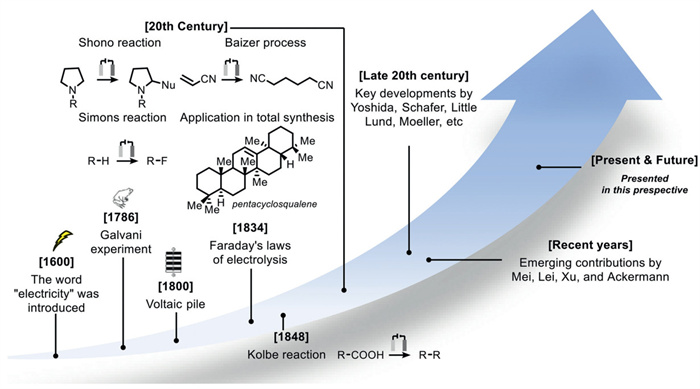
Figure 1.
History, current status and future trends in electrochemistry.
Advances in organoelectrochemical copper-catalyzed reactions
Pan Zhou , Ting Zou , Hong-Jian Song , Yu-Xiu Liu , Qing-Min Wang

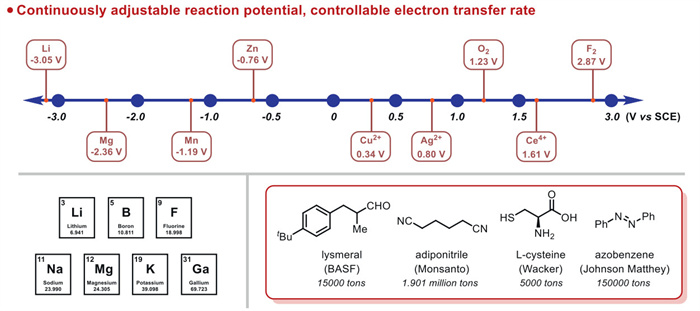
The term "electricus" was first cioned by William Gilbert in his 1600 work "On Magnetism" (Fig. 1). In 1786, Luigi Galvani performed the first electrochemical experiment on frog legs, but later Alessandro Volt refuted this research explanation while developing the first battery model - the Volta stack [1]. In 1834, Michael Faraday defined the main laws of electrolysis [2], and Hermann Kolbe conducted and analyzed the first organic electrosynthesis in 1848 [3], which set the stage for experimental scientists to introduce electricity into kinetic chemical reactions. Electrochemical technology enabled scientists such as Henry Moissan [4] and Humphrey Davy [5] to discover new elements such as Li, Na, and F, which were difficult or even impossible to obtain using chemical reagents at the time (Fig. 2). In the following century, electrosynthesis has undergone tremendous development (Fig. 1). Therefore, various challenging reactions have been achieved, ranging from industrial processes such as the Monsanto adiponitrile process developed by Professor Manuel Baizer in the United States [6], to basic conversion and total synthesis applications including Shono oxidation [7], electrofluorination [8,9].
Unlike conventional organic reactions, electrochemical transformations use inexpensive, safe, clean, and easily accessible electrons as "traceless" redox agents, rather than chemical oxidizing or reducing agents, which significantly improves environmental economics and reduces fossil fuel consumption. Organic electrosynthesis has the unique ability to modulate currents and potentials to achieve redox conversion with a precision rarely seen in conventional methods [10].
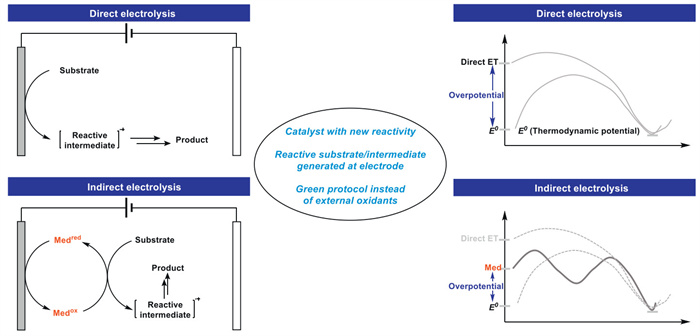
The electrochemical reaction modes are mainly categorized into direct and indirect electrolysis (Fig. 3) [11,12]. In the case of anodic oxidation, electrons shuttle between the electrode surface and the organic substrate, thereby driving electron transfer (ET). ET between electrodes is a heterogeneous process that may be hindered by kinetics, resulting in high potentials at the electrode surface. In addition, organic compounds will adsorb on the electrode surface, thereby reducing conductivity. If an insulating film is formed on the electrode surface, the terminal voltage will reach the limit of the constant current meter and no current can be generated. Indirect electrolysis is used to regulate the overpotential of the reaction by adding a redox mediator, thus generating an active intermediate [13,14]. In anodic oxidation, for example, in indirect electrolysis, redox catalysts are used to shuttle electrons between the anode surface and the organic substrate via outer or inner-sphere electron transfer, thus avoiding the limitations of direct electrolysis mentioned above. In addition, catalysts used in indirect electrolysis can also modulate chemicalselectivity, regioselectivity, and stereoselectivity.
In recent years, given the sustained strong demand for efficient molecular reactions, organic chemists have increased their efforts to design innovative electrosynthesis and have developed various transformations to achieve molecular functionalization in a sustainable manner, such as olefin difunctionalization [15], stereoselective heterocyclic synthesis [16], carboxylation reactions with CO2 [17], or applications in the synthesis of structurally complex natural products [18]. Meanwhile, innovative combinations with transition metal catalysis [19-21], photo-oxidation chemistry [22], and flow chemistry [23,24] have shown that electrochemical studies of inert bond activation outperform traditional methods.
Since the 1980s, transition metal catalysis has developed into one of the most important research areas in modern organic chemistry [25]. Transition metal catalysis has been recognized as a powerful tool for producing fine chemicals, pharmaceuticals, and functional materials. More importantly, it has been continuously demonstrating new chemical activation modes that can effectively utilize open-shell intermediates for traditionally challenging or even impossible transformations [26-29]. The prospect of transition metal catalysis mainly stems from the intrinsic properties of transition metals that are capable of having various oxidation states. Transition metals can form complexes with reactants and even intermediates in the reaction system, providing a versatile platform for designing new reaction modes and thus inventing valuable chemical reactions.
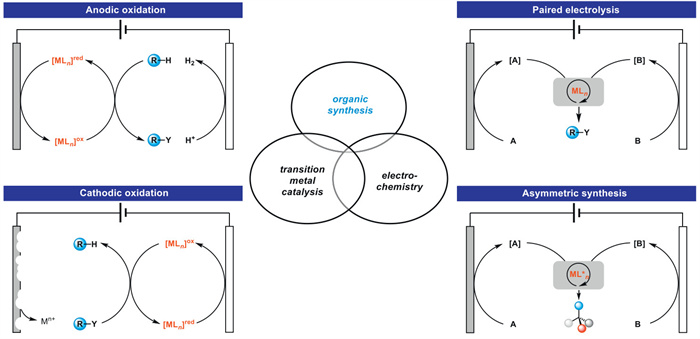
In a typical electrolysis process, substrates typically gain or lose electrons at the cathode or anode, and produce highly reactive free radical intermediates, making control of the chemistry, regioselectivity and enantioselectivity very challenging. Due to the rich and controllable valence states of transition metals, the combination of electrochemistry and metal catalysts has always been a hot research topic in the field of organic synthesis [30-32]. Electron transfer on electrodes can generate catalysts with more valence states [33]. The synthetic organometallic electrochemistry can be mainly divided into several reaction modes: oxidation, reduction, paired electrolysis, and asymmetric catalysis (Fig. 4). Compared with other catalysts, transition metals are attractive because: (1) The reactions catalyzed by transition metals are universal for electrochemical synthesis, with adjustable current and potential; (2) The redox potential of transition metal catalysts can be regulated through ligand modification; (3) The chemical, regional, and stereoselectivity can be determined by transition metal catalysts.
Among these transition metal catalysts, electrochemical transformations catalyzed by inexpensive and Earth-abundant copper metal has received considerable attention [34], such as enantioselective C–H cyanoformations [35], decarboxylative cyanoformations [36], and olefin difunctional reaction [15]. Copper can exist in the Cu0, CuⅠ, CuⅡ, and CuⅢ oxidation states, which allows for one-or two-electron conversion pathways. Thus, similar to palladium, both the radical pathways and strong two-electron bond-forming pathways via organometallic intermediates. In addition, the different oxidation states of copper can interact with Lewis acids or π-coordinate well with a large number of different functional groups [37]. The unique activity of the 3d transition metal copper makes it an attractive alternative to earlier established 4d and 5d transition metals such as palladium, rhodium and iridium. In this paper, we systematically investigated this field and reviewed electrochemical copper-catalyzed methods applied to organic synthesis in terms of different activation modes of substrates, which can be broadly classified into functionalization of C = C bonds, activation of C–H bonds, activation of C–C bonds, and activation of C–X bonds.
In 2019, Lin's group [38] developed a unique electrochemical copper-catalyzed approach for the highly enantioselective cyanation of vinyl aromatics in the asymmetric radical functionalization of olefins (Scheme 1). Two concurrent single electron transfer (SET) cycles between the anode and the metal are carefully combined to achieve in-situ formation of radical intermediates, which are ultimately captured by copper metal to achieve the desired production pathway. In the case of the cyanophosphorylation reaction, for example, the envisioned mechanism involves the parallel electrochemical generation of transient phosphonyl radicals and persistent [CuⅡ]–CN complexes provide CN•, and the subsequent reaction of these open-shell intermediates with olefin completes the difunctionalization.
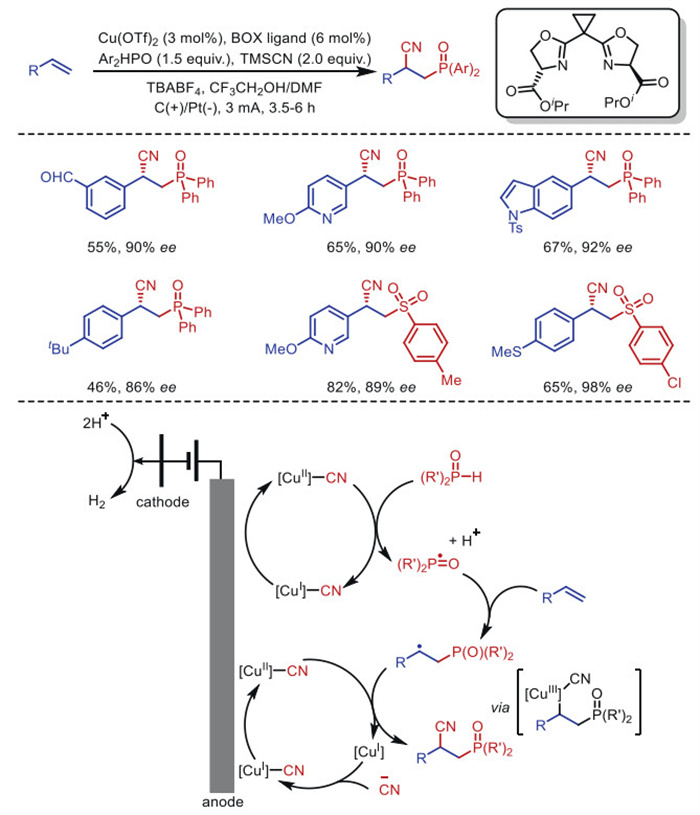
In this mechanism, the copper-mediated transfer of cyano radicals to carbon-centered radicals may occur in first-sphere manner, which is different from the second-sphere mechanism proposed for similar group transfer processes typically mediated by [MnⅢ]–X (X = N3, Cl) [39]. Specifically, [CuⅡ]–CN undergoes a single electron oxidative addition reaction with carbon radicals to form the formal [CuⅢ] intermediate, followed by reductive elimination to construct a C–CN bond. Sulfonic acid is also a suitable substrate for this reaction, allowing for the cyanosulfonation of vinyl aromatics.
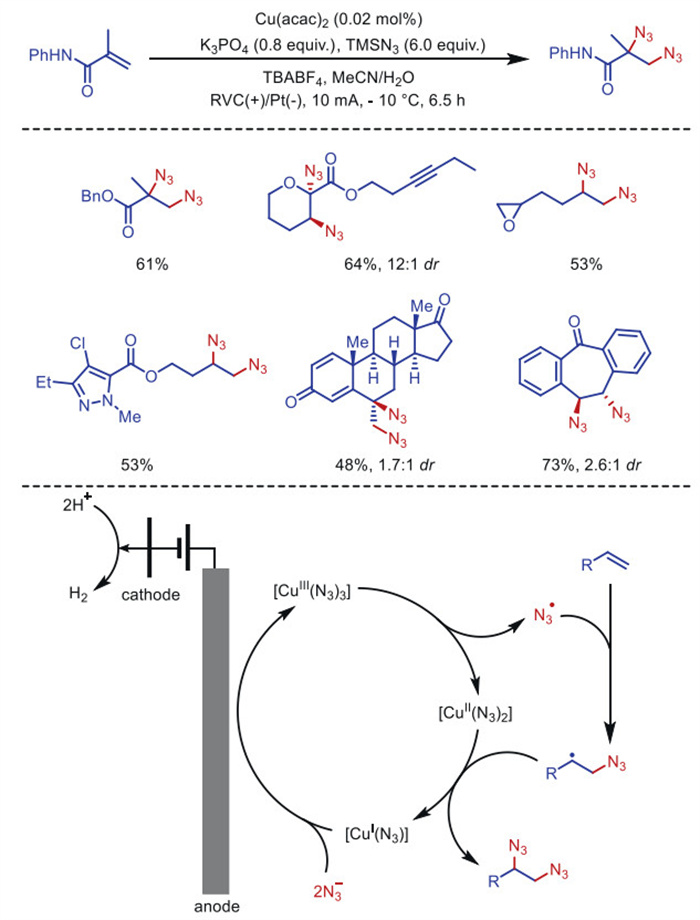
The successful development of enantioselective electrocatalysis further demonstrates the robustness of the electrocatalytic strategy as a potentially versatile mechanistic platform for the critical difunctionalization of simple olefins. In 2022, Xu's group [40] reported a scalable copper electrocatalytic diazotization reaction of olefins, in which 0.02 mol% copper acetylacetonate was used as a precatalyst without exogenous ligands (Scheme 2). In addition to using low catalyst loading, this electrocatalytic method is scalable and compatible with a wide range of functional groups for the diazotization of α, β-unsaturated carbonyl compounds as well as mono-, di-, tri- and tetra-substituted unactivated olefins.
Due to its favorable combination of lipophilicity and hydrogen bonding ability, difluoromethyl (CF2H) acts as an anesthetic bioisomer in drug discovery activities, thereby improving ADME properties. However, despite the high prevalence and importance of neighboring hydrogen bond donors in pharmaceuticals, there is no universal method for synthesizing adjacent-position difluoromethylated compounds. In 2024, Kim's group [41] developed a copper electrocatalytic strategy that can achieve ortho difluoromethylation of olefins. Zn(CF2H)2(DMPU)2 has been commonly suggested to act as an anion source for CF2H groups through metal transfer, but this method reveals its ability to act as a CF2H radical precursor under electrochemical oxidation (Scheme 3).
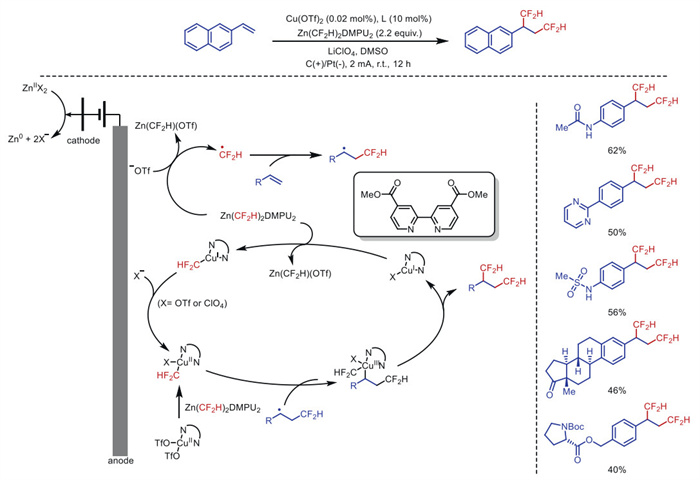
Mechanistic studies showed that the first CF2H group was introduced via radical addition to the terminal position of the olefin, followed by the capture of the remaining sec–alkyl group by the copper catalyst. Subsequent reductive elimination from [CuⅢ] was used to deliver the second CF2H group to the internal position. The practicality of this electrocatalytic 1,2-bis(difluoromethylation) strategy has been emphasized by the later bioisostatic substitution of drugs such as sotalol and dipivefrin.
Since 2019, a new field of exploration has emerged by combining the principles of photocatalysis and electrocatalysis, called photoelectrocatalysis [42-44]. This integration capitalizes on the complementary strengths of two fields, providing a comprehensive technology that is more versatile than either method alone. The field of photocatalysis has experienced rapid growth over the past few years. One of the key developments in this field is photoelectroasymmetric catalysis (PEAC), which combines molecular photoelectrocatalysis with asymmetric catalysis.
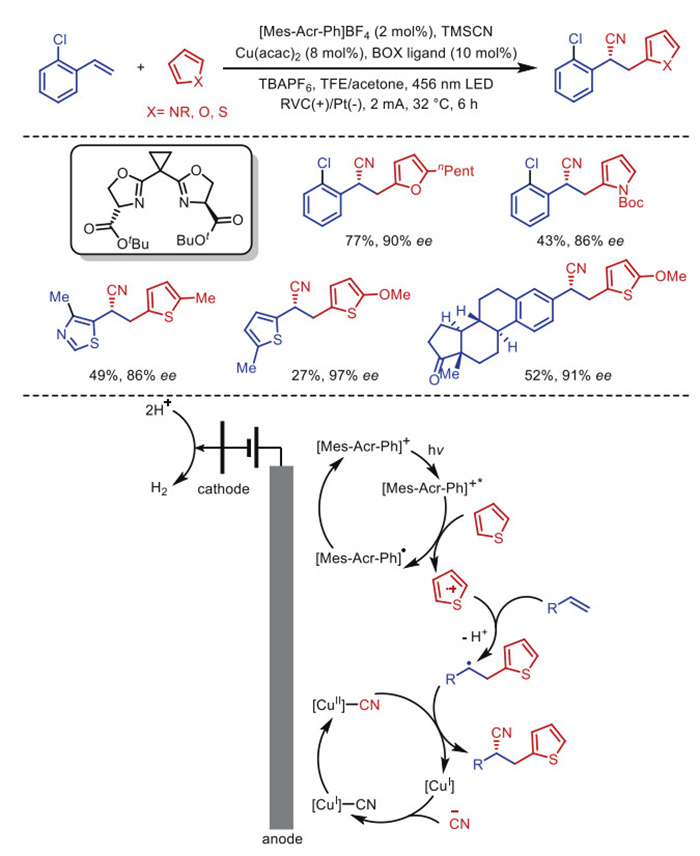
In 2023, Xu's group reported a PEAC strategy [45] for the enantioselective heteroaromatic nitrilation of aromatic olefins (Scheme 4). This method combines a dual catalytic system of photoredox catalysis and asymmetric electrochemistry to facilitate the formation of two C–C bonds via hydrogen release and eliminate the need for external chemical oxidants. The highly oxidized excited state, [Mes-Acr-Ph]+*, subsequently extracts electrons from the heterocyclic substrate, resulting in the formation of stable acridine radical [Mes-Acr-Ph]• and concomitant heteroaromatic cation. Heteroaromatic cation reacts with aromatic olefin substrate to generate distal radical cation, which subsequently binds to the electro-copper cycle and participates in a series of redoxes to give the corresponding enantioselective product.
Hydrofunctionalization reactions provide a complementary technique to difunctionalization, increasing the complexity of feedstock chemicals and complex bioactive molecules containing olefins. Enantioselective hydrogenation of olefins is a fundamental but challenging transfoemation in asymmetric catalysis, both in terms of scope and stereoselectivity [46-49]. Previous examples have mainly utilized the Ni-H process [50], but the widespread applications and highly enantioselective strategies for direct hydrogenation of olefins remain elusive.
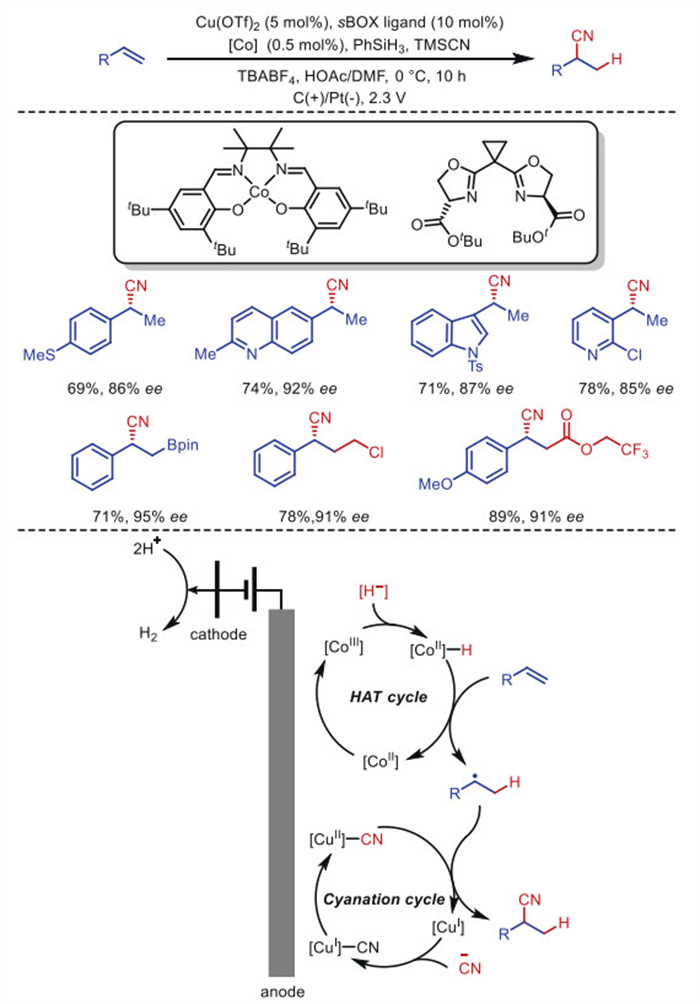
In 2020, Lin's group [51] ingeniously combined metal hydride hydrogen atom transfer catalysis with [CoⅢ]–salen and Cu (sBOX) to realize asymmetric radical hydrocyanation in an electrochemical environment (Scheme 5). Both redox-metal cycles are driven by anodic oxidation, converting H• and CN• addition to double bonds, respectively, with required electrode potentials as low as 0.2 V. This mild condition allows an unprecedented range of substrates to tolerate functional groups that are typically unstable under oxidative stress, such as nitrogen heterocycles, electron-rich aromatics, sulfides, and aldehydes, which are incompatible with chemical oxidation methods. From a synthetic perspective, this method is still limited to conjugated olefins, including alkenyl aromatics, dienes, and alkynes. Due to the versatility of its synthesis, it is necessary to extend it to include non-conjugated olefins.
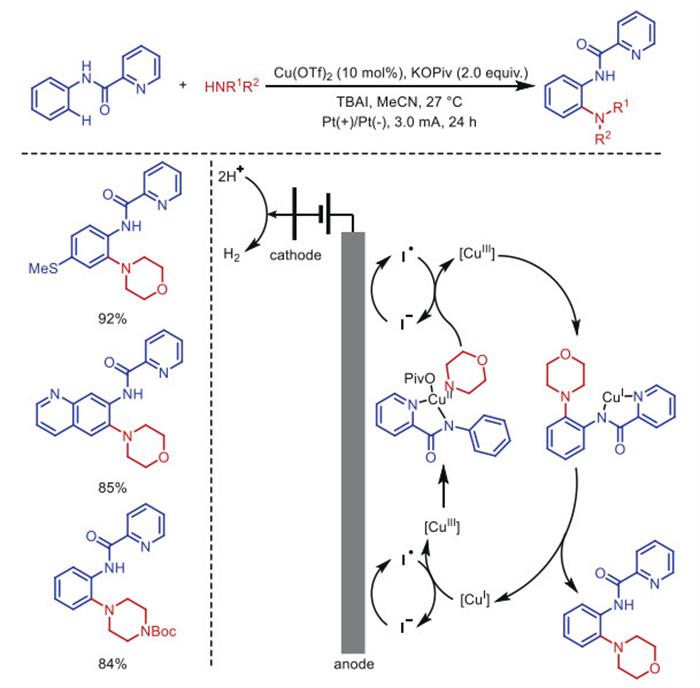
In 2018, Mei's group [52] developed an innovative electrochemical method to achieve cross-coupling between C(sp2)–H bonds in N-phenylpiperidinamide compounds and secondary amines, using copper as a catalyst (Scheme 6). This approach relies on iodide to facilitate a copper redox cycle (CuⅢ/CuⅡ/CuⅠ) that proceeds under relatively mild, low-voltage conditions. The key high-valent CuⅢ species enables efficient C–H amination at room temperature. Meanwhile, the reduction of the CuⅠ–substrate complex at the cathode, though less favorable than hydrogen evolution, given its reduction potential (Ered = –0.44 V vs. SHE), is effectively managed within this system. A notable advantage of employing iodide is that it overcomes diffusion-related challenges and reduces the need for costly copper catalysts: only a small amount of inexpensive iodide is required to keep the reaction going.
In 2019, the Nicholls's group [53] reported a similar electrochemical copper-catalyzed approach for the amination of C(sp2)–H bonds, wherein the quinoline moiety serves as a chelating group (Scheme 7). This strategy utilized 20 mol% copper catalyst loading, with direct anodic oxidation of the copper complex sufficient to drive the catalytic cycle.
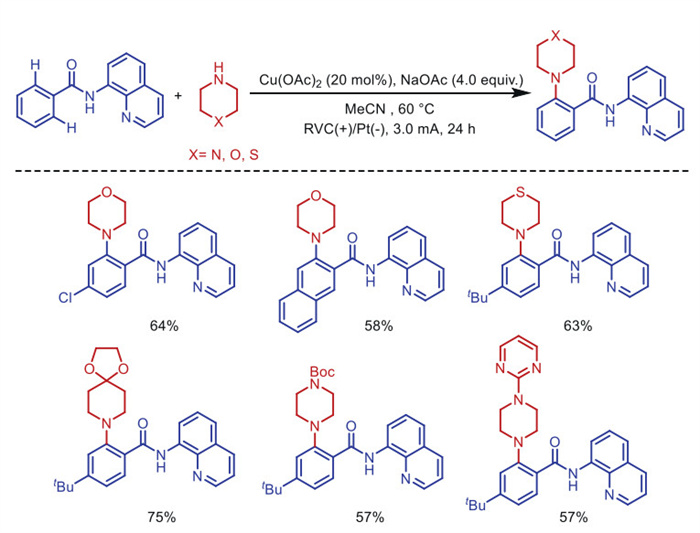
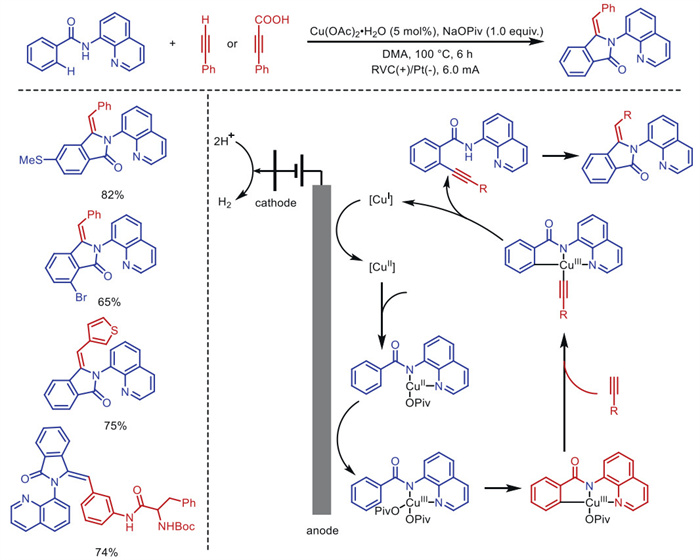
Moreover, Ackermann's group [54] achieved the first electrooxidative cascade cyclization for C(sp2)–H alkynylation using copper catalysis and a quinoline directing group, enabling the synthesis of isoindolinones (Scheme 8). This method allows broadly applicable C−H/N−H functionalization on both electron-rich and electron-deficient benzamides, featuring unique functional group tolerance and a broad substrate scope.
In 2020, Mei's group [55] also reported the application of this chelating directing group in copper-catalyzed electrochemical bromination, using NH4Br as the bromine source to achieve anodic dibromination at the quinoline C5 position (Scheme 9). The catalyst accelerates the bromination reaction through the formation of [CuⅠ] species.
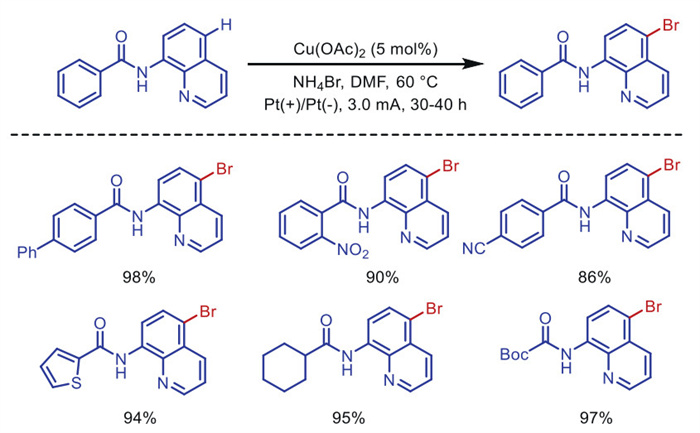
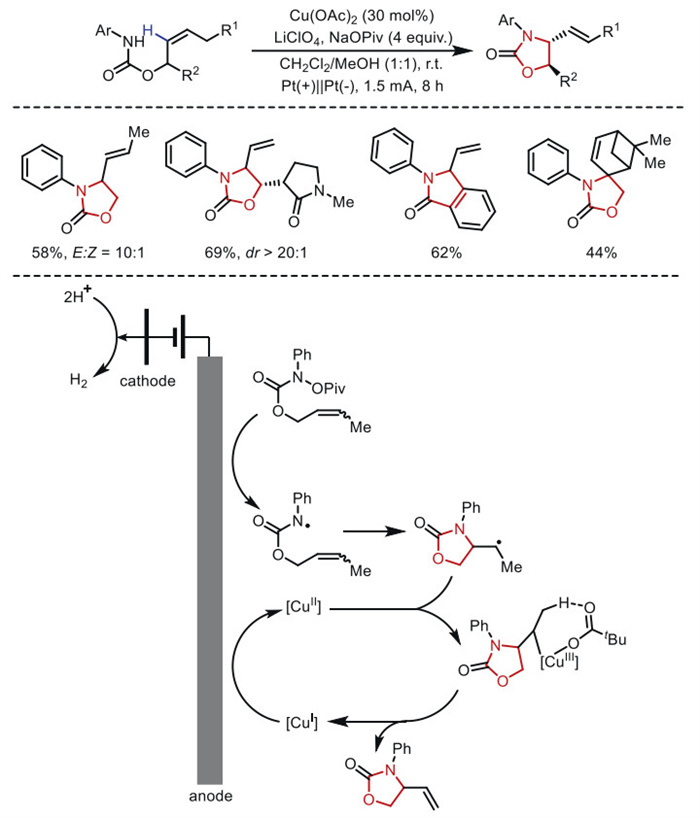
The intramolecular allylic amination reaction can also be achieved through a tandem of anodic oxidation and copper catalysis. In 2019, Hu's group [56] employed a divided cell to avoid the cathodic reduction of the [CuⅡ] catalyst, enabling the formal aza-Wacker cyclization of internal olefins (Scheme 10). Amide-based radicals are generated at the anode via PCET (proton-coupled electron transfer) using pivalate as the base. Subsequently, the cyclized alkyl group is oxidized to the final olefin product by [CuⅡ]. When the secondary alkyl group is efficiently converted into an olefin, the tertiary alkyl group is oxidized to a carbocation, leading to the formation of undesired MeOH trapping products. The copper catalyst is crucial for converting secondary and primary alkyl intermediates into olefins. Under mild conditions, various five-membered aza-heterocycles, including oxazolidinones, imidazolidinones, thiazolidinones, pyrrolidinones, and isoindolinones, can be prepared.
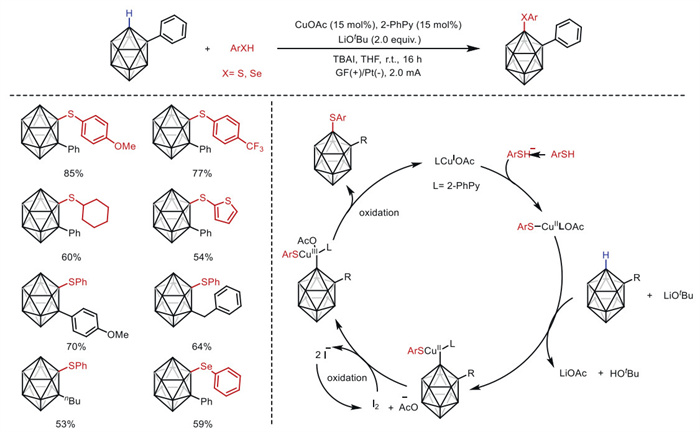
Carboranes are polyhedral molecular boron-carbon clusters with unique properties such as high boron content, icosahedral geometry, and three-dimensional electron delocalization [57]. These features render carboranes valuable building blocks for optoelectronic applications [58]. In 2021, Ackermann's group [59] achieved sustainable copper-catalyzed electrochemical C–H chalcogenation of o-carboranes with thiols and selenols at room temperature (Scheme 11). The C–H activation features mild reaction conditions and high functional group tolerance, enabling facile assembly of diverse o-carboranes. This establishes a transformative platform for the design of cage C–S and C–Se o-carboranes. Through detailed mechanistic studies, a plausible paired electrolysis mechanism was proposed.
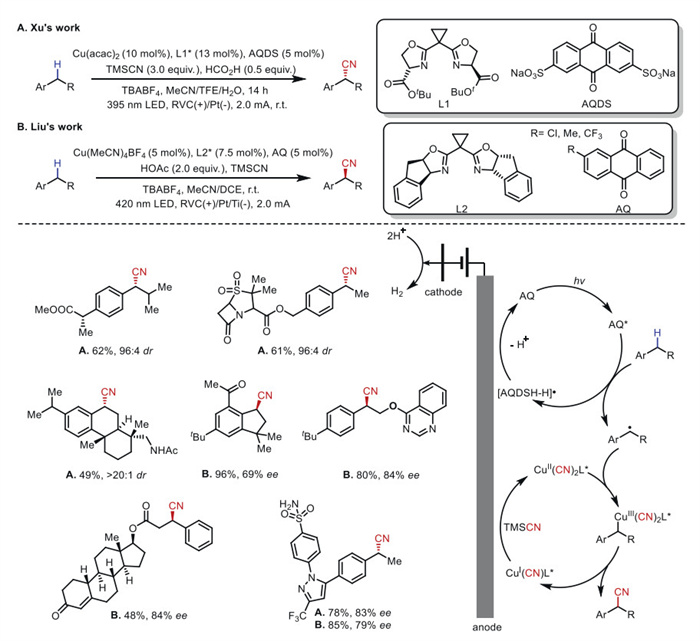
Asymmetric C(sp3)–H functionalization has been demonstrated to significantly streamline the late-stage modification of complex molecules [60-62]. In 2022, both Xu's group [63] and Liu's group [64] independently reported the first photoelectroasymmetric functionalization of C(sp3)–H bonds (Scheme 12). The asymmetric photoelectrocatalytic cyanation reaction employs a combination of copper catalyst and photocatalyst, featuring a broad substrate scope without the use of chemical oxidants. A series of late-stage cyanations of complex structures were achieved with excellent yields and high site and enantioselectivity. Mechanistic studies reveal that the photocatalyst AQDS forms an electronically excited state AQDS* under near-infrared irradiation, which undergoes single-electron oxidation to generate a benzyl radical. The benzyl radical then coordinates with the chiral copper complex (L*)CuⅡ(CN)2, followed by reductive elimination to release the desired cyanated product. The reduced intermediates (L*)CuⅠ(CN) and (AQDS−H)• are anodically oxidized to regenerate (L*)CuⅡ(CN)2 and AQDS, concomitantly releasing hydrogen gas. The integration of photoelectrocatalysis and asymmetric copper-catalyzed radical cyanation [35] enables modular control over diverse C–H functionalization processes via radical relay catalysis.
In 2020, Mei's group [65] developed a novel strategy for asymmetric Shono-type oxidative cross-coupling, leveraging a chiral bisoxazoline ligand to demonstrate the first example of Cu/TEMPO-cocatalyzed electrochemical enantioselective oxidative cross-coupling between cyclic tertiary amines and terminal alkynes (Scheme 13).
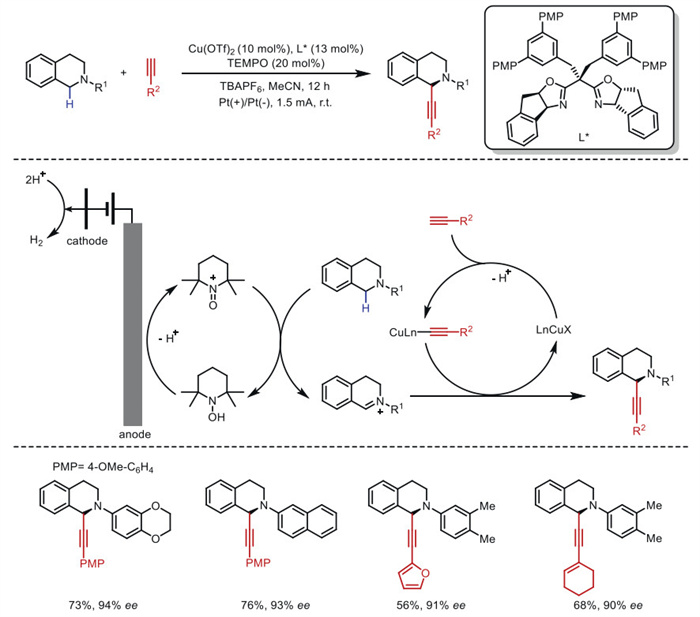
This approach afforded C1-alkynylated tetrahydroisoquinolines with good to excellent enantioselectivity. The use of TEMPO as a cocatalytic redox mediator was critical not only for oxidizing tetrahydroisoquinolines to iminium species but also for lowering the reaction's oxidation potential.
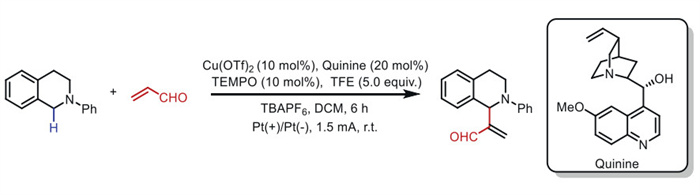
In 2023, Mei's group [66] developed a C(sp3)–H alkenylation method for N-aryl tetrahydroisoquinolines (THIQs) by replacing alkynes with enals (Scheme 14). This strategy employs TEMPO as the mediator and quinine as the catalyst ligand.
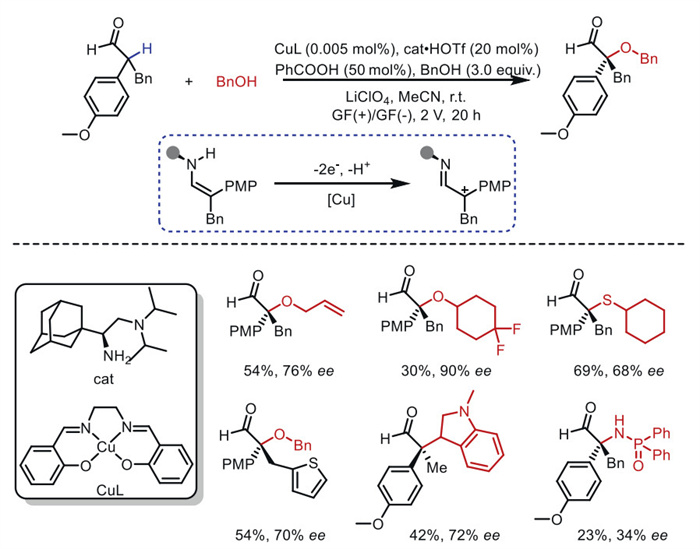
Carbocations serve as versatile reactive intermediates for constructing molecular frameworks and enabling functionalization in chemical and biological processes [67]. Among various generation methods, electrochemical C–H oxidation represents the most straightforward and atom-economical strategy, as it employs readily available unfunctionalized starting materials while avoiding stoichiometric oxidants [68]. In 2024, the Luo's group [69] developed chiral primary amine catalysts for asymmetric electrochemical catalysis in the oxidative functionalization of α-branched aldehydes, achieving the anodic generation of chiral α-iminium carbocation intermediates via enamine catalysis (Scheme 15). The critical SN1 step proceeds through a tertiary amine-mediated proton shuttle, allowing chiral carbocation intermediates to be stereoselectively trapped by diverse nucleophiles, such as alcohols, water, and thiols. Mechanistic studies confirm the carbocationic nature of the intermediates, with the tertiary amine-mediated N–H–X proton shuttle playing a pivotal role in determining the stereoselectivity of C–X bond formation.
Direct current (DC) electrosynthesis has undergone optimization over the past century and plays a pivotal role in various industrial processes [70-72]. Alternating current (AC) electrosynthesis, characterized by polarity reversal and periodic fluctuations, holds promise for enabling diverse chemical reactions [73], yet its apparatuses, underlying principles, and application scenarios remain underdeveloped. In 2024, Lei's group [74] introduced a strategy for programmed AC (pAC) electrosynthesis, achieving selective copper-catalyzed carbon-hydrogen bond cleavage in cross-coupling and difunctionalization reactions (Scheme 16). This protocol employs custom-tailored waveforms integrating asymmetric current profiles, duty ratios, and electrodes to facilitate C–H alkynylation and disproportionation in undivided electrolytic cells. Notably, comprehensive comparisons of copper catalyst dynamics and redox performance provided evidence for the fine-tuning of key copper species by pAC parameters.

To date, three activation modes of photocatalysts have been explored in electrochemical transition metal catalysis: hydrogen atom transfer (HAT), ligand-to-metal charge transfer (LMCT), and single-electron transfer (SET). Numerous complexes of high-valent Earth-abundant metals (CuⅡ, NiⅢ, FeⅢ, CeIV, CoⅢ, etc.) can absorb visible light and induce LMCT, leading to the homolytic cleavage of metal-substrate bonds [75,76].
Given the ready availability and non-toxicity of carboxylic acids, developing efficient and sustainable methods for decarboxylative transformations holds significant importance. Despite substantial efforts in this field, the direct development of enantioselective transformations from carboxylic acids remains challenging. Between 2022 and 2023, the groups of Xu [77], Zhang [78], and Fu [79] independently reported a photoelectrochemical asymmetric decarboxylative cyanation strategy, in which cerium salts facilitate catalytic decarboxylation, and chiral copper complexes enable stereoselective C–CN bond formation (Scheme 17).
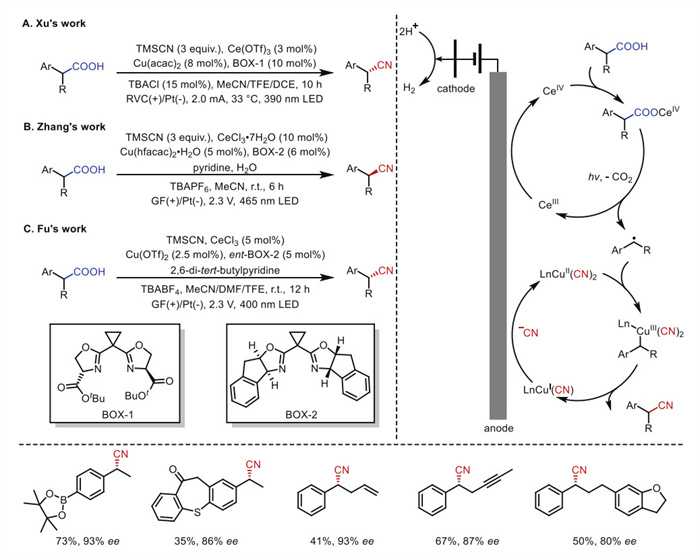
In these three systems, cerium-catalyzed radical decarboxylation generates benzyl radicals from α-substituted arylacetic acids via LMCT. The radicals are rapidly trapped by CuⅡ–CN species, followed by reductive elimination to produce the cyanated product. This synergistic approach eliminates the need for stoichiometric oxidants or substrate pre-functionalization, allowing the reaction to proceed under mild conditions with high enantioselectivity and a broad range of functional group compatibility.
The selective oxidation of hydroxyl groups to carbonyls is a timeless research topic in organic chemistry. In 2007, the Onomura's group [80] reported the chiral copper-catalyzed asymmetric oxidation of 1,2-diols to optically active α-ketols using NBS as a co-oxidant. One year later, the same group [81] achieved a similar asymmetric transformation by replacing NBS with anodic oxidation (Scheme 18). This oxidation method can be applied to the kinetic resolution of racemic cis-cycloalkane-1,2-diols, amino alcohols, and amino aldehydes, yielding optically active compounds with good to high enantioselectivity.
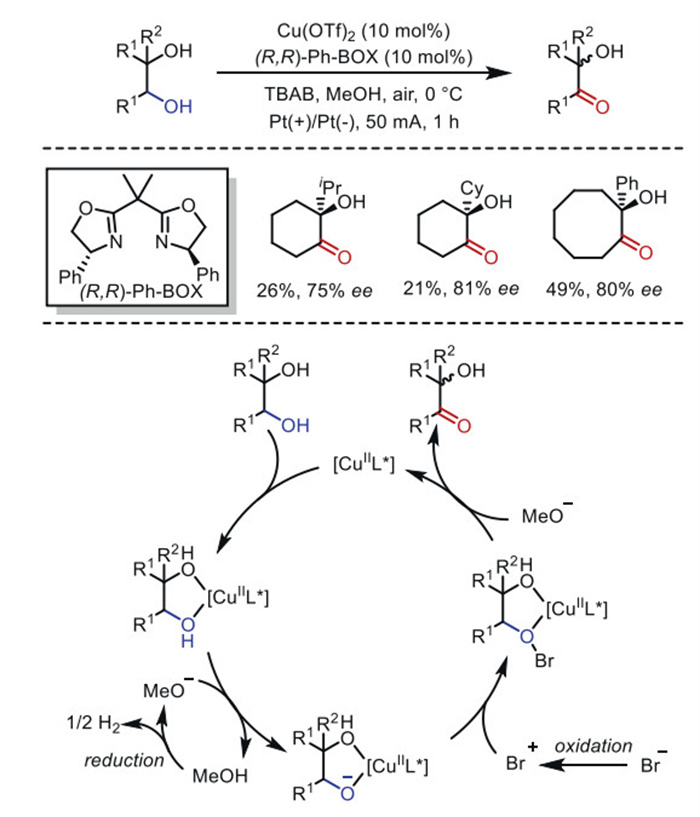
In the proposed catalytic cycle, the complex formed by the diol and [CuⅡ]L* is readily deprotonated by methoxide ions generated at the cathode, producing an alkoxide anion. This anion then reacts with Br⁺ species formed at the anode to generate a complex intermediate. Following the release of HBr, the optically active α-ketol is obtained, and the chiral copper catalyst is regenerated.
In 2024, Ruan's group [82] developed a mild and biocompatible paired electrolysis ring-opening strategy, utilizing copper electrocatalysis to synthesize various remote amino alcohols, including unnatural peptide alcohols (Scheme 19). Experimental results and density functional theory (DFT) calculations indicate that water serves as both a hydroxyl source and a solvent, facilitating the generation of CuH and CuⅠ at the cathode, thereby reducing the aldehyde intermediates formed during the reaction.

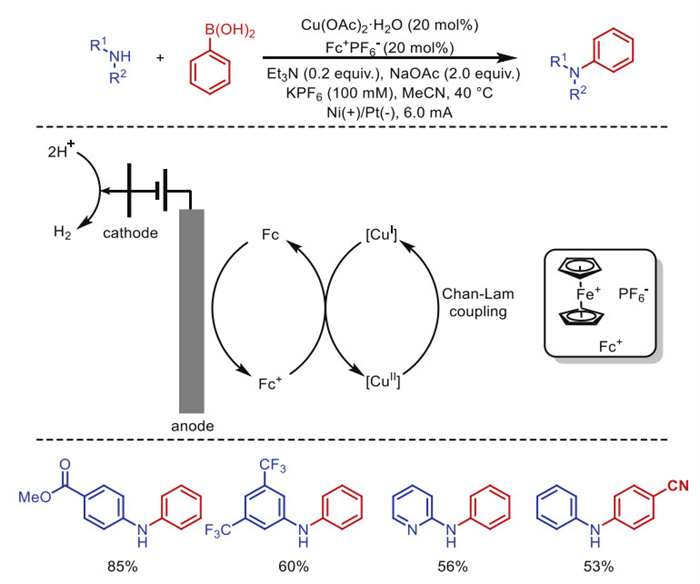
In 2021, Sevov and co-workers [83] reported a ligand-free copper salt catalysis for the electrochemical homogeneous Chan-Lam coupling reaction between amines and arylboronic acids, using Cu(OAc)2 as the catalyst and Fc⁺ as the anodic mediator, affording anilines in moderate to excellent yields (Scheme 20). The ferrocenium catalyst serves multiple functions: (1) It mediates the CuⅠ/CuⅡ cycle in the coupling reaction; (2) It protects the amine from anodic oxidation; (3) It regenerates CuⅡ from Cu0 deposited on the cathode surface.
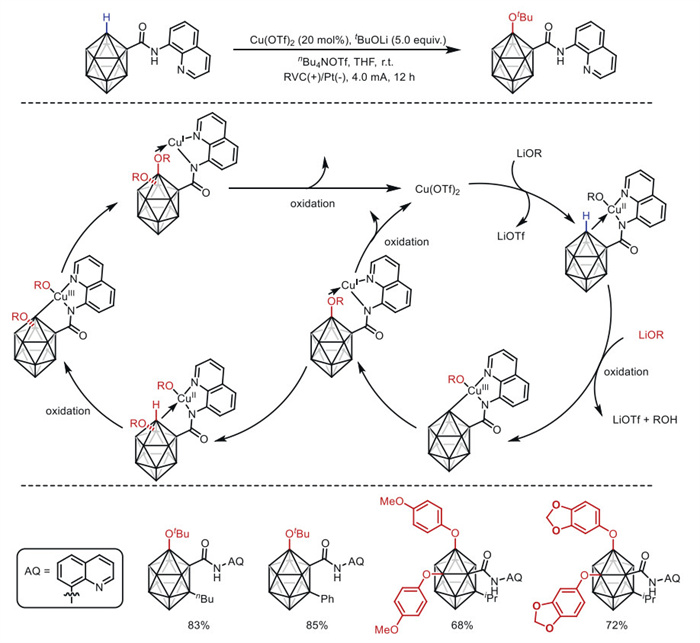
In 2020, Xie and co-workers [84] reported the first copper-catalyzed electrochemical B–H oxidation reaction of o-carborane by introducing 8-aminoquinoline as a directing group at room temperature (Scheme 21). Copper-catalyzed cross-coupling of carborane amides with lithium phenoxide led to the formation of 4,5-diphenol o-carborane via direct B–H activation, whereas the use of lithium tert–butoxide afforded the 4-monooxidation product. This reaction required no additional chemical oxidants and produced H2 and lithium salts as byproducts. Control experiments suggested that high-valent CuⅢ species might be involved in the reaction process. This study combines cheap metal catalysis with electrochemistry, opening up new avenues for the synthetic chemistry of carboranes and other boron clusters.
In recent years, electrosynthesis has been identified as an increasingly viable platform in molecular science. Organic electrosynthesis has emerged as a sustainable tool in modern synthesis, laying the foundation for innovative chemical transformations through new mechanistic pathways. Transition metal electrocatalysis is a rapidly growing field that has become a cutting-edge technology for opening up new synthetic routes. Relevant studies in the past have mainly focused on noble transition metals such as rhodium and ruthenium. Recent exploration of 3d transition metals such as copper has paved the way for the development of resource-economical and environmentally friendly processes. This paper presents a summary of recent advances in electrochemical copper catalysis in recent years (Fig. 5), focusing on the difunctionalization, C–H bond functionalization, and C–C bond activation of olefins by electro-copper catalysis.
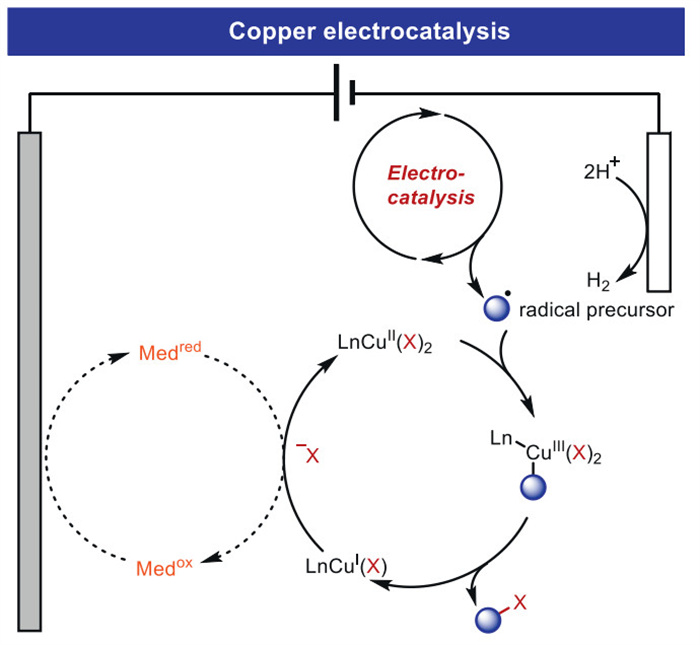
In most cases, direct oxidation at the anode or the addition of redox media achieve the CuⅠ/CuⅡ/CuⅢ catalytic cycle. The radical precursors generate radicals in addition to olefins through direct electrolysis, photoelectrocatalysis, HAT and other pathways to achieve difunctionalization of olefins. Distinguishing from the traditional coupling mode of C(sp2)–H bond activation, C(sp3)–H tends to generate carbon radicals, followed by oxidative addition to [CuⅡ] and reductive elimination to obtain the products. The C–C bond activation substrate is limited to carboxylic acids, which can generate carbon radicals via the LMCT pathway. In fact, there are still relatively few cases of C–C bond activation and C–X bond activation.
Although electrochemical copper-catalyzed has made considerable progress in recent years, this field still faces many challenges. A major challenge of electrochemical copper catalysis lies in the fact that copper ions have a low standard reduction potential and are sensitive to the electroreduction environment, leading to catalyst deactivation. Most copper-catalyzed electrochemical reactions are focused on oxidation at the anode, while reduction reactions or more efficient pairwise electrolysis reactions are still in the preliminary exploration stage; Enantioselective electrocatalysis is an emerging trend in asymmetric synthesis under very mild reaction conditions. Asymmetric electrochemical copper catalysis is an atomically efficient way to obtain α-aryl chiral cyanide. The unique stereocontrol of benzyl radicals may stem from attractive noncovalent interactions between benzyl radicals and copper ligands, which will determine the geometry of the transition state. In the future, the problem of stereoselective control of unstable alkyl radicals should be vigorously solved by designing new chiral ligands; The main types of electrocopper catalysis are functionalization of olefins and activation of C–H bonds. The activation of C–C bonds is also limited to benzylcarboxylic acids, and the development of more reaction types of electrochemical copper catalysis is the future development trend; Meanwhile, the acquisition of other chiral compounds through other radical precursors deserves special attention.
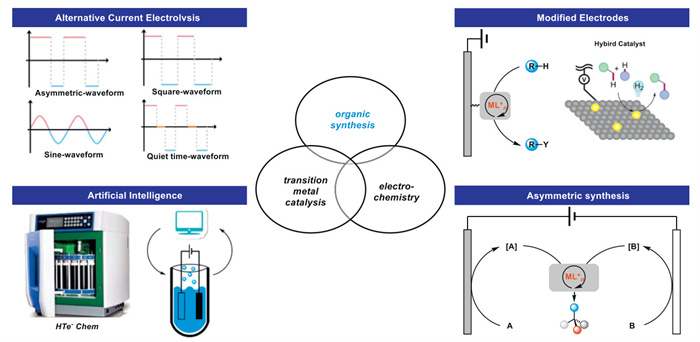
In terms of future trends of organic electrosynthesis, by modulating the overpotential of the reaction, new electrode materials will significantly change the limitations of electrosynthesis (Fig. 6). In fact, there are still few cases of electrochemical reactions that have been transformed from lab-scale to industrial applications. Reactors, electrode passivation, and mass transfer limitations are challenges to upscaling electrochemical reactions. The development of new electrode materials/catalysts and reactor design can both be trends in the future development of electrochemical industrialization. Reusable covalent catalysts will significantly enhance catalytic activity by directly connecting to the electrode surface, thereby greatly reducing catalyst loading. The combination of AI and electrochemistry also holds great potential, as recently demonstrated by Modestino's optimization of electrochemical adiponitrile synthesis through voltage-dose experiments [85]. In addition, automated electrochemical synthesis in flow, high-throughput experiments, and operational analysis [86] can effectively provide large datasets that, in combination with AI, constitute a viable platform.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Pan Zhou: Writing – original draft, Methodology, Investigation, Data curation. Ting Zou: Methodology, Investigation. Hong-Jian Song: Methodology, Investigation. Yu-Xiu Liu: Methodology, Investigation. Qing-Min Wang: Writing – review & editing, Project administration, Funding acquisition, Conceptualization.
This work is supported by the National Natural Science Foundation of China (No. 22271166) and the Frontiers Science Center for New Organic Matter, Nankai University (No. 63181206) for generous financial support for our programs.
A. Volta, Phil. Trans. R. Soc. Lond. 90 (1800) 403–431.
M. Faraday, Ann. Phys. 109 (1834) 433–451. doi: 10.1002/andp.18341092307
H. Kolbe, Ann. Chem. Pharm. 64 (1848) 339–341. doi: 10.1002/jlac.18480640346
H. Moissan, Comptesrendushebdomadaires des sé Ances de l'Académiedes Sciences 102 (1886) 1543–1544.
H.I. Davy, Phil. Trans. R. Soc. Lond. 98 (1808) 1–44.
M.M. Baizer, J. Electrochem. Soc. 111 (1964) 215. doi: 10.1149/1.2426086
T. Shono, Tetrahedron 40 (1984) 811–850. doi: 10.1016/S0040-4020(01)91472-3
J.H. Simons, J. Electrochem. Soc. 95 (1949) 47. doi: 10.1149/1.2776733
E.J. Corey, R.R. Sauers, J. Am. Chem. Soc. 81 (1959) 1739–1743. doi: 10.1021/ja01516a054
B.A. Frontana-Uribe, R.D. Little, J.G. Ibanez, et al., Green Chem. 12 (2010) 2099–2119. doi: 10.1039/c0gc00382d
C. Ma, P. Fang, Z.R. Liu, et al., Sci. Bull. 66 (2021) 2412–2429. doi: 10.1016/j.scib.2021.07.011
L.F.T. Novaes, J. Liu, Y. Shen, et al., Chem. Soc. Rev. 50 (2021) 7941–8002. doi: 10.1039/d1cs00223f
E.J. Horn, B.R. Rosen, P.S. Baran, ACS Cent. Sci. 2 (2016) 302–308. doi: 10.1021/acscentsci.6b00091
J. Yoshida, K. Kataoka, R. Horcajada, et al., Chem. Rev. 108 (2008) 2265–2299. doi: 10.1021/cr0680843
J.C. Siu, N. Fu, S. Lin, Acc. Chem. Res. 53 (2020) 547–560. doi: 10.1021/acs.accounts.9b00529
K. Yamamoto, M. Kuriyama, O. Onomura, Acc. Chem. Res. 53 (2019) 105–120. doi: 10.15261/serdj.26.105
N.W.J. Ang, J.C.A. Oliveira, L. Ackermann, et al., Angew. Chem. Int. Ed. 59 (2020) 12842–12847. doi: 10.1002/anie.202003218
B.R. Rosen, E.W. Werner, A.G. O'Brien, P.S. Baran, et al., J. Am. Chem. Soc. 136 (2014) 5571–5574. doi: 10.1021/ja5013323
K.J. Jiao, D. Liu, H.X. Ma, et al., Angew. Chem. Int. Ed. 59 (2020) 6520–6524. doi: 10.1002/anie.201912753
Z. Duan, L. Zhang, W. Zhang, et al., ACS Catal. 10 (2020) 3828–3831. doi: 10.1021/acscatal.0c00103
L. Niu, C. Jiang, Y. Liang, et al., J. Am. Chem. Soc. 142 (2020) 17693–17702. doi: 10.1021/jacs.0c08437
H. Yan, Z.W. Hou, H.C. Xu, et al., Angew. Chem. Int. Ed. 58 (2019) 4592–4595. doi: 10.1002/anie.201814488
M. Elsherbini, T. Wirth, Acc. Chem. Res. 52 (2019) 3287–3296. doi: 10.1021/acs.accounts.9b00497
C. Huang, X.X. Qian, H.C. Xu, Angew. Chem. Int. Ed. 58 (2019) 6650–6653. doi: 10.1002/anie.201901610
J. Hartwig (Ed.), Organotransition Metal Chemistry: from Bonding to Catalysis, University Science Books, New York, 2010.
K.L. Skubi, T.R. Blum, T.R. Yoon, Chem. Rev. 116 (2016) 10035–10074. doi: 10.1021/acs.chemrev.6b00018
J. Twilton, C. Le, P. Zhang, et al., Nat. Rev. Chem. 1 (2017) 0052.
J. Lu, Y. Wang, T. McCallum, N. Fu, et al., iScience 23 (2020) 101796. doi: 10.1016/j.isci.2020.101796
A.Y. Chan, I.B. Perry, N.B. Bissonnette, et al., Chem. Rev. 122 (2021) 1485–1542.
R.C. Samanta, T.H. Meyer, I. Siewert, et al., Chem. Sci. 11 (2020) 8657–8670. doi: 10.1039/d0sc03578e
X. Cheng, A. Lei, T. -S. Mei, et al., CCS Chem. 4 (2022) 1120–1152. doi: 10.31635/ccschem.021.202101451
C.A. Malapit, M.B. Prater, J.R. Cabrera-Pardo, et al., Chem. Rev. 122 (2022) 3180–3218. doi: 10.1021/acs.chemrev.1c00614
A. Jutand, Chem. Rev. 108 (2008) 2300–2347. doi: 10.1021/cr068072h
Y. Kim, W.J. Jang, Beilstein J. Org. Chem. 21 (2025) 155–178. doi: 10.3762/bjoc.21.9
F. Wang, P. Chen, G. Liu, Nat. Synth. 1 (2022) 107–116. doi: 10.1038/s44160-021-00016-x
L. Li, Y. Yao, N. Fu, Chem. Catal. 4 (2024) 100898.
S.E. Allen, R.R. Walvoord, R. Padilla-Salinas, et al., Chem. Rev. 113 (2013) 6234–6458. doi: 10.1021/cr300527g
N. Fu, L. Song, J. Liu, et al., J. Am. Chem. Soc. 141 (2019) 14480–14485. doi: 10.1021/jacs.9b03296
W. Liu, J.T. Groves, Acc. Chem. Res. 48 (2015) 1727–1735. doi: 10.1021/acs.accounts.5b00062
C.Y. Cai, Y.T. Zheng, J.F. Li, et al., J. Am. Chem. Soc. 144 (2022) 11980–11985. doi: 10.1021/jacs.2c05126
H. Kim, S. Kim, J. Am. Chem. Soc. 146 (2024) 22498–22508. doi: 10.1021/jacs.4c06207
S. Wu, J. Kaur, T.A. Karl, et al., Angew. Chem. Int. Ed. 61 (2022) e202107811. doi: 10.1002/anie.202107811
H. Huang, K.A. Steiniger, T.H. Lambert, J. Am. Chem. Soc. 144 (2022) 12567–12583. doi: 10.1021/jacs.2c01914
L. Qian, M. Shi, Chem. Commun. 59 (2023) 3487–3506. doi: 10.1039/d3cc00437f
X.L. Lai, H.C. Xu, J. Am. Chem. Soc. 145 (2023) 18753–18759. doi: 10.1021/jacs.3c07146
T.V. RajanBabu, A.L. Casalnuovo, J. Am. Chem. Soc. 114 (1992) 6265–6266. doi: 10.1021/ja00041a066
J. Wilting, M. Janssen, C. Müller, et al., J. Am. Chem. Soc. 128 (2006) 11374–11375. doi: 10.1021/ja064378u
A. Falk, A.L. Göderz, H.G. Schmalz, Angew. Chem. Int. Ed. 52 (2013) 1576–1580. doi: 10.1002/anie.201208082
X. Li, C. You, J. Yang, et al., Angew. Chem. Int. Ed. 58 (2019) 10928–10931. doi: 10.1002/anie.201906111
H. Zhang, X. Su, K. Dong, Org. Biomol. Chem. 18 (2020) 391–399. doi: 10.1039/c9ob02374g
L. Song, N. Fu, B.G. Ernst, et al., Nat. Chem. 12 (2020) 747–754. doi: 10.1038/s41557-020-0469-5
Q.L. Yang, X.Y. Wang, J.Y. Lu, et al., J. Am. Chem. Soc. 140 (2018) 11487–11494. doi: 10.1021/jacs.8b07380
S. Kathiravan, S. Suriyanarayanan, I.A. Nicholls, Org. Lett. 21 (2019) 1968–1972. doi: 10.1021/acs.orglett.9b00003
C. Tian, U. Dhawa, A. Scheremetjew, L. Ackermann, et al., ACS Catal. 9 (2019) 7690–7696. doi: 10.1021/acscatal.9b02348
X. Yang, Q.L. Yang, X.Y. Wang, et al., J. Org. Chem. 85 (2020) 3497–3507. doi: 10.1021/acs.joc.9b03223
X. Yi, X. Hu, Angew. Chem. Int. Ed. 58 (2019) 4700–4704. doi: 10.1002/anie.201814509
J. Poater, M. Solà, C. Viñas, F. Teixidor, Angew. Chem. Int. Ed. 53 (2014) 12191–12195. doi: 10.1002/anie.201407359
R. Núñez, M. Tarrés, A. Ferrer-Ugalde, et al., Chem. Rev. 116 (2016) 14307–14378. doi: 10.1021/acs.chemrev.6b00198
L. Yang, B.B. Jei, A. Scheremetjew, et al., Chem. Sci. 12 (2021) 12971–12976. doi: 10.1039/d1sc02905c
T. Cernak, K.D. Dykstra, S. Tyagarajan, et al., Chem. Soc. Rev. 45 (2016) 546–576. doi: 10.1039/C5CS00628G
Ł. Woźniak, J.F. Tan, Q.H. Nguyen, et al., Chem. Rev. 120 (2020) 10516–10543. doi: 10.1021/acs.chemrev.0c00559
H.M.L. Davies, R.E.J. Beckwith, Chem. Rev. 103 (2003) 2861–2904. doi: 10.1021/cr0200217
C.Y. Cai, X.L. Lai, Y. Wang, et al., Nat. Catal. 5 (2022) 943–951. doi: 10.1038/s41929-022-00855-7
W. Fan, X. Zhao, Y. Deng, et al., J. Am. Chem. Soc. 144 (2022) 21674–21682. doi: 10.1021/jacs.2c09366
P.S. Gao, X.J. Weng, Z.H. Wang, et al., Angew. Chem. Int. Ed. 59 (2020) 15254–15259. doi: 10.1002/anie.202005099
Z. He, H.L. Liu, Z.H. Wang, et al., J. Org. Chem. 88 (2023) 6203–6208. doi: 10.1021/acs.joc.3c00223
R.R. Naredla, D.A. Klumpp, Chem. Rev. 113 (2013) 6905–6948. doi: 10.1021/cr4001385
H. Wang, K. Liang, W. Xiong, et al., Sci. Adv. 6 (2020) eaaz0590. doi: 10.1126/sciadv.aaz0590
Q. Lin, Y. Duan, Y. Li, et al., Nat. Commun. 15 (2024) 6900. doi: 10.1038/s41467-024-50945-2
R. Francke, R.D. Little, Chem. Soc. Rev. 43 (2014) 2492–2521. doi: 10.1039/c3cs60464k
M. Yan, Y. Kawamata, P.S. Baran, Chem. Rev. 117 (2017) 13230–13319. doi: 10.1021/acs.chemrev.7b00397
A. Wiebe, T. Gieshoff, S. Möhle, et al., Angew. Chem. Int. Ed. 57 (2018) 5594–5619. doi: 10.1002/anie.201711060
L. Zeng, J. Wang, D. Wang, et al., Angew. Chem. Int. Ed. 62 (2023) e202309620. doi: 10.1002/anie.202309620
L. Zeng, Q. Yang, J. Wang, et al., Science 385 (2024) 216–223. doi: 10.1126/science.ado0875
Y. Abderrazak, A. Bhattacharyya, O. Reiser, Angew. Chem. Int. Ed. 60 (2021) 21100–21115. doi: 10.1002/anie.202100270
F. Juliá, ChemCatChem 14 (2022) e202200916. doi: 10.1002/cctc.202200916
X.L. Lai, M. Chen, Y. Wang, et al., J. Am. Chem. Soc. 144 (2022) 20201–20206. doi: 10.1021/jacs.2c09050
Y. Yuan, J. Yang, J. Zhang, Chem. Sci. 14 (2023) 705–710. doi: 10.1039/d2sc05428k
K. Yang, Y. Wang, S. Luo, N. Fu, Chem. Eur. J. 29 (2023) e202203962. doi: 10.1002/chem.202203962
O. Onomura, H. Arimoto, Y. Matsumura, Y. Demizu, Tetrahedron Lett. 48 (2007) 8668–8672. doi: 10.1016/j.tetlet.2007.10.014
D. Minato, H. Arimoto, Y. Nagasue, et al., Tetrahedron 64 (2008) 6675–6683. doi: 10.1016/j.tet.2008.05.015
X. Fang, X. Hu, Q.X. Li, et al., Angew. Chem. Int. Ed. 63 (2024) e202418277.
B.R. Walker, S. Manabe, A.T. Brusoe, C.S. Sevov, J. Am. Chem. Soc. 143 (2021) 6257–6265. doi: 10.1021/jacs.1c02103
Y.K. Au, H. Lyu, Y. Quan, Z. Xie, J. Am. Chem. Soc. 142 (2020) 6940–6945. doi: 10.1021/jacs.0c02490
D.E. Blanco, B. Lee, M.A. Modestino, Proc. Natl. Acad. Sci. U. S. A. 116 (2019) 17683–17689. doi: 10.1073/pnas.1909985116
M. Santi, J. Seitz, R. Cicala, et al., Chem. Eur. J. 25 (2019) 16230–16235. doi: 10.1002/chem.201904711

Figure 2 Electrochemical advantages, representative chemical elements and fine chemicals produced electrochemically.

Scheme 1 Electrochemical copper-catalyzed cyanophosphorylation cyanosulfonylation of olefins.

Scheme 4 Photoelectroasymmetric copper catalysis of heteroaromatic nitrification of olefins.

Scheme 6 Iodine-mediated electrochemical copper-catalyzed amination of C(sp2)–H bonds.

Scheme 8 Electrochemical copper-catalyzed cascade cyclization of C(sp2)–H bonds with alkynes.

Scheme 10 Electrochemical copper-catalyzed intramolecular allylic amination of C(sp2)–H bonds.

Scheme 11 Electrochemical copper-catalyzed sustainable electrocatalytic C–H chalcogenation of o-carborane with thiols and selenols.

Scheme 12 Photoelectrochemical copper-catalyzed asymmetric radical cyanation of C(sp3)–H bonds.

Scheme 16 Electrochemical copper-catalyzed alkynylation/alkenylation of C(sp3)–H via programmed alternating current.

Scheme 17 Photoelectrochemical copper-catalyzed asymmetric decarboxylative cyanation of C–C bonds.

Scheme 20 Electrochemical copper-catalyzed homogeneous Chan-Lam coupling reaction of amines and arylboronic acids.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:















 下载:
下载: