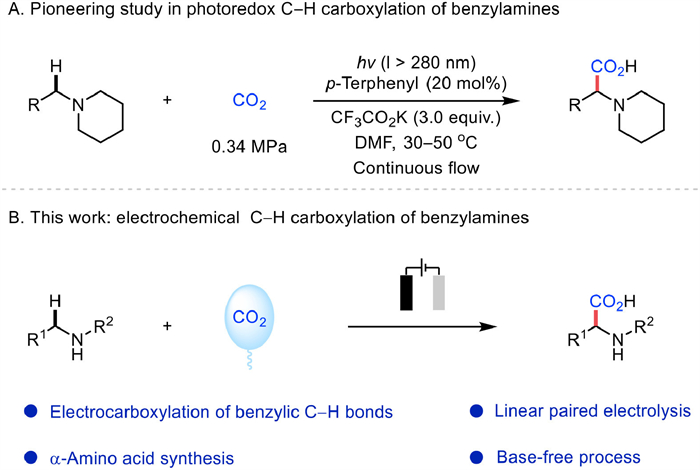
Figure 1.
Background and introduction. (A) Pioneering study in photoredox C—H carboxylation of benzylamines. (B) This work: electrochemical C—H carboxylation of benzylamines.

Carbon dioxide (CO2) serves as an ideal one-carbon (C1) synthon for synthesizing value-added chemicals [1-3]. However, the inherent inertness of CO2 seriously inhibits its application in important transformations [4-6]. So far, considerable efforts have been devoted to developing sustainable and efficient catalytic systems for CO2 activation, motivated by its non-toxicity and abundance. Recently, photochemistry [7-14] and electrochemistry [15-65] have shown unique strengths in the synthesis of carboxylic acids due to their new reactivity and mechanisms. In contrast to the state-of-the-art carboxylation reaction of (pseudo)halides [66-71], which typically involves the elimination of leaving groups (LGs), the direct carboxylation of benzylic C—H bonds offers a more atom-economical alternative. Nevertheless, this transformation is likely hindered by the inertness of benzylic C—H bonds, particularly the strong bond dissociation energy (BDE) and high electrochemical oxidation potential (Eox) [72,73]. Moreover, the conversion of benzylic C—H bonds to carbanions followed by CO2 incorporation often involves multiple processes, which poses another great challenge for such benzylic C—H carboxylation. Overcoming these obstacles to enable efficient electrochemical benzylic C—H carboxylation would therefore represent a significant strategic advance in the field.
Natural and unnatural α-amino acids find broad application in both academic research and industrial domains, particularly in pharmaceutical, biotechnology, and chemical sectors [74-80]. Traditional chemical methods for synthesizing α-amino acids mainly involve the Strecker and Gabriel syntheses, which generally require harsh reaction conditions and employ highly toxic reagents such as hydrogen cyanide (HCN) or hydrazine [75,77]. These drawbacks undoubtedly increase production costs, reduce reaction efficiency, and exacerbate environmental pollution. Of note, pioneering contributions to achieve carboxylation of benzylic C—H bonds date back to 2017, when Jamison and co-workers developed the photoredox carboxylation of tertiary N-benzylpiperidines to obtain α-amino acids (Fig. 1A) [81]. Several tertiary benzylamines could undergo carboxylation using ultraviolet light irradiation with 20 mol% p-terphenyl as the photocatalyst, 3 equiv. potassium trifluoroacetate (CF3CO2K) as the base and 0.34 MPa CO2 as the carboxyl source. Inspired by this elegant work and our continuous interest in electrochemical carboxylation [82-85], we envisioned that direct electrochemical benzylic C—H carboxylation of benzylamines could serve as an ideal approach to α-amino acids, combining enhanced atom economy with significantly improved reaction efficiency. This methodology represents a more sustainable and efficient alternative to conventional synthetic approaches, fully consistent with green chemistry principles. However, notable challenges include (i) how to solve the problems caused by paired electrolysis that benzylamines favor anodic reactions [86-88] while carboxylation usually occurs at the cathode [82-85] (ii) how to reach the relatively high oxidative potential of benzylamines and activate the inert CO2 molecule simultaneously; and (iii) how to achieve late-stage modification of benzylic C—H bonds of amines in a smooth manner. 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) proves to be an electrochemical oxidation catalyst for amines [89]. Thus, we considered it could promote this reaction efficiently.
In this work, we developed a DDQ-promoted electrochemical C—H carboxylation of benzylamines and their derivatives, providing a general, facilitated and sustainable method to obtain diverse α-amino acids (Fig. 1B). This electrocarboxylation of benzylic C—H bonds in benzylamines does not require prefunctionalization of substrates and it provides a novel retrosynthetic disconnection [90]. Notable advantages of this strategy include (a) providing a sustainable example of electrochemical C—H carboxylation of benzylamines, which maximizes atomic utilization; (b) linear paired electrolysis that fully utilizes the anode and the cathode in the reaction, improving electron efficiency; (c) easy and economic synthesis of valuable α-amino acids via a base-free process. Besides, this electrochemical protocol exhibits exceptional versatility, demonstrating broad substrate scope and excellent functional group compatibility. A wide range of benzylamine derivatives, including electron-deficient and electron-rich substrates, di- and multi-substituted variants, as well as heteroaryl-containing substrates, are all smoothly converted to the desired products. Importantly, the method successfully accommodates even structurally complex biorelevant molecules, underscoring its potential for late-stage functionalization applications.
We started our investigation on electrocarboxylation of benzylic C—H bonds using N-phenylbenzylamine 1a as the model substrate (Table 1). After careful optimization, the corresponding carboxylation product 1 could be obtained in 70% isolated yield when the following conditions were employed: DDQ (0.75 equiv.), nBu4NClO4/nBu4PPF6 = 1/1 as the mixed electrolyte, DMF/THF = 4/1 as the mixed solvent and cheap Mg plate and Ni plate as working electrodes with a constant current at 10 mA, using an undivided cell for 12 h at ambient temperature (Table 1, entry 1). Control experiments were conducted thereafter. According to the results, no reaction would occur without the electrical input, highlighting that it is an electro-assisted carboxylation (Table 1, entry 2). The significant inhibition of electrochemical performance and poor substrate consumption in the absence of DDQ clearly demonstrates its crucial role in promoting this reaction (Table 1, entry 3). Using nBu4PPF6 or nBu4NClO4 as the sole electrolyte would cause a high reaction voltage, which resulted in lower yields (Table 1, entries 4 and 5). Other common electrolytes, such as nBu4NBF4 and nBu4NBr were also tested, affording 1 in 21% and 34% yields, respectively (Table 1, entries 6 and 7). These studies demonstrated that an equal combination of nBu4PPF6 and nBu4NClO4 was the optimal electrolyte for this electrocarboxylation reaction. Compared to other types of ratios or solvents, a mixed solvent with DMF/THF = 4/1 exhibited the best-observed reactivity (Table 1, entries 8–11). Pt, GF and Pb were suitable cathodes for this electrosynthesis (Table 1, entry 12). Then, different active anodes, such as Al and Zn delivered the desired product in much lower yields (Table 1, entry 13). The inert GF anode was also investigated, but resulted primarily in unreacted starting material with only trace imine formation. Finally, increasing or decreasing the current was not conducive to the reaction (Table 1, entries 14 and 15).
 DownLoad:
CSV
DownLoad:
CSV
 |
||
| Entry | Variation | Yield (%)b |
| 1 | None | 70 |
| 2 | w/o electricity | NR |
| 3 | w/o DDQ | Trace |
| 4 | w/o nBu4NClO4 | 34 |
| 5 | w/o nBu4PPF6 | 58 |
| 6 | nBu4NBF4 instead of nBu4NClO4 | 21 |
| 7 | nBu4NBr instead of nBu4PPF6 | 34 |
| 8 | DMF as solvent | 38 |
| 9 | THF as solvent | 34 |
| 10 | DMF/DMSO = 4/1 as solvent | 55 |
| 11 | DMF/THF = 3/2 as solvent | 42 |
| 12 | Pt/GF/Pb(-) instead of Ni(-) | 65/13/30 |
| 13 | Al/Zn/GF(+) instead of Mg(+) | 42/38/ND |
| 14 | 5 mA instead of 10 mA | 34 |
| 15 | 15 mA instead of 10 mA | 20 |
| RT= room temperature; DDQ = 2,3-dichloro-5,6-dicyano-1,4-benzoquinone; DMF = N,N-dimethylformamide; THF = tetrahydrofuran; DMSO =dimethyl sulfoxide; w/o = without; NR = no reaction; ND = not detected. a Reaction conditions: Undivided cell, 1a (0.3 mmol), CO2 (balloon), nBu4NClO4 (0.3 mmol), nBu4PPF6 (0.3 mmol), DDQ (0.225 mmol), DMF/THF = 4/1 (5.0 mL) under 10 mA constant current at room temperature for 12 h with Mg plate (10 mm × 20 mm × 0.10 mm) as the anode and Ni plate (10 mm × 15 mm × 0.10 mm) as the cathode. b Isolated yield. |
||
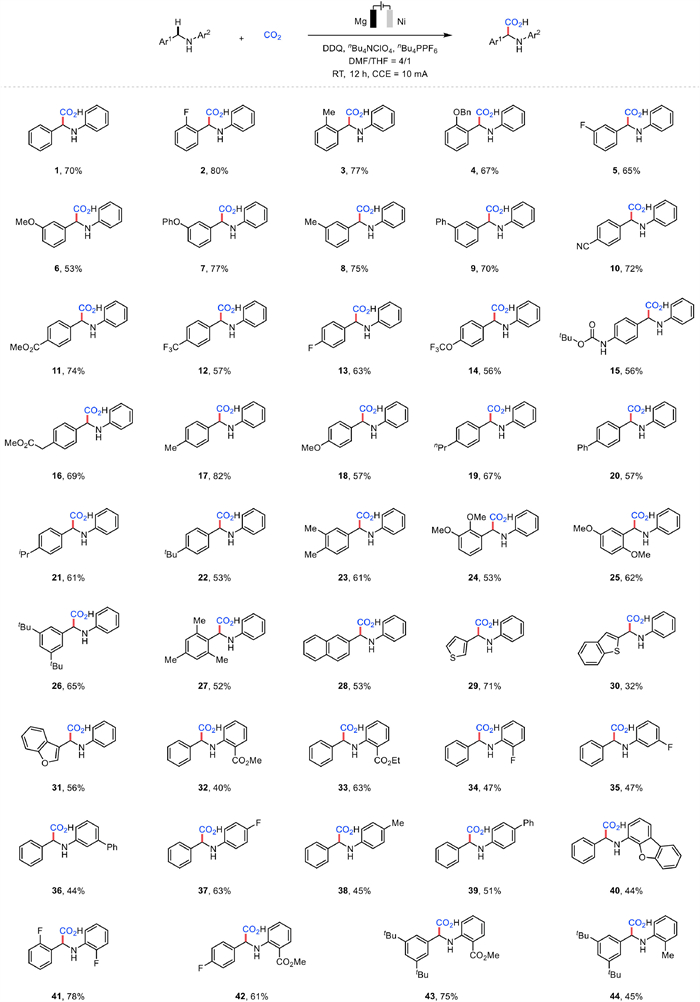
With the optimal conditions in hand, we sought to demonstrate the generality of this electrochemical C—H carboxylation of benzylamines. Overall, benzylamines bearing electron-withdrawing groups (EWGs) or electron-donating groups (EDGs) could be transformed smoothly to the desired corresponding α-AAs (Fig. 2). We investigated the scope of Ar1 first. To our delight, the presence of a fluoro, methyl or benzyloxy substituent in the ortho position on the benzene ring had a positive effect on the transformation, being evidenced by the production of α-AAs in 80%, 77% and 67% yields, respectively (2–4). We then turned our attention to the carboxylation of substrates with different meta-substituted groups on the aromatic ring (e.g., F, OMe, OPh, Me and Ph), and the desired products were obtained in moderate to excellent yields (5–9). To our delight, substrates with various functional groups in the para position worked successfully, showing high efficiency for such electrochemical carboxylation (10–22). It was worth mentioning that some functional groups, which are potentially sensitive to strongly reducing conditions, such as cyano, ester, trifluoromethyl, fluoro and amide groups, were tolerated and gave satisfactory results (10–13, 15 and 16). Encouragingly, the disubstituted substrates underwent smoothly to offer 23–26 in 53%−65% yields. Besides, N-(2,4,6-trimethylbenzyl)aniline was the ideal benzylamine substrate for this electrochemical synthesis (27). The reaction could also accommodate the naphthalene group, which provided the desired product in moderate yield (28). Moreover, heteroaryl-based benzylamines were suitable substrates for the benzylic C—H bond carboxylation reaction (29–31). Compared to the 3-thiophene-containing substrate, the 2-benzofuran-bearing substrate afforded a lower yield, likely due to the formation of an aromatic carboxylation product during electrolysis. Then, we extended the investigation to substituted aromatic rings connected to the nitrogen atom of benzylamines. When different functional groups were attached to the ortho, meta and para positions, the desired products were furnished in acceptable yields (32–39). Some highly reactive functional groups, for example, fluoro and ester groups, were tested and provided the corresponding α-amino acids in moderate yields (32–35 and 37). However, compared to EDGs, substrates containing EWGs seemed more compatible with the standard conditions, resulting in higher yields. For instance, the substrate with an electron-deficient fluoro substituent (37) performed better than those connecting with an electron-rich functional group (Me and Ph, 38 and 39). Besides, N-benzyldibenzofuran-4-amine was suitable to be converted into 40 in moderate yield. Further exploration was taken when two aromatic rings were simultaneously substituted by substituents, affording the corresponding carboxylated products 41–44 in 45%−78% yields. Overall, the low yields of partial products were primarily due to incomplete conversion of the starting materials, alongside the formation of imine and homo-coupling byproducts.
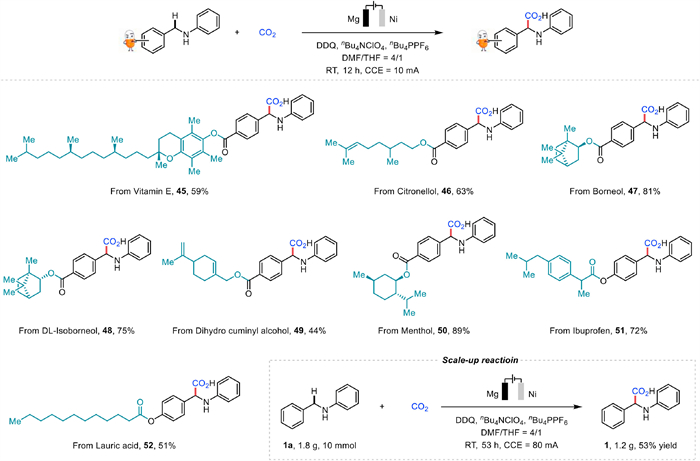
Bearing the crucial role of α-amino acids in pharmaceutical science in mind, late-stage modification was carried out with this electrosynthesis (Fig. 3). There is no doubt that late-stage site-selective activation of benzylic C—H bonds in complex molecules is a significant and challenging task. Gratifyingly, benzylamine compounds derived from vitamin E (45), citronellol (46), borneol (47), dl-isoborneol (48), dihydro cuminyl alcohol (49), menthol (50), ibuprofen (51) and lauric acid (52), all showed great compatibility under this electrochemical carboxylation. Notably, substrate with an electrochemically sensitive alkene was suitable for this reaction (46 and 49). Moreover, a scale-up experiment was conducted and it showcased the potential application and synthetic utility of this carboxylation method.
Several mechanistic experiments were carried out to gain insight into the reaction mechanism. Firstly, kinetic isotope effect experiments were conducted between benzylamine 1a and its deuterated derivative 1a-d2 or 1a-d1 (Fig. 4A). According to the intermolecular and intramolecular competitive experiments, the proportions of 1 and 1′ were 2 and 6, respectively. Besides, the ratio of kH/kD for the parallel KIE experiment was 3. The above results suggested that C—H bond cleavage may be involved in the rate-determining step of this carboxylation reaction. Then, to verify if S1 was the key intermediate in this electrocarboxylation reaction, benzylamines 1a was replaced with imine S1 to couple with CO2, and 91% yield of product 1 was obtained under the standard conditions, showing that amine might be oxidized to imine during the reaction (Fig. 4B). However, when the reaction was carried out in the absence of DDQ, the yield of the product showed significantly reduced, possibly due to incomplete conversion of S1. Moreover, the divided-cell electrolysis reaction displayed that it was a linear paired electrolysis system because when the starting material and DDQ were in the anodic chamber, no product could be detected neither in the anodic chamber nor the cathodic chamber (Fig. 4C). Finally, several CV studies were performed under CO2 atmosphere (Fig. 4D). The oxidation peak of 1a was +1.13 V vs. Ag/AgCl, which could be oxidized at the anode. DDQ displayed a reversible oxidation peak at +0.75 V vs. Ag/AgCl, demonstrating a more easily oxidizable ability than benzylamine. This was further supported by Fig. 4D, image b, when 18a was combined with DDQ, an increased oxidation current of DDQ was observed with the increased amount of 18a. Meanwhile, cyclic voltammetry experiments revealed that under an Ar atmosphere, the reduction potential of S1 was −2.06 V vs. Ag/AgCl. Notably, under a CO2 atmosphere, the reduction peak of S1 remained, indicating that the imine S1 was reduced preferentially over CO2.
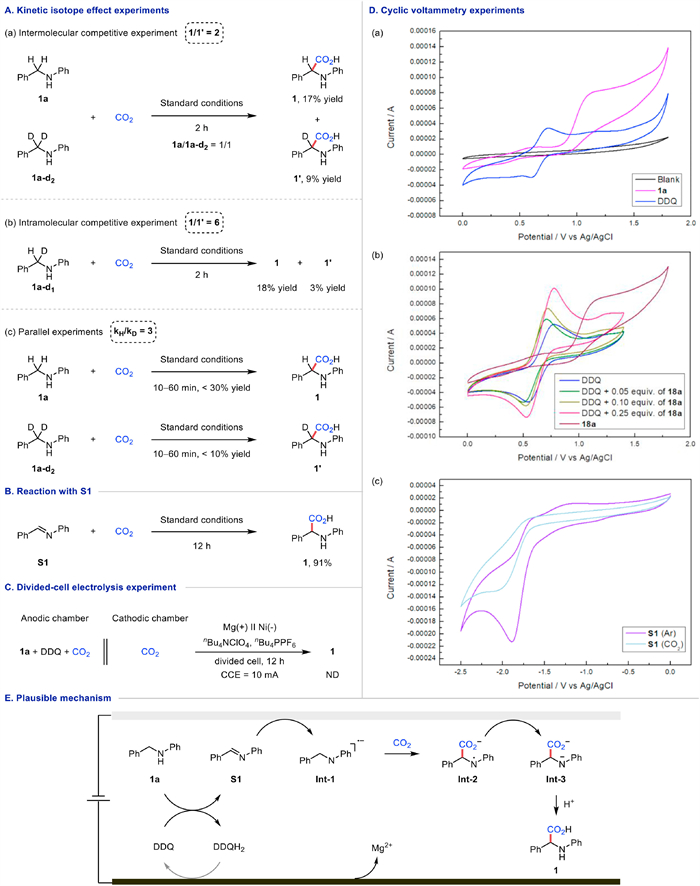
Based on our experimental data and previous literature reports [77-79,89], a plausible mechanism for this electrochemical carboxylation of C—H bonds is proposed (Fig. 4E). DDQ could oxidize benzylamine 1a to S1 at the anode, accompanied by two-electron/two-proton transfer to generate DDQH2. The key intermediate S1, formed by the second single electron transfer with DDQ, is prone to gain an electron to form Int-1 by cathodic reduction. The following carboxylation might occur between Int-1 and CO2 to give intermediate Int-2, which is able to undergo the second reduction at the cathode to afford Int-3. Final protonation with aqueous HCl offers the corresponding product 1.
In conclusion, we have reported a general and novel strategy for the synthesis of α-amino acids via electrocarboxylation of benzylic C—H bonds in benzylamines under mild conditions. A series of value-added α-amino acids were generated through linear paired electrolysis in a base-free process. This approach successfully achieved the carboxylation of various substituted benzylamines containing a diverse range of functional groups and even highly reactive ones. Besides, it has a broad substrate scope, including electro-deficient/rich, multi-substituted and heteroaryl-based benzylamines. The extension to late-stage modification of natural product derivatives and scale-up experiment showed potential prospect of the method. Mechanistic studies such as a radical trapping experiment, KIE tests and cyclic voltammetry studies provided insight and evidence for the proposed mechanistic pathway. Further exploration of the electrocarboxylation of benzylic C—H bonds is currently ongoing in our laboratory.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Weimei Zeng: Writing – original draft, Methodology, Investigation, Formal analysis, Conceptualization. Youai Qiu: Writing – review & editing, Supervision, Methodology, Investigation.
Financial support from National Key R&D Program of China (No. 2023YFA1507203), National Natural Science Foundation of China (Nos. 22371149 and 22188101), the Fundamental Research Funds for the Central Universities (No. 63224098), Frontiers Science Center for New Organic Matter, Nankai University (No. 63181206) and Nankai University are gratefully acknowledged. We thank the Haihe Laboratory of Sustainable Chemical Transformations for financial support.
Supplementary material associated with this article can be found, in the online version, at doi:
K. Huang, C.L. Sun, Z.J. Shi, Chem. Soc. Rev. 40 (2011) 2435–2452. doi: 10.1039/c0cs00129e
Y. Cao, X. He, N. Wang, et al., Chin. J. Chem. 36 (2018) 644–659. doi: 10.1002/cjoc.201700742
J.H. Ye, T. Ju, H. Huang, et al., Acc. Chem. Res. 54 (2021) 2518–2531. doi: 10.1021/acs.accounts.1c00135
B. Cai, H.W. Cheo, T. Liu, et al., Angew. Chem. Int. Ed. 60 (2021) 18950–18980. doi: 10.1002/anie.202010710
M. He, Y. Sun, B. Han, Angew. Chem. Int. Ed. 61 (2022) e202112835. doi: 10.1002/anie.202112835
Y. Liu, P. Li, Y. Wang, et al., Angew. Chem. Int. Ed. 62 (2022) e202306679.
J. Hou, J.S. Li, J. Wu, Asian J. Org. Chem. 7 (2018) 1439–1447. doi: 10.1002/ajoc.201800226
C.S. Yeung, Angew. Chem. Int. Ed. 58 (2019) 5492–5502. doi: 10.1002/anie.201806285
Q.Y. Meng, T.E. Schirmer, A.L. Berger, et al., J. Am. Chem. Soc. 141 (2019) 11393–11397. doi: 10.1021/jacs.9b05360
N. Ishida, Y. Masuda, Y. Imamura, et al., J. Am. Chem. Soc. 141 (2019) 19611–19615. doi: 10.1021/jacs.9b12529
H. Wang, Y. Gao, C. Zhou, et al., J. Am. Chem. Soc. 142 (2020) 8122–8129. doi: 10.1021/jacs.0c03144
M. Schmalzbauer, T.D. Svejstrup, F. Fricke, et al., Chem 6 (2020) 2658–2672. doi: 10.1016/j.chempr.2020.08.022
Y. Liu, G.-H. Xue, Z. He, et al., J. Am. Chem. Soc. 146 (2024) 28350–28359.
J.L. Li, S.S. Zhang, L.L. Jiang, et al., Angew. Chem. Int. Ed. 64 (2024) e202420852.
H. Komatsu, Y. Fujimura, H. Senboku, et al., Synlett 3 (2001) 418–420.
C. Li, G. Yuan, H. Jiang, Chin. J. Chem. 28 (2010) 1685–1689. doi: 10.1002/cjoc.201090285
K.J. Jiao, Z.M. Li, X.T. Xu, et al., Org. Chem. Front. 5 (2018) 2244–2248. doi: 10.1039/c8qo00507a
S. Bazzi, G.L. Duc, E. Schulz, et al., Org. Biomol. Chem. 17 (2019) 8546–8550. doi: 10.1039/c9ob01752f
A. Alkayal, V. Tabas, S. Montanaro, et al., J. Am. Chem. Soc. 142 (2020) 1780–1785. doi: 10.1021/jacs.9b13305
W. Zhang, S. Lin, J. Am. Chem. Soc. 142 (2020) 20661–20670. doi: 10.1021/jacs.0c08532
N.W.J. Ang, J.C.A. Oliveira, L. Ackermann, Angew. Chem. Int. Ed. 59 (2020) 12842–12847. doi: 10.1002/anie.202003218
S. Tang, S. Wang, D. Zhang, et al., Chin. Chem. Lett. 35 (2024) 108660. doi: 10.1016/j.cclet.2023.108660
H. Senboku, Chem. Rec. 21 (2021) 2354–2374. doi: 10.1002/tcr.202100081
M.A. Stalcup, C.K. Nilles, H.J. Lee, et al., ACS Sustainable Chem. Eng. 9 (2021) 10431–10436. doi: 10.1021/acssuschemeng.1c03073
K. Zhang, B.H. Ren, X.F. Liu, et al., Angew. Chem. Int. Ed. 61 (2022) e202207660. doi: 10.1002/anie.202207660
G.Q. Sun, P. Yu, W. Zhang, et al., Nature 615 (2023) 67–72. doi: 10.1038/s41586-022-05667-0
S. Chen, A. Shi, G. Yang, et al., Chin. Chem. Lett. 36 (2025) 110810. doi: 10.1016/j.cclet.2024.110810
N. Fu, G.S. Sauer, A. Saha, et al., Science 357 (2017) 575–579. doi: 10.1126/science.aan6206
T. Shen, T.H. Lambert, Science 371 (2021) 620–626. doi: 10.1126/science.abf2798
D.S. Chung, S.H. Park, S. Lee, et al., Chem. Sci. 12 (2021) 5892–5897. doi: 10.1039/d1sc00760b
S.Z. Sun, Y.M. Cai, D.L. Zhang, et al., J. Am. Chem. Soc. 144 (2022) 1130–1137. doi: 10.1021/jacs.1c12350
R. Li, Y. Wu, C. Wang, et al., Nat. Commun. 13 (2022) 5951. doi: 10.1038/s41467-022-33779-8
J. Zhang, B. Das, O. Verho, et al., Angew. Chem. Int. Ed. 61 (2022) e202212131. doi: 10.1002/anie.202212131
Y. Yu, Y. Jiang, S. Wu, et al., Chin. Chem. Lett. 33 (2022) 2009–2014. doi: 10.1016/j.cclet.2021.10.016
Z.J. Shen, C. Zhu, X. Zhang, et al., Angew. Chem. Int. Ed. 62 (2023) e202217244. doi: 10.1002/anie.202217244
T.V. Münchow, S. Dana, Y. Xu, et al., Science 379 (2023) 1036–1042. doi: 10.1126/science.adg2866
B. Zhang, J. He, Y. Gao, et al., Nature 623 (2023) 745–751. doi: 10.1038/s41586-023-06677-2
J. Lu, Y. Yao, L. Li, et al., J. Am. Chem. Soc. 145 (2023) 26774–26782. doi: 10.1021/jacs.3c08839
Y.T. Zheng, H.C. Xu, Angew. Chem. Int. Ed. 63 (2024) e202313273. doi: 10.1002/anie.202313273
C. Huang, Y. Tao, X. Cao, et al., J. Am. Chem. Soc. 146 (2024) 1984–1991. doi: 10.1021/jacs.3c10194
L. Zhang, M. Li, Y. Yang, et al., Green Chem. 26 (2024) 2059–2066. doi: 10.1039/d3gc03832g
A. Shi, Y. Liu, R. Zhang, et al., eScience 4 (2024) 100255. doi: 10.1016/j.esci.2024.100255
L. Li, X. Wang, N. Fu, Angew. Chem. Int. Ed. 63 (2024) e202403475. doi: 10.1002/anie.202403475
J. Sheng, J. Cheng, X. Cheng, Tetrahedron Chem. 10 (2024) 100074. doi: 10.1016/j.tchem.2024.100074
G. Kou, P. Li, G. Yang, et al., CCS Chem. 7 (2025) 3005–3014. doi: 10.31635/ccschem.024.202404697
Y. Li, Y. Peng, W. Dong, et al., J. Am. Chem. Soc. 146 (2024) 14194–14202. doi: 10.1021/jacs.4c03218
Y. Cao, J. Chen, C. Ding, et al., Chem. Catal. 4 (2024) 101158.
J. Xiang, M. Shang, Y. Kawamata, et al., Nature 573 (2019) 398–402. doi: 10.1038/s41586-019-1539-y
H. Liang, L.J. Wang, Y.X. Ji, et al., Angew. Chem. Int. Ed. 60 (2020) 1839–1844.
Y. You, W. Kanna, H. Takano, et al., J. Am. Chem. Soc. 144 (2022) 3685–3695. doi: 10.1021/jacs.1c13032
X. Cheng, A. Lei, T.S. Mei, et al., CCS Chem. 4 (2022) 1120–1152. doi: 10.31635/ccschem.021.202101451
C.J. Long, H. Cao, B.K. Zhao, et al., Angew. Chem. Int. Ed. 61 (2022) e202203666. doi: 10.1002/anie.202203666
X. Wang, S. Wu, Y. Zhong, et al., Chin. Chem. Lett. 34 (2023) 107537. doi: 10.1016/j.cclet.2022.05.051
Y. Liu, Y. Sun, Y. Deng, et al., Angew. Chem. Int. Ed. 64 (2025) e202504459. doi: 10.1002/anie.202504459
S.H. Park, G. Bae, A. Choi, et al., J. Am. Chem. Soc. 145 (2023) 15360–15369. doi: 10.1021/jacs.3c03172
L. Zeng, J. Wang, D. Wang, et al., Angew. Chem. Int. Ed. 62 (2023) e202309620. doi: 10.1002/anie.202309620
S. Fang, K. Zhong, S. Zeng, et al., Chem. Commun. 59 (2023) 11425–11428. doi: 10.1039/d3cc02852f
C. Feng, X. Liu, Y. She, et al., Chin. Chem. Lett. 34 (2023) 107935. doi: 10.1016/j.cclet.2022.107935
W. Zeng, Y. Wang, C. Peng, et al., Chem. Soc. Rev. 54 (2025) 4468–4501. doi: 10.1039/d4cs01142b
P. Li, Z. Zhu, C. Guo, et al., Nat. Catal. 7 (2024) 412–421. doi: 10.1038/s41929-024-01118-3
M. Liu, Y. Wang, C. Gao, et al., Angew. Chem. Int. Ed. 64 (2025) e202425634. doi: 10.1002/anie.202425634
Y. Li, J. Xu, J.C.A. Oliveira, et al., ACS Catal. 14 (2024) 8160–8167. doi: 10.1021/acscatal.4c01886
Q. Hu, B. Wei, M. Wang, et al., J. Am. Chem. Soc. 146 (2024) 14864–14874. doi: 10.1021/jacs.4c04211
X. Fang, Y. Zeng, Y. Huang, et al., Nat. Commun. 15 (2024) 5181. doi: 10.1038/s41467-024-49223-y
H. Li, Y. Li, J. Chen, et al., Angew. Chem. Int. Ed. 63 (2024) e202407392. doi: 10.1002/anie.202407392
B.L. Chen, H.W. Zhu, Y. Xiao, et al., Electrochem. Commun. 42 (2014) 55–59. doi: 10.1016/j.elecom.2014.02.009
H.P. Yang, Y.N. Yue, Q.L. Sun, et al., Chem. Commun. 51 (2015) 12216–12219. doi: 10.1039/C5CC04554A
D.T. Yang, M. Zhu, Z.J. Schiffer, et al., ACS Catal. 9 (2019) 4699–4705. doi: 10.1021/acscatal.9b00818
S. Bazzi, E. Schulz, M. Mellah, Org. Lett. 21 (2019) 10033–10037. doi: 10.1021/acs.orglett.9b03927
L. Muchez, D.E.D. Vos, M. Kim, ACS Sustainable Chem. Eng. 7 (2019) 15860–15864. doi: 10.1021/acssuschemeng.9b04612
H. Senboku, K. Yoneda, S. Hara, Tetrahedron Lett. 56 (2015) 6772–6776. doi: 10.1016/j.tetlet.2015.10.068
N. Sauermann, T.H. Meyer, Y. Qiu, et al., ACS Catal. 8 (2018) 7086–7103. doi: 10.1021/acscatal.8b01682
Y. Zhang, T. Zhang, S. Das, Chem 8 (2022) 3175–3201. doi: 10.1016/j.chempr.2022.10.005
J.A. Ma, Angew. Chem. Int. Ed. 42 (2003) 4290–4299. doi: 10.1002/anie.200301600
G. Li, Y. Liang, J.C. Antilla, J. Am. Chem. Soc. 129 (2007) 5830–5831. doi: 10.1021/ja070519w
A.M. Fournier, R.A. Brown, W. Farnaby, et al., Org. Lett. 12 (2010) 2222–2225. doi: 10.1021/ol100627c
Y. Qu, C. Tsuneishi, H. Tateno, et al., React. Chem. Eng. 2 (2017) 871–875. doi: 10.1039/C7RE00149E
Y. Naito, Y. Nakamura, N. Shida, et al., J. Org. Chem. 86 (2021) 15953–15960. doi: 10.1021/acs.joc.1c00821
K. Zhang, X.F. Liu, W.Z. Zhang, et al., Org. Lett. 24 (2022) 3565–3569. doi: 10.1021/acs.orglett.2c01267
A. Dmitrieva, J.J. Medvedev, X.V. Medvedeva, et al., J. Electrochem. Soc. 170 (2023) 075501. doi: 10.1149/1945-7111/ace0dc
H. Seo, M.H. Katcher, T.F. Jamison, Nat. Chem. 9 (2017) 453–456. doi: 10.1038/nchem.2690
Y. Wang, S. Tang, G. Yang, et al., Angew. Chem. Int. Ed. 61 (2022) e202207746. doi: 10.1002/anie.202207746
Y. Wang, Z. Zhao, D. Pan, et al., Angew. Chem. Int. Ed. 61 (2022) e202210201. doi: 10.1002/anie.202210201
Z. Zhao, Y. Liu, S. Wang, et al., Angew. Chem. Int. Ed. 62 (2023) e202214710. doi: 10.1002/anie.202214710
W. Zeng, C. Peng, Y. Qiu, J. Am. Chem. Soc. 147 (2025) 13461–13470. doi: 10.1021/jacs.5c00259
N. Fu, L. Li, Q. Yang, et al., Org. Lett. 19 (2017) 2122–2125. doi: 10.1021/acs.orglett.7b00746
P.S. Gao, X.J. Wang, Z.H. Wang, et al., Angew. Chem. Int. Ed. 59 (2020) 15254–15259. doi: 10.1002/anie.202005099
Z.H. Wang, P.S. Gao, X. Wang, et al., J. Am. Chem. Soc. 143 (2021) 15599–15605. doi: 10.1021/jacs.1c08671
O.R. Luca, T. Wang, S.J. Konezny, et al., New J. Chem. 35 (2011) 998–999. doi: 10.1039/c0nj01011a
K.J. Jiao, Y.K. Xing, Q.L. Yang, et al., Acc. Chem. Res. 53 (2020) 300–310. doi: 10.1021/acs.accounts.9b00603

Figure 1 Background and introduction. (A) Pioneering study in photoredox C—H carboxylation of benzylamines. (B) This work: electrochemical C—H carboxylation of benzylamines.

Figure 2 Substrate scope. Reaction conditions: undivided cell, benzylamines (0.3 mmol), CO2 (balloon), nBu4NClO4 (0.3 mmol), nBu4PPF6 (0.3 mmol), DDQ (0.225 mmol), DMF/THF = 4/1 (5.0 mL) under 10 mA constant current at room temperature for 12 h with Mg plate (10 mm × 20 mm × 0.10 mm) as the anode and Ni plate (10 mm × 15 mm × 0.10 mm) as the cathode. Isolated yield.

Figure 3 Substrate scope of complex compounds and scale-up experiment. Reaction conditions: undivided cell, benzylamines (0.3 mmol), CO2 (balloon), nBu4NClO4 (0.3 mmol), nBu4PPF6 (0.3 mmol), DDQ (0.225 mmol), DMF/THF = 4/1 (5.0 mL) under 10 mA constant current at room temperature for 12 h with Mg plate (10 mm × 20 mm × 0.10 mm) as the anode and Ni plate (10 mm × 15 mm × 0.10 mm) as the cathode. Isolated yield. For scale-up reaction: 1a (10 mmol), CO2 (balloon), nBu4NClO4 (10 mmol), nBu4PPF6 (10 mmol), DDQ (7.5 mmol), DMF/THF = 4/1 (100.0 mL) under 80 mA constant current at room temperature for 53 h with Mg plate (25 mm × 50 mm × 0.10 mm) as the anode and Ni plate (25 mm × 50 mm × 0.20 mm) as the cathode. Isolated yield.

Figure 4 Mechanistic studies. (A) Kinetic isotope effect experiments. (B) Reaction with S1. (C) Divided-cell electrolysis experiment. (D) Cyclic voltammetry experiments. (E) Plausible mechanism.
Table 1. Optimization of reaction conditions.a
|
||
| Entry | Variation | Yield (%)b |
| 1 | None | 70 |
| 2 | w/o electricity | NR |
| 3 | w/o DDQ | Trace |
| 4 | w/o nBu4NClO4 | 34 |
| 5 | w/o nBu4PPF6 | 58 |
| 6 | nBu4NBF4 instead of nBu4NClO4 | 21 |
| 7 | nBu4NBr instead of nBu4PPF6 | 34 |
| 8 | DMF as solvent | 38 |
| 9 | THF as solvent | 34 |
| 10 | DMF/DMSO = 4/1 as solvent | 55 |
| 11 | DMF/THF = 3/2 as solvent | 42 |
| 12 | Pt/GF/Pb(-) instead of Ni(-) | 65/13/30 |
| 13 | Al/Zn/GF(+) instead of Mg(+) | 42/38/ND |
| 14 | 5 mA instead of 10 mA | 34 |
| 15 | 15 mA instead of 10 mA | 20 |
| RT= room temperature; DDQ = 2,3-dichloro-5,6-dicyano-1,4-benzoquinone; DMF = N,N-dimethylformamide; THF = tetrahydrofuran; DMSO =dimethyl sulfoxide; w/o = without; NR = no reaction; ND = not detected. a Reaction conditions: Undivided cell, 1a (0.3 mmol), CO2 (balloon), nBu4NClO4 (0.3 mmol), nBu4PPF6 (0.3 mmol), DDQ (0.225 mmol), DMF/THF = 4/1 (5.0 mL) under 10 mA constant current at room temperature for 12 h with Mg plate (10 mm × 20 mm × 0.10 mm) as the anode and Ni plate (10 mm × 15 mm × 0.10 mm) as the cathode. b Isolated yield. |
||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载: