@M and its application in the combined treatment of ferroptosis and apoptosis for cervical cancer. The designed and synthesized biomimetic Fe-based metallacycle nanoplatforms can specifically target tumor cells, and internalize into tumor cells to consume intracellular overexpressed H2O2 and GSH, leading to intracellular redox imbalance and GPX4 inactivation, thereby synergistically enhancing LPO-induced tumor cell ferroptosis, which is an effective treatment strategy for cervical cancer.
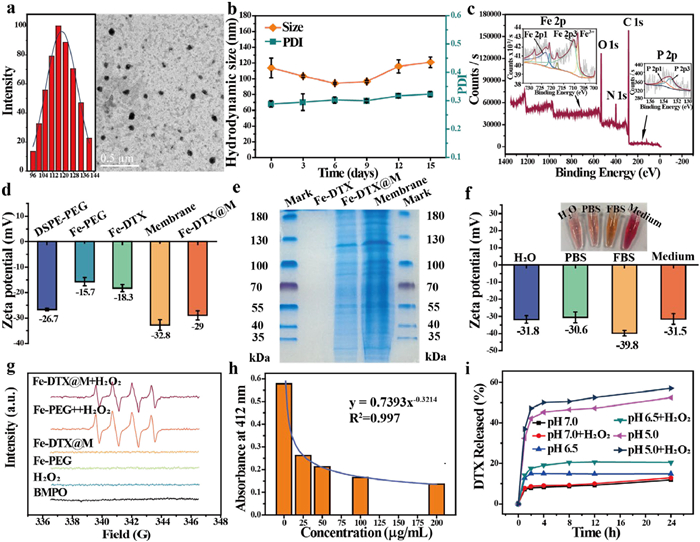
Fe-based metallacycles (Fe12ⅢL2(AcO)18(EtO)4O6, ligand L: 5-(1H-Imidazo[4,5-f][1,10]phenanthrolin-2-yl)picolinic acid) were synthesized via a hydrothermal method, and the synthesis process was characterized by nuclear magnetic resonance hydrogen spectroscopy (1H NMR) and crystal data (Figs. S1–S6 and Table S2 in Supporting information). To enhance the biocompatibility of hydrophobic Fe-based metallacycles, a hydrophilic nanopolymer, Fe-PEG, with a size of approximately 116.4 nm by transmission electron microscopy (TEM), was obtained through hydrophobic polymerization with DSPE-PEG 2000 (Fig. 1a). And the Fe-PEG showed good physiological stability in phosphate-buffered saline (PBS) within 20 days as determined by dynamic light scattering (DLS) (Fig. 1b), indicating that the employment of DSPE-PEG 2000 encapsulation could improve the stability and water dispersion of Fe-based metallacycles to avoid agglomeration [38,39], further increasing the cell membrane encapsulation efficiency by biomimetic approach. Given the significant chemotherapeutic effect of DTX on cervical cancer, but its inherent poor drug solubility of DTX and the non-specificity of solvent-based administration, we chose to load DTX into nanopolymers of Fe-PEG, endowing the DTX-loaded Fe-PEG nanopolymers (Fe-DTX) to improve its chemotherapeutic effect. The typical absorption peak of DTX appears in Fe-DTX (Fig. S7 in Supporting information), with the encapsulation efficiency of DTX reaching 50.3% (6.26% loading efficiency), demonstrating the successful loading of DTX. Additionally, X-ray photoelectron spectroscopy (XPS) of the Fe-based metallacycles and Fe-DTX, as depicted in Fig. 1c and Figs. S8 and S9 (Supporting information), exhibited identical peaks for Fe, C, N, and O, along with the characteristic P absorption peak attributed to DSPE-PEG 2000, confirming the successful synthesis of the nanopolymers. The characteristic peaks at 2890 and 1100 cm−1 in the Fourier transform infrared spectroscopy (FT-IR) spectrum (Fig. S10 in Supporting information) were attributed to the stretching vibrations of C-C and C-O of DSPE-PEG 2000, respectively. And the Fe-DTX nanomaterials also exhibited the characteristic peaks of DTX, further confirming the successful PEG functionalization of the Fe-based metallacycles and the loading of DTX.
Figure 1
Figure 1.
Characterization and catalytic performance of Fe-DTX@M. (a) TEM image and size distribution of Fe-PEG. (b) Size distribution and polymer dispersity index (PDI) of Fe-PEG at different time points. (c) Survey, Fe 2p, and P 2p XPS spectra of Fe-DTX. (d) Zeta potential of the different materials. (e) SDS-PAGE protein analysis. Samples were stained with Coomassie blue. (f) Zeta potential of Fe-DTX@M dispersed in different media. (g) ESR spectra of •OH generation after different treatments. (h) Degradation of DTNB treated with Fe-DTX@M at 412 nm. (i) The percentage of DTX released from Fe-DTX@M under different conditions. Data are presented as mean ± SD (n = 3).
To improve Fe-DTX nanopolymer delivery efficiency, enhance tumor treatment effects, and reduce toxicity, tumor cell membrane fragments were extracted and encapsulated onto Fe-DTX by ultrasound and physical extrusion strategies. As shown in Fig. S11 (Supporting information), TEM images confirmed the presence of a membrane coating on the surface of Fe-DTX, forming Fe-DTX@M, with a particle size of about 151 nm. As shown in Fig. 1d, the zeta potential shift to negative for the Fe-based metallacycles further indicated the successful PEG coating, while the increased negative charge of DTX and tumor cell membrane indicated the successful Fe-DTX@M coating. Furthermore, protein gel electrophoresis revealed similar protein profiles in Fe-DTX@M and the extracted pure tumor cell membrane, demonstrating the successful tumor cell membrane coating (Fig. 1e). Additionally, Fe-DTX@M is stable without aggregation or sedimentation in the dispersants needed for bioexperiments, such as deionized water, PBS, fetal bovine serum (FBS), and cell culture medium (Fig. 1f). The Fe content of Fe-DTX@M nanomaterials measured by ICP-MS exceeds 26.4 wt%, which is higher after being encapsulated into nanoparticles and higher than the 23% iron content of ferritin that induces ferroptosis [24].
To explore the potential of Fe-DTX@M as a ferroptosis inducer for cervical cancer treatment, we first accessed its catalytic activity in producing hydroxyl radicals (•OH) from H2O2 using methylene blue (MB) as a probe, which can be degraded by •OH. As shown in Fig. S12 (Supporting information), when MB was incubated with Fe-DTX@M and H2O2 solution, the absorbance of MB decreased significantly, whereas in the solution without Fe-DTX@M, the absorbance of MB remained unchanged. These results indicate that Fe-DTX@M can drive Fenton-like reaction to produce ROS. Then, we evaluated the peroxidase-like catalase activity of Fe-DTX@M based on the chromogenic reaction between hydrogen peroxide and o-phenylenediamine (OPD) under the catalysis of Fe2+. No reaction occurred when only Fe-DTX@M was added. However, with the addition of H2O2, the reaction produced a purple product with a maximum absorption at 448 nm (Fig. S13 in Supporting information), and the amount of product increased with the concentration of H2O2 solution. This demonstrates that Fe-DTX@M can catalyze H2O2 to produce ROS. The characteristic peak of •OH generated by the Fe-DTX@M under H2O2 was further verified using the BMPO probe via electron spin resonance (ESR) (Fig. 1g). Additionally, the oxidizing GSH ability of Fe-DTX@M was assessed with Ellman's reagent (DTNB). In the negative control group, GSH can react with DTNB to produce a detectable 5-thio-2-nitrobenzoic acid (TNB) at 412 nm without Fe-DTX@M. The absorption peak of the reaction decreased with increasing concentrations of Fe-DTX@M, indicating that Fe3+ in Fe-DTX@M can consume reducing GSH to form Fe2+ and glutathione disulfide (GSSG), with higher concentrations resulting in greater consumption (Fig. 1h). These experiments collectively indicate that Fe-DTX@M can not only produce •OH but also consume reducing GSH, thereby providing the necessary conditions for ferroptosis. To evaluate the potential of Fe-DTX as a gatekeeper for controlled drug release, the hydrophobic anticancer drug DTX was employed as a model guest and loaded into Fe-PEG. As depicted in Fig. 1i, the DTX-loaded Fe-DTX@M exhibited negligible drug release in PBS at pH 7.0. In contrast, under acidic conditions at pH 5.0 with H2O2, a sustained pH/H2O2-dependent release profile of DTX was observed, achieving a cumulative drug release efficiency of 57.1% within 24 h. The drug release kinetics study in Fig. S14 (Supporting information) further confirmed the sustained release behavior of Fe-DTX@M under different acidic and H2O2 environments, indicating its pH and H2O2-dependent controlled release kinetics of DOX, consistent with the previously reported work [40,41]. These results indicate that Fe-DTX holds the promise for precise and efficient TME-targeted cancer therapy.
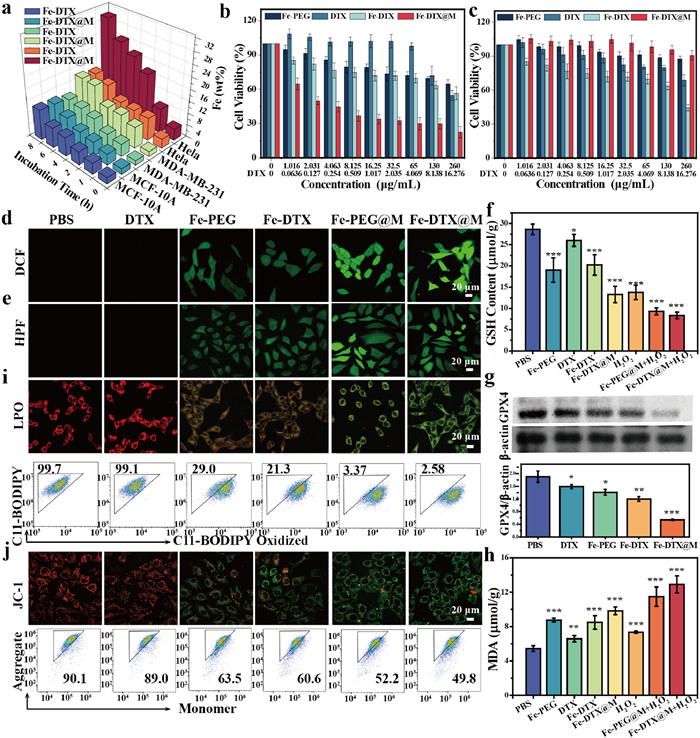
Inspired by the successful modification of cancer cell membrane, we first evaluated the cellular uptake efficiency of Fe-DTX@M in tumor cells and its cytotoxicity. The in vitro uptake efficiency of Fe-DTX@M was evaluated by analyzing the iron content using inductively coupled plasma mass spectrometry (ICP-MS) to detect the uptake of Fe-DTX and Fe-DTX@M (HeLa cell membrane coating) by HeLa, MDA-MB-231, and MCF-10A cells (Fig. 2a). Compared to the Fe-DTX group, the uptake of iron by HeLa cells treated with Fe-DTX@M was significantly increased at the same iron concentration, attributed to the homologous targeting conferred by the HeLa cell membrane coating. Conversely, the cellular uptake efficiency in MCF-10A cells or MDA-MB-231 cells was much lower than that of Fe-DTX without membrane coating, further indicating that the modification with HeLa cell membranes endowed Fe-DTX@M with active targeting capability towards HeLa cells, while reducing the uptake of Fe-DTX@M by other cell types including normal cell lines. To evaluate its intracellular endocytosis in detail, Fe-DTX@M was labeled with DiD probe (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine, 4-chlorobenzenesulfonate), a far-red plasma membrane fluorescent probe. The confocal laser scanning microscopy (CLSM) and flow cytometry results revealed the time-dependent cellular uptake behavior (Fig. S15 in Supporting information) and lysosome targeting capabilities (Fig. S16 in Supporting information) of Fe-DTX@M.
Figure 2
Figure 2.
Mechanism of Fe-DTX@M-induced ferroptosis. (a) Cellular uptake rate of Fe-DTX@M by different cells at various time points using ICP-MS. Cell viability of HeLa (b) and MCF-10A (c) cells with different treatment groups (mean ± SD, n ≥ 3). CLSM images of HeLa cells after different treatments and staining with (d) intracellular ROS probe DCFH-DA and (e) •OH probe HPF, respectively. (f) GSH and (h) MDA levels in HeLa cells after different treatments (mean ± SD, n ≥ 3). (g) Western blot results and quantitative analysis of GPX4 protein expression under different treatment conditions. (i) CLSM images and flow cytometry of C11-BODIPY581/591-dye-stained HeLa cells after different treatments. (j) CLSM images and flow cytometry of changes in MMP of HeLa cells after different treatments detected using JC-1 probe. Scale bar: 20 µm. HPF and DCFH-DA Probe: λex = 488 nm, λem = 520 ± 20 nm. C11-BODIPY581/591 (Red): λex = 561 nm, λem = 591 ± 20 nm. C11-BODIPY581/591 (Oxidation form, Green): λex = 488 nm, λem = 520 ± 20 nm. JC-1 (monomer, Green): λex = 488 nm, λem = 529 ± 20 nm. JC-1 (aggregate, red): λex = 561 nm, λem = 590 ± 20 nm. P < 0.05, **P < 0.01, ***P < 0.001 of (c–e) compared with the PBS group.
Subsequently, the cytotoxicity of HeLa cells was evaluated by standard MTT assay. Compared to the free DTX group, Fe-DTX exhibited increased cytotoxicity of HeLa cells due to the enhanced cellular uptake of the Fe-DTX nanopolymer (Fig. 2b). In contrast, the Fe-DTX@M group exhibited the strongest killing ability towards HeLa cells, with the relative survival rate significantly reduced to 22.5%. This dose-dependent cytotoxicity is mainly due to the tumor cell membrane coating, which could promote the specific uptake of Fe-DTX@M by homologous targeting. To further elucidate the homologous targeting effect of Fe-DTX@M, we synthesized Fe-DTX@M (U14, murine cervical cancer cell line) coated with U14 cell membrane to study its cytotoxicity to U14 cells (Fig. S17 in Supporting information). The results were similar to the cytotoxicity of Fe-DTX@M (HeLa) in HeLa cells. These results further confirm the homologous targeting capability of Fe-DTX@M. To address the need for comparative analysis with established ferroptosis inducers and elucidate detailly the biomimetic mechanism, we employed hemin as a control, a famous ferroptosis inducer [42,43]. Our comparative studies in Fig. S18 (Supporting information) revealed that Fe-DTX@M demonstrates significantly higher cytotoxicity against HeLa cells than hemin at equivalent Fe3+ concentrations, and the biomimetic Fe-DTX@M achieves comparable cytotoxicity at lower effective concentrations than hemin. These results further indicated that the biomimetic approach in this work facilitates targeted delivery while minimizing systemic exposure, contributing to both therapeutic enhancement and toxicity reduction. In addition, Fe-DTX@M coated with HeLa cell membranes also showed minimal cytotoxicity in normal cells MCF-10A even at a high concentration of 260 µg/mL (Fig. 2c), which was attributed to the low cellular uptake efficiency of MCF-10A, ingeniously ensuring good biocompatibility of Fe-DTX@M.
Prior to evaluating the in vitro ferroptosis efficacy of Fe-DTX@M, intracellular ROS and •OH generation were observed using DCFH-DA and HPF probes, respectively. As shown in Figs. 2d and e, Negligible green fluorescence in the PBS and DTX treatment groups contrasted with moderate signals in Fe-PEG and Fe-DTX groups, while the fluorescence in HeLa cells treated with Fe-PEG@M and Fe-DTX@M was significantly stronger, confirming that specific targeting by the cell membrane coating significantly increased the ROS and •OH levels. These results suggest that Fe-DTX@M induces a substantial accumulation of ROS and •OH in tumor cells, demonstrating its potential as a nanocatalyst for promoting the accumulation of LPO. Ferroptosis efficacy is often greatly attenuated by enriched GSH levels in the tumor environment [44]. The intracellular GSH levels assays in Fig. 2f demonstrated that the decrease of GSH in Fe-DTX@M-incubated cells amplifies oxidative stress in tumor cells, leading to the increased accumulation of ROS and lipid peroxide [25]. Additionally, we also investigated the expression levels of the GSH-related enzyme GPX4 that can convert toxic phospholipid peroxides (L-OOH) into benign lipid alcohols (L-OH) [3]. Western blot analysis confirmed a reduction in GPX4 protein expression in Fe-DTX@M incubated cells (Fig. 2g).
Given the demonstrated capabilities of Fe-DTX@M in generating high ROS generation, depleting GSH, and inactivating GPX4, the LPO level in HeLa cells was further evaluated. To investigate whether the accumulated ROS produced by the Fe-DTX@M nanoplatforms could induce LPO and subsequently trigger ferroptosis, we measured the levels of malondialdehyde (MDA) to evaluate lipid degradation caused by oxidative damage. As shown in Fig. 2h, Fe-DTX@M-treated cells exhibited a 2.4-fold upregulation of MDA levels compared to the PBS group, indicating an increased level of LPO that significantly amplified the ferroptosis effect. Additionally, the membrane LPO was assessed using C11-BODIPY581/591, a specific fluorescent probe for detecting ferroptosis. Notably, the probe emitted green fluorescence upon oxidation, and Fe-DTX@M significantly increased the membrane LPO level in HeLa cells, collectively demonstrating that Fe-DTX@M can induce ferroptosis. Flow cytometry results further confirmed that Fe-DTX@M can activate the ferroptosis pathway (Fig. 2i and Fig. S19 in Supporting information). As shown in Fig. S20 (Supporting information), the increased release of lactate dehydrogenase (LDH) in HeLa cells treated with Fe-DTX@M suggests significant membrane disruption.
Ferroptosis in cells is often associated with the disruption of mitochondrial regulatory signaling [45]. Therefore, we evaluated the effect of treatment on mitochondrial membrane potential (MMP), which is easily lost during the redox process of cellular damage. The MMP-specific probe JC-1 was used to monitor potential changes in MMP. JC-1 stain assay showed that incubating HeLa cells with Fe-DTX or Fe-DTX@M led to a decrease in MMP, consistent with the results observed by flow cytometry (Fig. 2j and Fig. S21 in Supporting information). The mitochondrial morphology in Fe-DTX@M incubated HeLa cells further exhibited distinct mitochondrial shrinkage and a reduction in cristae, characteristic features of ferroptosis-induced mitochondrial dysfunction (Fig. S22 in Supporting information). Moreover, the MTT assay results showed that these typical ferroptosis inhibitors could effectively alleviate the damage to cancer cells caused by Fe-DTX@M treatment (Fig. S23 in Supporting information). The Fe-DTX@M nanoplatform, by simulating catalase-like activity, can induce ferroptosis in cancer cells through a cascade of reactions that catalyze the consumption of GSH and the generation of ROS, as well as the inactivation of intracellular GPX4 due to GSH depletion. To better understand this process, a ferroptosis mechanism diagram was provided in Fig. S24 (Supporting information). The ferroptosis cascade is initiated when Fe-DTX undergoes intracellular reduction by tumor-overexpressed GSH, generating Fe2+ and GSSG through a redox-reversible process. The free Fe2+ subsequently catalyzes H2O2 decomposition via Fenton reaction, producing •OH that induces LPO. Concurrently, GSH depletion weakens GPX4 activity by limiting its essential cosubstrate availability, leading to significant GPX4 downregulation and inhibited detoxification of lipid hydroperoxides (L-OOH). This dual mechanism synergistically amplifies ferroptotic cell death. The above results demonstrated that Fe-DTX@M induces MMP decrease, which can evoke the activation of cellular caspases, an important protein family involved in the apoptosis process [46]. The effect of Fe-DTX@M treatment on caspase 3/7 activity in cells was studied using the caspase 3/7 Glo assay. Cells treated with DTX or Fe-DTX, as well as Fe-DTX@M, showed enhanced activity of caspase 3/7 (Fig. S25 in Supporting information), indicating that DTX in Fe-DTX@M can induce cell death through apoptosis.
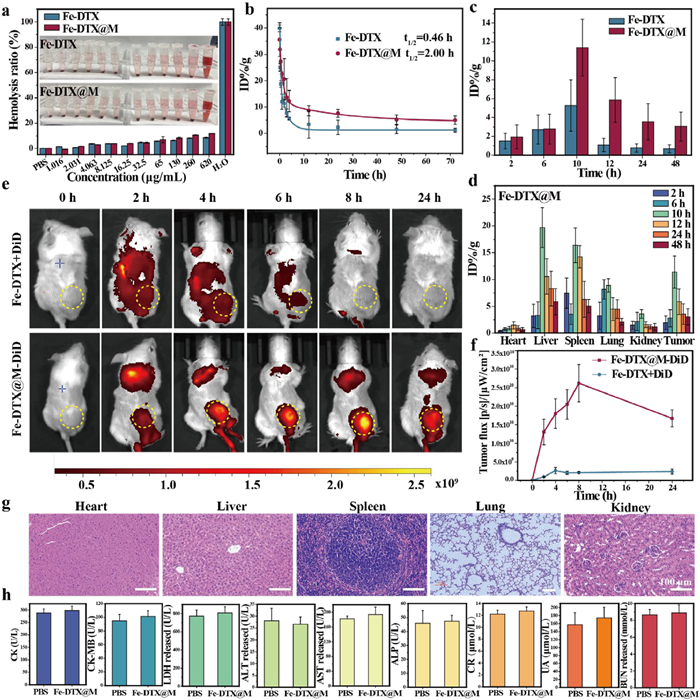
The excellent in vitro performance of Fe-DTX@M (U14) prompted us to further evaluate its in vivo tumor therapeutic effect on mice with cervical cancer models. All the animal experiments were performed following the guidelines of the Experimental Animal Center of Sun Yat-sen University (approval No. SYSU-IACUC-2023-002059). The results of in vitro hemolysis experiments showed that even at a high iron concentration of 620 µg/mL, Fe-DTX@M did not cause significant erythrocyte hemolysis (Fig. 3a), indicating its good biocompatibility. The pharmacokinetic profile of Fe-DTX@M in mice was determined by measuring the time-dependent concentrations of iron in the blood by ICP-MS. The results showed that the blood circulation half-life of Fe-DTX was 0.46 h, while that of Fe-DTX@M was 2.0 h (Fig. 3b), indicating that cell membrane-camouflaged Fe-DTX can increase its blood circulation time. The interleukin-6 (IL-6) and IL-1β, key inflammatory factors mediating inflammatory responses [47,48], were detected by enzyme linked immunosorbent assay (ELISA) method. As shown in Fig. S26 (Supporting information), compared with the control group, no significant changes in the concentrations of interleukin IL-6 and IL-1β were observed within 48 h after injection of Fe-DTX@M, which indicates that Fe-DTX@M materials confer immune-evasive properties and intrinsic tumor targeting. The distribution in the five internal organs (heart, liver, spleen, lung, and kidney) and tumors of mice showed that the highest enrichment rate was reached around 10 h in the tumor. Compared with conventional Fe-DTX, Fe-DTX@M demonstrated a 5.4-fold increase in tumor accumulation, achieving significantly enhanced lesion enrichment while simultaneously reducing systemic toxicity (Fig. 3c). Additionally, the iron concentration in the liver, kidney, and spleen increased, which may be metabolized within 24 h and cleared. Importantly, when the same dose was injected, the accumulation of Fe-DTX@M in the tumor was higher than that of Fe-DTX nanopolymers (Fig. 3d and Fig. S27 in Supporting information), indicating that Fe-DTX@M exhibits specific targeting capabilities toward homologous tumor cells while demonstrating excellent biocompatibility.
Figure 3
Figure 3.
Evaluation of in vivo biodistribution and tumor targeting of Fe-DTX@M. (a) Hemolysis analysis of Fe-DTX and Fe-DTX@M dispersed at different concentrations. Deionized water and PBS were used as positive and negative controls, respectively. Inset: Photograph of the mixture after centrifugation to observe the presence of hemoglobin in the supernatant. (b) Pharmacokinetic profile of Fe-DTX and Fe-DTX@M. (c) Comparison of biodistribution of Fe-DTX and Fe-DTX@M in tumors. (d) Quantitative biodistribution of Fe-DTX@M in major organs and tumors at different time points after injection. (e) In vivo fluorescence image of U14-tumor-bearing mice at different time points after intravenous injection of DiD-loaded Fe-DTX@M. The yellow circles indicate the tumor sites. (f) Quantitative analysis of fluorescence intensity in tumor area. (g) H&E staining of heart, liver, spleen, lung, and kidney of normal mice 30 days after Fe-DTX@M injection. Scale bar: 100 µm. (h) Serum CK, CK-MB, LDH, ALT, AST, ALP, CR, UA, and BUN levels in normal mice 30 days after Fe-DTX@M injection. Data are presented as mean ± SD (n = 3).
To visualize the biodistribution and tumor accumulation of Fe-DTX@M in vivo, DiD-labeled Fe-DTX@M was intravenously injected into tumor-bearing mice. Using a mouse intravital imager, the fluorescence of DiD-labeled Fe-DTX@M was significantly enriched in the tumor area over time, with the fluorescence signal reaching a maximum value at 8 h time point after injection (Figs. 3e and f), indicating a specific targeting effect. In the control group, the cell membrane probe was not specifically enriched and was quickly metabolized out of the body. Consequently, tumors and major organs were excised 24 h after injection for ex vivo imaging (Fig. S28 in Supporting information). Tumors from Fe-DTX@M-treated mice showed the strongest fluorescence signal among all resected tissues, indicating that Fe-DTX@M was heavily enriched in tumor sites at 24 h after injection.
To further verify the long-term in vivo safety, Fe-DTX@M was intravenously injected into normal mice. Over a period of 30 days, the body weight of the mice remained stable, as evidenced by the consistent weight measurements shown in Fig. S29 (Supporting information). Furthermore, the blood biochemical indicators, including heart, liver, and kidney function markers showed no obvious fluctuations, indicating no systemic toxicity (Fig. 3h). Histopathological analysis of the main organs, such as the heart, liver, spleen, lungs, and kidneys, exhibited no obvious morphological damage or signs of toxicity (Fig. 3g), further confirming the biocompatibility and safety of Fe-DTX@M. These results collectively indicate that the Fe-DTX@M possesses significant biosafety and excellent tumor-specific targeting properties.
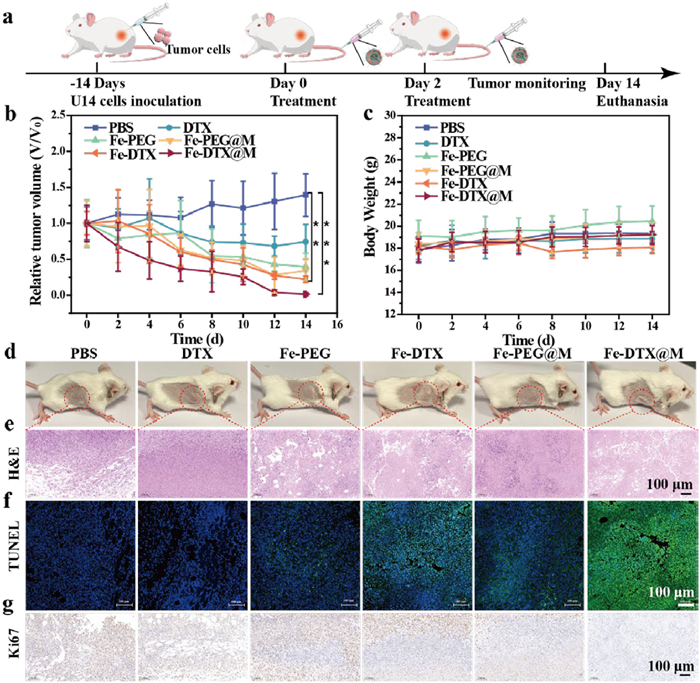
To determine whether Fe-DTX@M-induced ferroptosis-induced tumor inhibition is also applicable to in vivo tumor models, we utilized mouse cervical cancer cell U14 as a cell model and randomly divided tumor-bearing mice into 6 groups for treatment. PBS, DTX, Fe-PEG, Fe-DTX, Fe-PEG@M, and Fe-DTX@M were injected into the tail vein on days 0 and 2, respectively (Fig. 4a). The mice were monitored for 2 weeks, with body weight and tumor volume recorded every 2 days. The tumors in the control group and DTX-treated group grew rapidly, whereas those in the Fe-PEG, Fe-DTX, and Fe-PEG@M-treated groups grew slowly. Notably, the Fe-DTX@M group exhibited the best therapeutic effect, with the tumor almost completely eradicated, due to the synergistic effect of DTX-induced cell apoptosis (Figs. 4b and d). Additionally, there was no significant change in the body weight of mice in all groups during the treatment period (Fig. 4c), indicating that all treatments had limited adverse reactions in vivo. Fe-DTX@M with a small injection dose of 3.3 mg/kg showed excellent anti-tumor effects in vivo. By requiring a significantly lower dose of iron compared to traditional iron-based nanomaterials, Fe-DTX@M minimizes the risk of long-term toxicity and accumulation in the body. Moreover, the targeted accumulation of Fe-DTX@M within tumor tissues ensures that the therapeutic effects are localized, reducing potential harm to healthy cells. This targeted approach, combined with the low dosage requirement, makes Fe-DTX@M a safer and more efficient alternative for treating cervical cancer through the combined treatment of ferroptosis and apoptosis.
Figure 4
Figure 4.In vivo antitumor efficacy. (a) The scheme illustrating the experiment regimen. (b) Relative tumor volume changes of mice during treatments. (c) Body weight of mice during treatments. (d) Photos of tumors collected on day 14. (e-g) H&E staining, TUNEL staining, and Ki67 assays of tumor sections from different treatment groups. Scale bar: 100 µm. Data are shown as mean ± SD (n ≥ 3). P-values were calculated using one-way ANOVA with Tukey’s post-hoc test. **P < 0.01, ***P < 0.001.
The effects of different treatment groups on tumor tissues were further evaluated. Two weeks after the end of treatment, the collected tumor tissues were stained with hematoxylin and eosin (H&E) for histological analysis. No pathological changes were observed in the mice in the PBS group (Fig. 4e), while the other groups had varying degrees of nuclear damage, especially the tumor tissue in the Fe-DTX@M group was severely damaged. Simultaneously, TdT-mediated dUTP Nick-End Labeling (TUNEL) staining was used to further study the apoptosis of tumor tissues. The results showed that green fluorescence is the nucleus with double-strand breaks in DNA, and high levels of cell apoptosis were found in tumor tissues treated with Fe-DTX@M (Fig. 4f). The above results demonstrate that Fe-DTX@M has a strong anti-tumor effect. Ki67 staining is a protein that reflects the proliferation of tumor cells. The reduction of brown in the Fe-DTX@M treatment group indicates that the inhibitory effect on cell proliferation is the greatest (Fig. 4g). The above experiments show that Fe-DTX@M can treat tumors and inhibit tumor proliferation. The main organs and serum of mice in each group after treatment were collected, the H&E section (Fig. S30 in Supporting information) and biochemical analysis (Fig. S31 in Supporting information) revealed no obvious toxicity of Fe-DTX@M. The above research results consistently showed the biosafety of the biomimetic Fe-DTX@M nanoplatform for synergistic ferroptosis/apoptosis anticancer therapy.
In summary, we designed a cancer cell membrane-camouflaged Fe-based metallacycles nanoplatform (Fe-DTX@M) and demonstrated that Fe-DTX@M can precisely target tumor locations to release drugs and act as a ferroptosis amplifier in response to TME for synergistic ferroptosis/apoptosis anticancer therapy. The biomimetic Fe-DTX@M nanoplatform can homologously target tumor lesions to release Fe-DTX, causing redox dyshomeostasis by Fe2+-induced H2O2 generation of •OH and Fe3+-mediated GSH consumption. Enhanced ROS and inactivated GPX4 co-induced efficient ferroptosis, which synergized with DTX-elicited apoptosis to kill tumor cells. Compared with traditional iron-based nanomaterials, the experimental results demonstrated that a lower dose of Fe-DTX@M achieved safe and excellent tumor suppression capability without obvious side effects. This study not only synthesized a safe and effective ferroptosis inducer of Fe-based metallacycles, but also provided a biomimetic membrane-camouflaged Fe-based metallacycles drug delivery system for the combined treatment of cervical cancer via ferroptosis and apoptosis, which is expected to pave the way for a safe and effective anti-cervical cancer mechanisms in the future.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by National Key Research and Development (NKRD) Program of China (No. 2021YFA1500401), National Natural Science Foundation of China (NSFC, Nos. 22171291, 92261114, 92461302), and Guangdong Basic and Applied Basic Research Foundation (No. 2023A1515110702).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.111692.
Y. Tian, Z. Gao, N. Wang, et al., J. Am. Chem. Soc. 144 (2022) 18419–18428. doi: 10.1021/jacs.2c06877
[39]
H. Zhao, Y. Wang, Q. Chen, et al., Adv. Sci. 11 (2024) 2309131. doi: 10.1002/advs.202309131
[40]
H. Shang, D. Xia, R. Geng, et al., ACS Nano 19 (2025) 19599–19621. doi: 10.1021/acsnano.4c17444
[41]
M. Wang, Y. Li, M. Wang, et al., Acta Biomater. 138 (2022) 453–462.
[42]
X. Zhang, Y. Zhang, X. Lv, et al., ACS Nano 18 (2024) 33531–33544. doi: 10.1021/acsnano.4c11275
[43]
W. Xuan, Y. Xia, T. Li, et al., J. Am. Chem. Soc. 142 (2020) 937–944. doi: 10.1021/jacs.9b10755
[44]
Z. Zhou, H. Liang, R. Yang, et al., Angew. Chem. Int. Ed. 61 (2022) e202202843.
[45]
W. Feng, Z. Liu, L. Xia, et al., Angew. Chem. Int. Ed. 61 (2022) e202212021. doi: 10.1002/anie.202212021
[46]
Y. Zhu, D. Jin, M. Liu, et al., Small 18 (2022) 2200116.
[47]
V. Pop, A. Seicean, O. Soritau, et al., Ann. Oncol. 30 (2019) iv15–iv16.
[48]
M. Bruchard, M. Geindreau, A. Perrichet, et al., Nat. Immunol. 23 (2022) 262–274. doi: 10.1038/s41590-021-01120-y
Scheme 1
Schematic illustration of the formation of Fe-DTX@M and its application in the combined treatment of ferroptosis and apoptosis for cervical cancer. The designed and synthesized biomimetic Fe-based metallacycle nanoplatforms can specifically target tumor cells, and internalize into tumor cells to consume intracellular overexpressed H2O2 and GSH, leading to intracellular redox imbalance and GPX4 inactivation, thereby synergistically enhancing LPO-induced tumor cell ferroptosis, which is an effective treatment strategy for cervical cancer.
Figure 1
Characterization and catalytic performance of Fe-DTX@M. (a) TEM image and size distribution of Fe-PEG. (b) Size distribution and polymer dispersity index (PDI) of Fe-PEG at different time points. (c) Survey, Fe 2p, and P 2p XPS spectra of Fe-DTX. (d) Zeta potential of the different materials. (e) SDS-PAGE protein analysis. Samples were stained with Coomassie blue. (f) Zeta potential of Fe-DTX@M dispersed in different media. (g) ESR spectra of •OH generation after different treatments. (h) Degradation of DTNB treated with Fe-DTX@M at 412 nm. (i) The percentage of DTX released from Fe-DTX@M under different conditions. Data are presented as mean ± SD (n = 3).
Figure 2
Mechanism of Fe-DTX@M-induced ferroptosis. (a) Cellular uptake rate of Fe-DTX@M by different cells at various time points using ICP-MS. Cell viability of HeLa (b) and MCF-10A (c) cells with different treatment groups (mean ± SD, n ≥ 3). CLSM images of HeLa cells after different treatments and staining with (d) intracellular ROS probe DCFH-DA and (e) •OH probe HPF, respectively. (f) GSH and (h) MDA levels in HeLa cells after different treatments (mean ± SD, n ≥ 3). (g) Western blot results and quantitative analysis of GPX4 protein expression under different treatment conditions. (i) CLSM images and flow cytometry of C11-BODIPY581/591-dye-stained HeLa cells after different treatments. (j) CLSM images and flow cytometry of changes in MMP of HeLa cells after different treatments detected using JC-1 probe. Scale bar: 20 µm. HPF and DCFH-DA Probe: λex = 488 nm, λem = 520 ± 20 nm. C11-BODIPY581/591 (Red): λex = 561 nm, λem = 591 ± 20 nm. C11-BODIPY581/591 (Oxidation form, Green): λex = 488 nm, λem = 520 ± 20 nm. JC-1 (monomer, Green): λex = 488 nm, λem = 529 ± 20 nm. JC-1 (aggregate, red): λex = 561 nm, λem = 590 ± 20 nm. P < 0.05, **P < 0.01, ***P < 0.001 of (c–e) compared with the PBS group.
Figure 3
Evaluation of in vivo biodistribution and tumor targeting of Fe-DTX@M. (a) Hemolysis analysis of Fe-DTX and Fe-DTX@M dispersed at different concentrations. Deionized water and PBS were used as positive and negative controls, respectively. Inset: Photograph of the mixture after centrifugation to observe the presence of hemoglobin in the supernatant. (b) Pharmacokinetic profile of Fe-DTX and Fe-DTX@M. (c) Comparison of biodistribution of Fe-DTX and Fe-DTX@M in tumors. (d) Quantitative biodistribution of Fe-DTX@M in major organs and tumors at different time points after injection. (e) In vivo fluorescence image of U14-tumor-bearing mice at different time points after intravenous injection of DiD-loaded Fe-DTX@M. The yellow circles indicate the tumor sites. (f) Quantitative analysis of fluorescence intensity in tumor area. (g) H&E staining of heart, liver, spleen, lung, and kidney of normal mice 30 days after Fe-DTX@M injection. Scale bar: 100 µm. (h) Serum CK, CK-MB, LDH, ALT, AST, ALP, CR, UA, and BUN levels in normal mice 30 days after Fe-DTX@M injection. Data are presented as mean ± SD (n = 3).
Figure 4In vivo antitumor efficacy. (a) The scheme illustrating the experiment regimen. (b) Relative tumor volume changes of mice during treatments. (c) Body weight of mice during treatments. (d) Photos of tumors collected on day 14. (e-g) H&E staining, TUNEL staining, and Ki67 assays of tumor sections from different treatment groups. Scale bar: 100 µm. Data are shown as mean ± SD (n ≥ 3). P-values were calculated using one-way ANOVA with Tukey’s post-hoc test. **P < 0.01, ***P < 0.001.

 DownLoad:
DownLoad:

 下载:
下载:






 下载:
下载:
