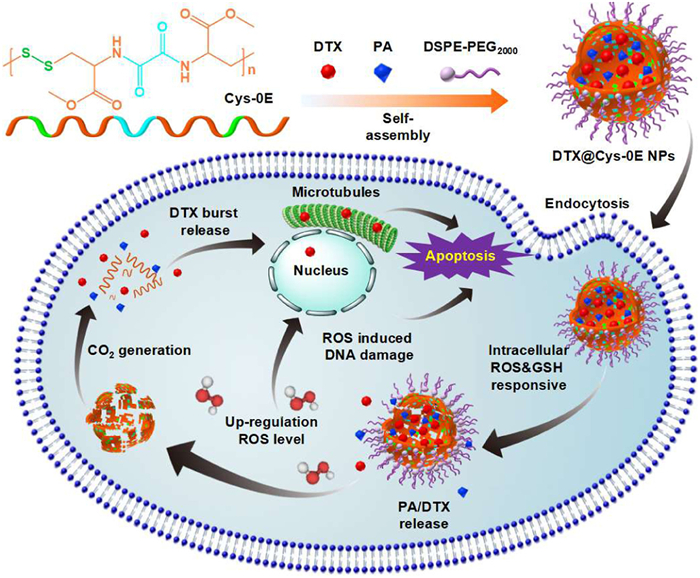
Scheme 1.
Schematic illustration of the novel dual-redox-sensitive CO2-generating polymer (Cys-0E), the corresponding self-assembly of drug-loaded nanoparticles (DTX@Cys-0E NPs), and their mechanism of tumor inhibition in vivo.
Dual redox-responsive CO2-generating nanoparticles assembled from one-step synthesized L-cystine-based biodegradable polymers for enhanced chemotherapy of tumors
Ruhe Zhang , Dandan Li , Ting Liang , Xinyu Zhang , Jingyi Hou , Yang Kang , Dongjun Lin , Jun Wu

Malignant tumors remain a critical challenge to human health and survival [1]. Conventional cancer treatments, including surgery, radiotherapy, chemotherapy, and immunotherapy, are frequently applied in combination to improve therapeutic outcomes [2–4]. Despite its widespread application, chemotherapy suffers from drawbacks such as systemic toxicity, poor drug selectivity, low bioavailability, hydrophobic drug properties, and the development of drug resistance [5–7]. Therefore, there is an urgent need for innovative drug delivery systems (DDSs) to improve the precision and effectiveness of cancer treatment [8].
With the emergence and progress of nanotechnology, batches of DDSs have been developed for the delivery of antitumor drugs, ranging from conventional liposomes to advanced smart nanoparticles [9–11]. At present, a variety of smart nanoparticles have been developed by modified with various groups sensitive to tumor endogenous or exogenous stimuli including redox substances, enzymes, temperature, etc. [12–17]. It is necessary to note that the tumor microenvironment has unusual redox potential, including a higher level of glutathione (GSH) or reactive oxygen species (ROS) expression in cancer tissues compared to normal tissues [18]. Generally, the concentration of H2O2 in normal tissues is 20 nmol/L, while that in cancer tissues is as high as 50–100 µmol/L due to excessive production and accumulation of H2O2 [19,20]. Therefore, nanocarriers incorporating ROS-responsive functional groups, particularly peroxalate ester linkages, thioethers, and thioketal moieties, demonstrate controlled drug release in response to elevated ROS levels in tumor cells [21–23]. It is also reported that the level of GSH in tumor cells (10–40 mmol/L) is more than 1000 times higher than that in normal blood environments (2–20 µmol/L) and 4 times higher than that of healthy cells, respectively [24–27]. These significant concentration differences provide the mechanism for intracellular controlled drug release in cancer sites. Among GSH-responsive DDSs, disulfide bonds are frequently used groups because their exchanges with intracellular glutathione degrade the polymer and lead to drug-controlled release [28,29]. Besides, this group is easily modified both in the main chain and the side chain in different polymers [30,31].
In our previous works, biocompatible and redox-sensitive poly(disulfide amide)s were synthesized based on natural disulfide-containing L-cystine ester [32–34]. Compared with other poly(disulfide)s-containing systems, this system exhibited the advantages of simple synthesis, adjustability, and good biocompatibility [12,33]. However, these systems are only single-reduction responses and are insufficient to cope with the complex tumor microenvironment. Based on this research foundation, we designed a novel GSH and ROS dual-sensitive polymer by a simple one-step synthesis of disulfide bond-containing L-cystine dimethyl ester and peroxalate ester-containing oxalyl chloride to improve the responsiveness [35]. The prepared polymer is named Cys-0E, with E representing the methyl ester on the side chain, and 0 indicating no methylene groups in the oxalyl chloride unit [33]. Based on this polymer, dual redox-responsive and uniformly stable nanoparticles (DTX@Cys-0E NPs) with high drug-loading content were prepared by the optimal nanoprecipitation method. 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy-(polyethylene glycol)-2000] (DSPE-PEG2000) and 6-o-palmitoyl ascorbic acid (PA) were also involved in the construction of NPs which could both improve the stability of nanoparticles in the blood circulation. Besides, PA was also able to generate more H2O2 to stimulate the breakage of peroxalate ester-like linkages in the tumor microenvironment [36–38]. Once the NPs accumulated in tumor cells, the controlled release of docetaxel (DTX) and the enhanced anticancer effect was realized both in vitro and in vivo (Scheme 1).
First, Cys-0E was synthesized through a one-step polycondensation reaction. Disulfide bond-containing L-cystine dimethyl ester and peroxalate ester-containing oxalyl chloride were used in the reaction at a molar ratio of 1:1.2, as illustrated in Fig. 1A. The structural characteristics of Cys-0E were validated by proton nuclear magnetic resonance (1H NMR), Fourier transform infrared spectroscopy (FT-IR), and gel permeation chromatography (GPC). As shown in Fig. 1B, a characteristic peak originating from the hydrogen proton of the amide unit appeared at 9.22–9.34 ppm, indicating the successful synthesis of Cys-0E. According to the FT-IR spectrum of Cys-0E (Fig. 1C), the characteristic vibration of the amide bond is visible. Among them, the N—H stretching vibration in the amide bond (amide A band) appears at about 3287 cm−1, the C=O stretching vibration (amide Ⅰ band) appears at about 1664 cm−1, and the C—N stretching vibration (amide Ⅱ band) appears at 1506 cm−1, and these characteristic peaks further prove the successful formation of Cys-0E [39]. The molecular weight (Mw) of Cys-0E was measured to be 14,800 Da (Table S1 and Fig. S1 in Supporting information).
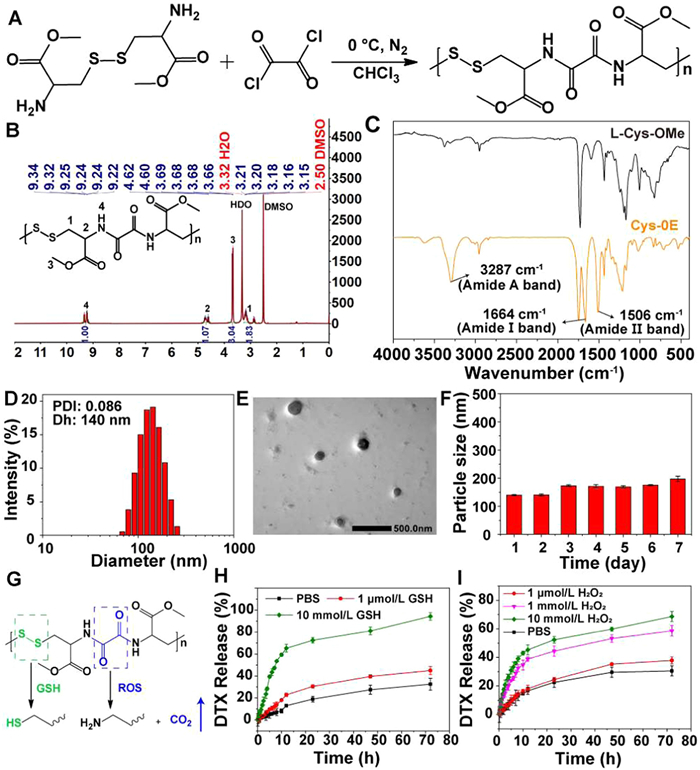
Then, the yielded redox-sensitive biodegradable polymer Cys-0E was utilized to prepare polymeric micelles according to an optimized nanoprecipitation method [40]. The nanoparticles obtained from the assembly of Cys-0E, DSPE-PEG2000, and DTX showed obvious precipitation with low stability. However, the nanoparticles (DTX@Cys-0E NPs) formed from the co-assembly of Cys-0E, DSPE-PEG2000, PA, and DTX exhibited good colloidal stability. As measured by dynamic light scattering (DLS), the mean diameter of DTX@Cys-0E NPs was 140 ± 2 nm with a low polydispersity index (PDI) of 0.086 (Fig. 1D), and the zeta potential was about −38.4 mV (Fig. S2 in Supporting information). Transmission electron microscope (TEM) imaging exhibited that DTX@Cys-0E NPs presented spherical morphology and the size of NPs roughly observed was about 137 nm, which was consistent with the result detected by DLS (Fig. 1E). Fig. 1F showed that the size of DTX@Cys-0E NPs underwent slight variation and was always less than 200 nm and lasted for one week in RPMI 1640 medium containing 10% fetal bovine serum (FBS), which indicates that DTX@Cys-0E NPs have good stability in the physiological environment. DTX was utilized as a model drug and dissolved in CH3CN to configure a series of concentrations as a working fluid. Then the working fluid was measured by HPLC to obtain a standard curve (Fig. S3 in Supporting information). According to the HPLC results, the drug loading content (DL) and entrapment efficiency (EE) of DTX@Cys-0E NPs are 13.1% and 83.2%, respectively. Blank Cys-0E NPs (without PA and DTX added) were also obtained and measured with a size of 111 nm and a PDI of 0.153 (Fig. S4 in Supporting information).
To study the redox-responsive release of DTX@Cys-0E NPs, the ROS-responsive degradation of Cys-0E was first evaluated with ATR infrared spectroscopy since peroxalate ester bonds can be oxidated by H2O2 to generate CO2 (Fig. 1G) [41]. As shown in Fig. S5 (Supporting information), the transmittance of the characteristic absorption peaks of CO2 at 2340 and 2360 cm−1 gradually decreased over time, signifying the gradual degradation of Cys-0E and concurrent production of CO2 [42,43]. This CO2 generation may enhance the release of DTX in a high-ROS environment. Consequently, the drug release from DTX@Cys-0E NPs was assessed by the dialysis method to explore the dual redox-responsive behavior. As we can see from Figs. 1H and I, in dialysate without or with low concentrations of GSH or H2O2, DTX@Cys-0E NPs exhibited a slow release. Under these circumstances, DTX released within 24 h was almost below 30%. This indicates that the drug-loaded nanoparticles have better drug encapsulation ability in normal blood circulation, and will not cause serious drug leakage to affect normal tissues. In Fig. 1H, under the condition of high GSH concentration, DTX showed obvious rapid release behavior within 24 h, which was more than 50%. After that, the drug release curve tended to be flat. At 24 h, the cumulative release of DTX from nanoparticles in 10 mmol/L GSH release media was 72.5%. Over 72 h, the cumulative release of DTX was more than 90%. These results fully demonstrate that the drug encapsulated in nanoparticles can be released efficiently under the condition of high GSH levels in tumors. The H2O2-responsive behavior of nanoparticles resembled the results of the GSH response. After exposure to a high concentration of H2O2 (1 mmol/L and 10 mmol/L) for 72 h, the release rate was 55.6% and 68.6%, respectively (Fig. 1I). Therefore, the incorporation of the H2O2-responsive group will assist the GSH-responsive disulfide bond to achieve more controlled, accurate, and efficient drug release. The above experimental outcomes show that DTX@Cys-0E NPs prepared in this study hold considerable promise for use in advanced drug-controlled and sustained release systems.
To track the uptake kinetic of Cys-0E NPs in CT26 cells, we used C6 to label Cys-0E NPs for flow cytometry analysis [44]. The results in Figs. S6A and B (Supporting information) exhibited that the fluorescence intensity gradually increased over time during incubation with CT26 cells, indicating that the C6@Cys-0E NPs can be effectively absorbed by tumor cells and the cellular uptake of C6@Cys-0E NPs is time-dependent.
Confocal laser scanning microscopy was applied to further observe the cellular uptake of Cys-0E NPs. Lipophilic dye Dil was encapsulated in Cys-0E NPs as a marker for nanoparticles and fluorescein 5-isothiocyanate (FITC)-phalloidin was used to label cytoskeleton. In the CT26 cells treated with Dil@Cys-0E NPs in Fig. S6C (Supporting information), when the culture time was prolonged from 0.5 h to 6 h, the red fluorescence of nanoparticles increased significantly, signifying the gradual uptake of Dil@Cys-0E NPs. The above results consistent with the flow cytometry analysis result, indicated that CT26 cells could efficiently capture Cys-0E NPs, providing reliable evidence for DTX@Cys-0E NPs in the treatment of colon cancer.
To investigate the cytotoxicity of DTX@Cys-0E NPs, 3-(4,5-dimethyl thiazol-2-yl)-2,5diphenyl tetrazolium bromide (MTT) assay was first applied on CT26 cells, NIH 3T3 cells, and RAW264.7 cells. Cells were incubated with free DTX, DTX@Cys-0E NPs, and Cys-0E NPs at different concentrations for 48 h. Fig. 2A showed that almost all CT26 cells treated with Cys-0E NPs survived and all DTX formulations exhibited apparent cytotoxicity which depended on concentration. The half-maximum inhibitory concentration (IC50) was 3.80 and 2.31 µg/mL for CT26 cells treated with free DTX and DTX@Cys-0E NPs, respectively. These results indicate that DTX@Cys-0E NPs have stronger cytotoxicity to tumor cells than free DTX. Under the same concentration of different formations, free DTX also caused obvious cytotoxicity to normal cells (NIH 3T3 cells and RAW264.7 cells), while DTX@Cys-0E NPs had lower toxicity and side effects on normal cells (Fig. 2B and Fig. S7 in Supporting information). These results suggest that this nanocarrier realizes the controlled release of anticancer drugs in tumor sites with low toxicity to normal tissues compared with the non-selective cytotoxic effect of free DTX, which is associated with elevated GSH and ROS levels in tumor cells. Therefore, this nano platform exhibits exceptional potential as an effective nano-drug delivery carrier due to its advantages including targeting release, high efficiency, and low toxicity to normal cells.
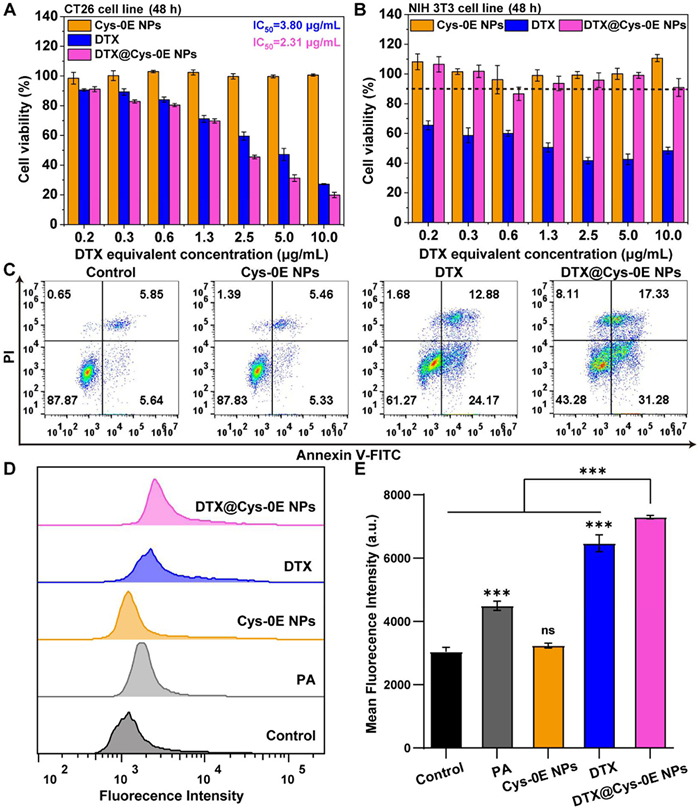
Furthermore, Annexin V-FITC/propidium iodine (PI) was applied for an in vitro apoptosis study to quantitatively analyze the inhibitory effect of DTX@Cys-0E NPs on CT26 cells. Analysis of Fig. 2C revealed that CT26 cells treated with Cys-0E NPs showed a total apoptotic percentage of 10.8%, which was comparable to the control group. This observation indicates that the biodegradable carrier material has no inhibitory effect on tumor cells. Besides, the apoptosis of CT26 cells treated with free DTX and DTX@Cys-0E NPs was mainly early apoptosis, with apoptosis rates of 24.2% and 31.3%, respectively. Notably, the DTX@Cys-0E NPs demonstrated superior apoptotic induction, achieving a total apoptotic rate of 48.6%, which was 1.3-fold higher than that of free DTX (37.0%). These data confirm that DTX@Cys-0E NPs have superior antitumor effects.
To verify that DTX@Cys-0E NPs can improve the ROS level and further promote the oxidative response disintegration of the carrier, the intracellular ROS was observed by FCM with the use of the ROS Assay Kit. This detection kit uses 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) as a fluorescent probe, which generates quantifiable fluorescent DCF through the reaction between DCFH and ROS in cells [45]. As shown in Figs. 2D and E, PA treatment resulted in a significant elevation of intracellular ROS level, showing a 1.48-fold increase compared to the control group, which provided favorable conditions for the intracellular ROS-responsive release of DTX. A slightly higher ROS level was observed in Cys-0E NPs-treated cells because ROS-responsive peroxalate ester linkages in Cys-0E consumed ROS and GSH-responsive disulfide bonds consumed GSH to produce ROS. Direct addition of DTX could also elevate the ROS level, with a twofold increase. Moreover, the ROS level of DTX@Cys-0E-treated cells was even marked higher (2.40 fold increase compared to the control group), which stemmed from the direct H2O2 production by PA and the subsequent oxidative stress induced by released DTX.
Due to the good in vitro anticancer effect of the DTX@Cys-0E NPs, we then conducted studies on their performance in vivo. Blood compatibility of the Cys-0E NPs was firstly evaluated using the hemolysis test. According to the hemolysis result in Fig. S8 (Supporting information), Cys-0E NPs exhibited low hemolysis rate of less than 5% at concentration of 1 mg/mL, indicating good blood compatibility for in vivo applications.
To study the in vivo targeting ability of DTX@Cys-0E NPs, DiR was selected as the probe and DiR@Cys-0E NPs were prepared and injected intravenously into CT26 tumor-bearing BALB/c mice. All animal procedures in this manuscript were approved by the Animal Ethics Committee of Sun Yat-sen University (approval No. SYSU-IACUC-2021–000901). Then IVIS imaging system was applied to detect the distribution of nanoparticles in mice over time [46]. An equivalent dose of free DiR was used as a blank control. The biodistribution of free DiR and DiR@Cys-0E NPs was displayed in Fig. 3A. Compared to the group of free DiR, which mainly accumulated in the liver, stronger fluorescence signals in tumors were observed in the DiR@Cys-0E NPs group, indicating that nanoparticles could accumulate more efficiently in tumors. The accumulation in the tumor site was increased with time after injection. Figs. 3B and C showed the fluorescent images of excised major organs and tumors and the corresponding quantitative results. Compared with the negligible fluorescence signal of free DiR, DiR@Cys-0E NPs had obvious fluorescence intensity at the tumor, which further verified that DTX@Cys-0E NPs can enhance tumor targeting ability and the accumulation of DTX in tumor tissues, showing potential to improve the therapeutic effect of chemotherapy.
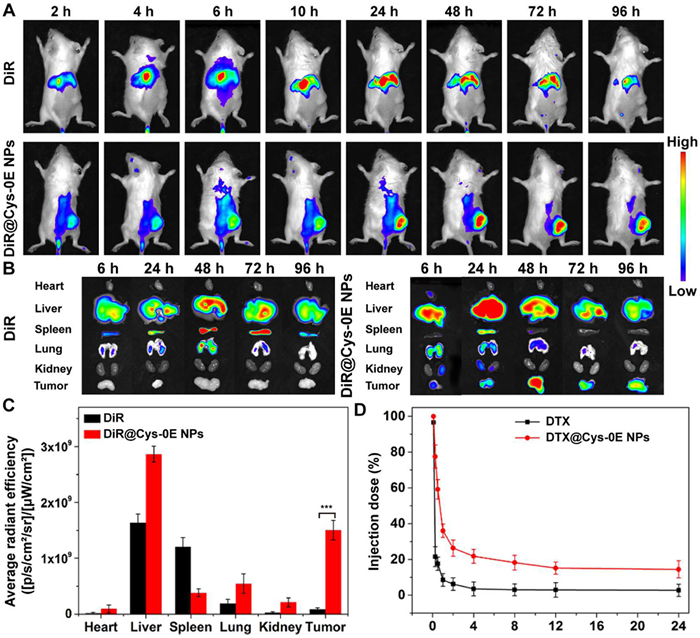
For the in vivo pharmacokinetic study, the time-dependent DTX blood concentration level was monitored with the application of HPLC-MS. As shown in Fig. 3D, it was obvious that DTX@Cys-0E NPs had longer circulation time in vivo than free DTX. Simultaneously, the quantitative data results obtained from Table S2 (Supporting information) were consistent with this situation, which showed that the elimination half-lives (t1/2β) and area under the curve (AUC) of DTX@Cys-0E NPs were separately 4.9-folds and 6.5-folds that of free DTX. The longer t1/2β and higher AUC prove that the stability of nanoparticles can be effectively improved in the blood circulation and the premature leakage of the encapsulated drug can also be prevented.
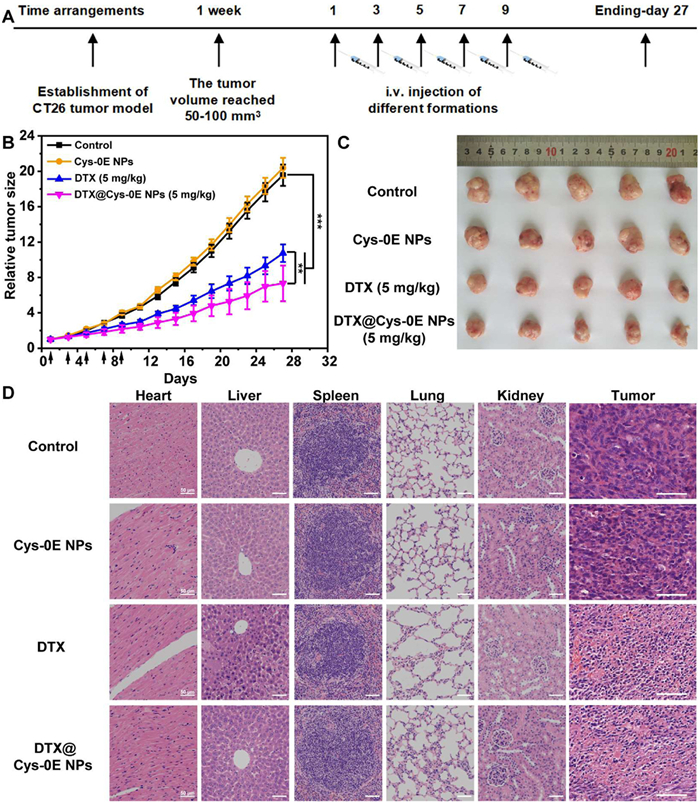
To further verify the anticancer effect of DTX@Cys-0E NPs, CT26 tumor-bearing BALB/c male mice were separately treated with the equivalent dose of saline, Cys-0E NPs, free DTX, and DTX@Cys-0E NPs, with saline as a blank control. Meanwhile, the tumor volume and weight changes in each group were recorded for nearly a month (Fig. 4A). As demonstrated by the tumor growth curves (Fig. 4B) and the excised tumor tissues (Fig. 4C), all DTX formulations exhibited effective antitumor activity. Notably, the DTX@Cys-0E NPs showed superior therapeutic outcomes compared to free DTX. According to the weight curves shown in Fig. S9 (Supporting information), there were no significant changes in the body weight of mice injected with DTX@Cys-0E NPs. In contrast to the slight weight loss observed in free DTX treatment, the stability in body weight indicates that the DTX@Cys-0E NPs can reduce the systemic toxicity of DTX. Finally, the tumors and major organs in the above experimental groups were collected and sliced and then stained with hematoxylin and eosin to further demonstrate the excellent antitumor activity of DTX@Cys-0E NPs. The results of hematoxylin and eosin (H&E) staining in Fig. 4D showed that no difference was shown in mice injected with Cys-0E and saline, demonstrating that Cys-0E had no inhibitory effect on CT26 tumor-bearing BALB/c mice. DTX@Cys-0E NPs caused more obvious necrosis of tumor cells without damage to the major normal organs. On the contrary, although DTX had a certain lethal effect on tumor tissues, its apparent liver damage could not be ignored. The above results indicate that DTX@Cys-0E NPs not only enhance the inhibitory effect on tumors but also solve the systemic toxicity of free DTX, which provides a novel delivery platform for insoluble anticancer drugs to achieve more effective cancer treatment.
In summary, we designed and synthesized the dual-redox-sensitive polymer for DTX encapsulation, which formed stable, uniformly sized particles with high drug loading content. Besides, the DTX@Cys-0E NPs could release DTX rapidly under the stimulation of high GSH or ROS. In cell and animal experiment results demonstrated that compared with free DTX, DTX@Cys-0E NPs exhibited better cellular uptake capacity, higher cytotoxicity and apoptosis in tumor cells, and higher antitumor efficacy. This antitumor drug delivery platform has three advantages: (1) The preparation of dual-redox-sensitive Cys-0E and the construction of DTX@Cys-0E NPs are simple; (2) The DTX@Cys-0E NPs have better biocompatibility and higher biological safety thanks to the natural sulfur-containing material and non-toxic substances after decomposition of Cys-0E; (3) The DTX@Cys-0E NPs can overcome the complex redox microenvironment in vivo to achieve more accurate controlled release due to the ROS and GSH dual responsive of Cys-0E, the generation of CO2 after peroxalate ester bond degradation, and addition of PA as potent H2O2 generator in tumor cells. Therefore, the novel dual redox-responsive platform can be a promising therapeutic agent for various cancer therapies.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Ruhe Zhang: Writing – review & editing, Visualization, Methodology, Funding acquisition, Data curation. Dandan Li: Writing – original draft, Visualization, Software, Methodology, Formal analysis, Data curation. Ting Liang: Writing – review & editing, Visualization, Validation, Methodology, Formal analysis, Data curation. Xinyu Zhang: Validation, Methodology, Formal analysis. Jingyi Hou: Validation, Methodology, Formal analysis. Yang Kang: Writing – review & editing, Supervision, Project administration, Conceptualization. Dongjun Lin: Writing – review & editing, Supervision, Resources. Jun Wu: Writing – review & editing, Resources, Project administration, Funding acquisition, Conceptualization.
This project was supported by the National Natural Science Foundation of China (No. 52173150), the Guangzhou Science and Technology Program City-University Joint Funding Project (No. 2023A03J0001), the Affiliated Qingyuan Hospital of Guangzhou Medical University Open Project Fund (No. 202301-211), Science and Technology Projects in Guangzhou (No. 2023A03J0704), Sanming Project of Medicine in Shenzhen (No. SZSM202411022), and Research Start-up Fund of Post-doctoral of SAHSYSU (No. ZSQYRSFPD0074). We sincerely acknowledge the funding and generous support from these foundations.
Supplementary material associated with this article can be found, in the online version, at doi:
F. Bray, M. Laversanne, H. Sung, et al., CA Cancer J. Clin. 74 (2024) 229–263. doi: 10.3322/caac.21834
Y. Jin, J. Jiang, W. Mao, et al., Cancer Lett. 591 (2024) 216858. doi: 10.1016/j.canlet.2024.216858
A. Audisio, R. Fazio, V. Daprà, et al., Cancer Treat. Rev. 123 (2024) 102676. doi: 10.1016/j.ctrv.2023.102676
M. Chalabi, Y.L. Verschoor, P.B. Tan, et al., N. Engl. J. Med. 390 (2024) 1949–1958. doi: 10.1056/nejmoa2400634
C. Holohan, S. Van Schaeybroeck, D.B. Longley, et al., Nat. Rev. Cancer 13 (2013) 714–726. doi: 10.1038/nrc3599
C. Frederiks, S. Lam, H. Guchelaar, et al., Cancer Treat. Rev. 41 (2015) 935–950. doi: 10.1016/j.ctrv.2015.10.010
C. Zhang, L. Yan, X. Wang, et al., Nano Today 35 (2020) 101008. doi: 10.1016/j.nantod.2020.101008
T.H. Baryakova, B.H. Pogostin, R. Langer, et al., Nat. Rev. Drug Discov. 22 (2023) 387–409. doi: 10.1038/s41573-023-00670-0
M. Chehelgerdi, M. Chehelgerdi, O.Q.B. Allela, et al., Mol. Cancer 22 (2023) 169.
W. Chen, K. Shi, Y. Yu, et al., Chin. Chem. Lett. 35 (2024) 109159. doi: 10.1016/j.cclet.2023.109159
P.R. Cullis, P.L. Felgner, Nat. Rev. Drug Discov. 23 (2024) 709–722. doi: 10.1038/s41573-024-00977-6
R.H. Zhang, T.Q. Nie, Y.F. Fang, et al., Biomacromolecules 23 (2022) 1–19.
R. Zhang, T. Nie, L. Wang, et al., Biomater. Sci. 11 (2023) 4254–4264. doi: 10.1039/d3bm00461a
D.D. Li, R.H. Zhang, G.T. Liu, et al., Adv. Healthc. Mater. 9 (2020) 2000605. doi: 10.1002/adhm.202000605
Y. Liu, M. Jiang, Z. Zhao, et al., Acta Biomater. 166 (2023) 567–580. doi: 10.32604/jrm.2022.023037
J. Liu, D. He, T. Hao, et al., Chin. Chem. Lett. 35 (2024) 109296. doi: 10.1016/j.cclet.2023.109296
W. Zhong, X. Zhang, X. Duan, et al., Acta Biomater. 144 (2022) 67–80.
S. Bai, X. Ma, X. Shi, et al., ACS Appl. Mater. Interfaces 11 (2019) 36130–36140. doi: 10.1021/acsami.9b13214
C. de Gracia Lux, S. Joshi-Barr, T. Nguyen, et al., J. Am. Chem. Soc. 134 (2012) 15758–15764. doi: 10.1021/ja303372u
Y.T. Chiang, Y.W. Yen, C.L. Lo, Biomaterials 61 (2015) 150–161. doi: 10.1016/j.biomaterials.2015.05.007
C. Tapeinos, A. Pandit, Adv. Mater. 28 (2016) 5553–5585. doi: 10.1002/adma.201505376
J. Liu, X. You, L. Wang, et al., Small 20 (2024) e2401438.
X. Dong, Z. Zhang, R. Wang, et al., Small 20 (2024) e2309529. doi: 10.1002/smll.202309529
R.S. Navath, Y.E. Kurtoglu, B. Wang, et al., Bioconjug. Chem. 19 (2008) 2446–2455. doi: 10.1021/bc800342d
A. Russo, W. DeGraff, N. Friedman, et al., Cancer Res. 46 (1986) 2845–2848.
R. Cheng, F. Feng, F. Meng, et al., J. Control. Release 152 (2011) 2–12.
A. Bansal, M.C. Simon, J. Cell Biol. 217 (2018) 2291–2298. doi: 10.1083/jcb.201804161
C.J. Prange, N.Y.B. Sayed, B. Feng, et al., J. Control. Release 368 (2024) 251–264.
Z.Q. Zhang, X.X. Xu, J.W. Du, et al., Nat. Commun. 15 (2024) 1118. doi: 10.1038/s41467-024-44963-3
F.Z. Yu, Y.L. Tu, S.W. Luo, et al., Nano Lett. 21 (2021) 2216–2223. doi: 10.1021/acs.nanolett.0c05028
Z.M. Shu, I. Tanaka, A. Ota, et al., Angew. Chem. Int. Ed. 58 (2019) 6611–6615. doi: 10.1002/anie.201900993
X.D. Xu, J. Wu, S.S. Liu, et al., Small 14 (2018) e1802565.
J. Wu, L.L. Zhao, X.D. Xu, et al., Angew. Chem. Int. Ed. 54 (2015) 9218–9223. doi: 10.1002/anie.201503863
Y. Tao, C. Dai, Z. Xie, et al., Chin. Chem. Lett. 35 (2024) 109170.
L. Zhang, S. Zhang, M. Li, et al., Mater. Sci. Eng. C: Mater. Biol. Appl. 123 (2021) 111956.
J. Li, W. Ke, L. Wang, et al., J. Control. Release 225 (2016) 64–74. doi: 10.17647/jss.2016.08.64.64
Q. Chen, M.G. Espey, A.Y. Sun, et al., Proc. Natl. Acad. Sci. U. S. A. 104 (2007) 8749–8754. doi: 10.1073/pnas.0702854104
Q. Chen, M.G. Espey, A.Y. Sun, et al., Proc. Natl. Acad. Sci. U. S. A. 105 (2008) 11105–11109. doi: 10.1073/pnas.0804226105
H. Liu, J. Xiao, S. Wang, et al., Chem. Eng. J. 482 (2024) 149167.
X. Yan, J. Bernard, F. Ganachaud, Adv. Colloid Interface Sci. 294 (2021) 102474.
W. Cao, W. Wei, B. Qiu, et al., Chem. Eng. J. 483 (2024) 149187.
Ⅲ M.F. Baruch, J.E. Pander, J.L. White, et al., ACS Catal. 5 (2015) 3148–3156. doi: 10.1021/acscatal.5b00402
S. Mukhopadhyay, M.S. Naeem, G. Shiva Shanker, et al., Nat. Commun. 15 (2024) 3397.
K.Y. Ou, X.J. Xu, S.Y. Guan, et al., Adv. Funct. Mater. 30 (2020) 1907857.
C. Li, W. Jia, Z. Guo, et al., J. Mater. Chem. B 12 (2024) 800–813. doi: 10.1039/d3tb02288a
S.L. Yu, R.H. Zhang, Z.X. Xie, et al., ACS Biomater. Sci. Eng. 10 (2024) 4336–4346. doi: 10.1021/acsbiomaterials.4c00345

Scheme 1 Schematic illustration of the novel dual-redox-sensitive CO2-generating polymer (Cys-0E), the corresponding self-assembly of drug-loaded nanoparticles (DTX@Cys-0E NPs), and their mechanism of tumor inhibition in vivo.

Figure 1 (A) Specific synthesis process of Cys-0E. (B) 1H NMR spectrum of Cys-0E. (C) The FT-IR spectra of L-cystine dimethyl ester (L-cys-OMe) and Cys-0E. (D) The particle size distribution of DTX@Cys-0E NPs. (E) The TEM image of DTX@Cys-0E NPs (scale bar: 500 nm). (F) The particle size of DTX@Cys-0E NPs monitored by DLS for one week in RPMI 1640 medium containing 10% FBS. (G) Mechanism of GSH and ROS responsive degradation of Cys-0E. Cumulative release of DTX from the DTX@Cys-0E NPs treated with a variable concentration of (H) GSH or (I) H2O2. Data are presented as mean ± standard deviation (SD) (n = 3).

Figure 2 (A) CT26 cells and (B) NIH 3T3 cells viability after treatment with free DTX, DTX@Cys-0E NPs, and Cys-0E at different concentrations for 48 h. (C) Apoptosis assay of CT26 cells after treatment with Cys-0E, free DTX, and DTX@Cys-0E NPs after 48 h at an equal DTX concentration of 10 µg/mL. (D) Representative fluorescence intensity histograms of CT26 cells after treatment with PA, Cys-0E, free DTX, and DTX@Cys-0E NPs for 12 h and subjected to DCFH-DA incubation for 30 min. (E) Mean fluorescence intensity calculated by flow cytometry analysis in (D). Data are presented as mean ± SD (n = 3). ***P < 0.001 vs. control group. ns, not significant.

Figure 3 (A) Live animal imaging of free DiR and DiR@Cys-0E NPs in CT26 tumor-bearing BALB/c mice. (B) Representative fluorescent images of excised major organs and tumors at different time points. (C) Quantification of DiR fluorescent signal in different ex-vivo tissues in tumor-bearing mice after intravenous injections of DiR and DiR@Cys-0E NPs for 96 h. ***P < 0.001 vs. DiR group. (D) In vivo pharmacokinetics of free DTX and DTX@Cys-0E NPs after administration in Sprague-Dawley rats. Data are presented as mean ± SD (n = 3).

Figure 4 In vivo antitumor efficacy of DTX@Cys-0E NPs. (A) Arrangement of antitumor experiments. (B) Tumor growth curves, and (C) macro views of excised tumors after treatment with saline, Cys-0E (equal DTX concentration of 5 mg/kg), free DTX, and DTX@Cys-0E NPs respectively. (D) H&E stained the histological section of CT26 tumor tissue and major organs collected from every group at the end of the antitumor experiment. Scale bar: 50 µm. All data are presented as mean ± SD (n = 5). Control was mice without any treatment (***P < 0.001 treated groups vs. control, **P < 0.01 free DTX vs. DTX@Cys-0E NPs).

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
 下载:
下载: