Scheme 1.
Reaction modes of metal carbynes and hydride reagents.
Synthesis, characterization and hydride reduction of PNP-pincer molybdenum and tungsten carbyne complexes
Xiwen Yu , Haiqi Xu , Tao Xu , Tongling Liang , Yunhui Yang , Congyang Wang

Since the synthesis of transition metal carbyne/alkylidyne complexes [LnM(≡CR)] were reported by Fischer and Schrock [1,2], these species have been broadly applied in the fields of organic synthesis and material chemistry [3-8]. For instances, metal carbyne species had great potentials in catalytic hydrocarbon reforming reactions [9-12]. Also, high-valence metal carbyne complexes showed catalytic reactivity in alkyne metathesis reactions, thereby attracting extensive attentions [13-16]. Molybdenum and tungsten carbynes were widely studied in oxidative decarbonylation reactions [17-19] and construction of varied metal-carbon-bond containing compounds upon treatments with either nucleophiles or electrophiles [20-23].
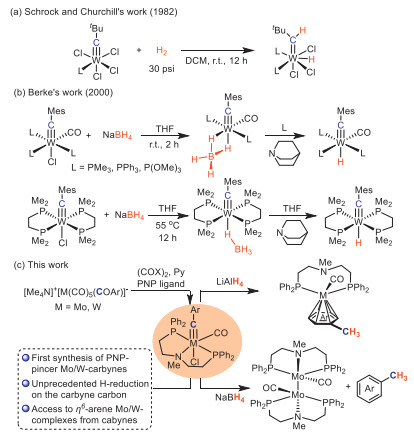
It is noteworthy that metal carbyne species were identified as putative intermediates in Fischer–Tropsch process. Thus, reactions of metal carbyne complexes with dihydrogen or hydride reagents might give clues to the underlying mechanism of CO reduction [24,25]. However, explorations on the reduction reactions of metal carbynes were rather limited. Agapie et al. reported that a molybdenum carbyne carbonyl intermediate was gradually reduced by strongly reducing agent of KC8 to give a coupling product of alkyne [26]. Only few reductive reactions of metal carbynes with milder hydrogen-based reducing agents were reported. Schrock, Churchill and coworkers showed that tungsten carbynes [W(≡C-tBu)Cl3L2 (L = PMe3)] reacted with molecular hydrogen to yield the tungsten alkylidene hydride complexes [W(=CH-tBu)(H)Cl3L2] (Scheme 1a) [27]. When sodium borohydride (NaBH4) was used as a reducing reagent to react with monodentate phosphine and/or CO supported tungsten carbynes, the corresponding dihydride-bridged W(≡CMes)(η2-H2BH2)(CO)nL3-n compounds were separated by Berke's group, which was further transformed with quinuclidine to tungsten carbyne hydride species (Scheme 1b) [28]. Bidentate-phosphine-coordinated tungsten carbyne complex, trans-W(≡CMes)Cl(dmpe)2, reacted with NaBH4 to form a monohydride-bridged complex, [trans-W(≡CMes)(η1-BH4)(dmpe)2], and further removal of BH3 with quinuclidine yielded the tungsten carbyne hydride complex trans-W(≡CMes)H(dmpe)2 [29]. Of note, the formation of metal-hydride bonds was favoured towards electrophilic addition of the hydride to the carbyne carbon in the above-mentioned reactions. So far, dissociation of the carbyne moiety from the metal center has been rare due to the high bonding energy of metal carbynes [3,30-32], and complete reduction of the carbyne carbon by hydrides remains elusive.
In order to achieve the reactivity on the carbyne carbon of metal carbynes with hydride reagents, we speculated that tridentate PNP-pincer ligands instead of previously-reported mono- and bidentate ones might tune the reactive sites of metal carbynes with hydrides from the metal center to the carbyne carbon. Herein, we reported the first synthesis of a series of molybdenum and tungsten carbyne complexes bearing PNP-pincer ligands (Scheme 1c). Contrary to previous reactivity on the metal centers of mono- and bidentate tungsten carbynes with NaBH4, tridentate PNP-pincer molybdenum and tungsten carbyne complexes underwent complete reduction of the carbyne carbon by hydride reagents. Specifically, PP-bidentate η6-arene molybdenum and tungsten complexes [(η6-arene)(PP)M(CO)] (M = Mo, W) were selectively accessed when LiAlH4 was used as the reductant. On the other hand, treatment with NaBH4 eventually gave toluene product and the dimerized dimolybdenum carbonyl complex [(PNP)Mo(CO)]2, which showcased the diverse reactivity of PNP-pincer molybdenum and tungsten carbynes with hydride reagents.
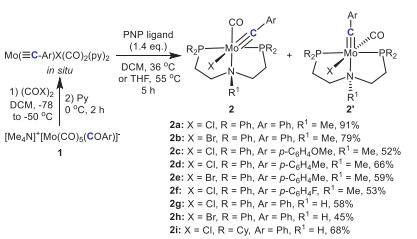
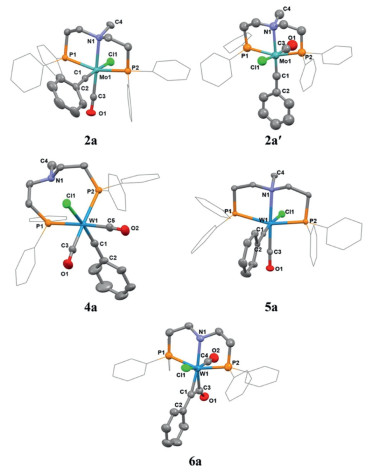
According to Mayr's method [33], treatment of [NMe4]+[Mo(CO)5(CO)Ar]- 1 (Ar = Ph, 4-fluorphenyl, 4-tolyl, 4-methoxyphenyl) with a slight excess of oxalyl halide (COX)2 led to formation of trans-halotetracarbonylmetal carbyne complexes [Mo(≡CAr)X(CO)4], which was converted to carbyne complexes [Mo(≡CAr)X(CO)2(py)2] (X = Cl, Br) upon addition of pyridine. Subsequent addition of a PNP-pincer ligand [Ph2P(CH2)2N(CH3)(CH2)2PPh2; R2P(CH2)2N(H)(CH2)2PR2, R = Ph, Cy] smoothly produced the corresponding PNP-pincer coordinated molybdenum carbyne complexes [(PNP)Mo(≡CAr)X(CO)], which contained two interconvertible stereoisomers 2 and 2′ (Scheme 2). The reaction yields (45%−91%) of these complexes were robust across various aromatic substituents (Ar) with electron-withdrawing or electron-donating groups. For 2a, the characteristic carbon signals (Mo≡C and CO groups) in the 13C NMR spectrum appeared at 271.68 ppm and 243.06 ppm, respectively (Fig. S7 in Supporting information). The two phosphorus atoms of the PNP-pincer ligand in the molybdenum carbyne product had the same chemical environment, exhibiting only one peak at 38.60 ppm in 31P NMR. Meanwhile, the single crystal X-ray diffraction analysis confirmed the product structure and revealed its eight-coordinate, distorted octahedral geometry (Fig. 1). Specifically, the Mo-C1 bond length of 2a was determined to be 1.808(4) Å (Table 1), which was in agreement with the Mo≡C bond length [3,34]. The bond angle of C1-Mo1-Cl1 was 173.14(11)°, indicating the chlorine atom was in a trans configuration to the carbyne ligand. This configuration predominated in the products presumably due to its more favorable electronic structure. Single crystal X-ray diffraction analysis of isomeric complex 2a′ showed that the bond angle of C1-Mo-Cl1 was 101.20(8)°, and the carbyne ligand was nearly perpendicular to the Mo-Cl bond (Fig. 1 and Table 1). In the 13C NMR spectrum of 2a′, the 'Mo≡C' and 'CO' group showed signals at 288.97 ppm and 237.80 ppm, respectively (Fig. S26 in Supporting information), and the phosphorus signal of the PNP-pincer ligand was shown at 45.60 ppm in 31P NMR.
 DownLoad:
CSV
DownLoad:
CSV
| Complex | M1-C1 (Å) | M1-C3 (Å) | M1-Cl (Å) | M1-N1 (Å) | C1-C2 (Å) | C1-M1-X1 (deg) | C1-M1-C3 (deg) |
| 2a | 1.808(4) | 1.978(4) | 2.5698(9) | 2.355(3) | 1.432(6) | 173.14(11) | 85.19(18) |
| 2a′ | 1.796(3) | 1.964(3) | 2.5269(7) | 2.464(2) | 1.443(3) | 101.20(8) | 85.02(11) |
| 4a 5a 6a |
1.821(3) 1.817(3) 2.007(3) |
2.023(3) 1.963(3) 2.138(3) |
2.5504(7) 2.5445(7) 2.5482(8) |
- 2.346(2) 2.313(3) |
1.433(4) 1.443(4) 1.455(5) |
169.33(9) 172.55(9) 90.29(10) |
85.37(13) 85.27(12) 38.48(13) |
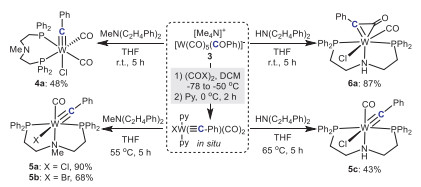
Interestingly, when the congener tungsten precursor [W(≡CAr)X(CO)2(py)2] was treated with the pincer ligand of Ph2P(CH2)2N(CH3)(CH2)2PPh2 at room temperature, a bright-yellow tungsten carbyne complex 4a was obtained in 48% isolated yield, in which the pincer ligand acted only as a bidentate PP-ligand replacing two pyridine ligands of the precursor (Scheme 3). The single crystal X-ray diffraction analysis of 4a indicated that the chlorine atom adopted a trans configuration relative to the carbyne ligand and two W-P bonds were perpendicular to each other, different from the linear configuration of PNP-pincer mode (Fig. 1). The bond length of the tungsten-carbon bond was 1.821(3) Å (Table 1), confirming its carbyne structure [35]. The 13C NMR spectrum of 4a revealed a triplet signal for the 'Mo≡C' group at 266.24 ppm, accompanied by two signals for the carbonyl carbons (CO) at 210.37 ppm and 210.02 ppm (Fig. S40 in Supporting information). The 31P NMR spectrum of 4a showed that the phosphine signal was at 8.58 ppm. When the reaction temperature was raised to 55 ℃, the tridentate PNP-pincer tungsten carbyne complexes 5a and 5b were isolated in high yields. Characterization via 13C NMR of 5a displayed significant shifts of characteristic carbon signals (W≡C and CO), observed at 259.92 ppm and 241.79 ppm, respectively in comparison with those of 4a (Fig. S43 in Supporting information). Surprisingly, similar PP-bidentate biscarbonyl tungsten carbyne species as 4a could not be afforded when Ph2P(CH2)2N(H)(CH2)2PPh2 was used in the reaction at room temperature. Instead, a PNP-pincer η2-ketenyl complex 6a was obtained as purple microcrystals. The single-crystal structure of 6a revealed that the ketene group (C=C=O) lied in the equatorial coordination plane of tungsten (Fig. 1), with W-C1 bond elongated to 2.007 Å and the bond angle of Cl1-W1-C1 almost vertical [90.29(2)o] (Table 1). It was further found that complex 6a slowly extruded CO to provide PNP-pincer tungsten carbyne complex 5c when warmed up to 65 ℃, which was in agreement with previous results reported by Hill and coworkers [36,37]. When the reaction temperature was directly raised to 65 ℃, complex 5c was obtained via a one-pot procedure in 43% isolated yield.
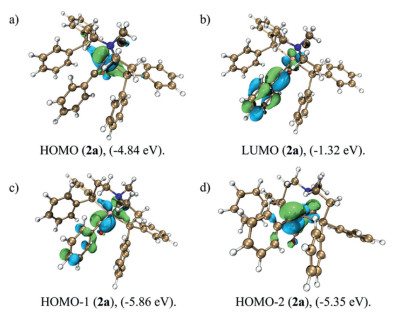
To get more insights of the bonding nature and the reactivity properties of the carbyne carbon, computational studies of 2a was carried out using Gaussian16 program (version RevC.01) [38]. The analysis of computation result was realized by Multifwn (Ver. 3.8) [39]. The optimized structures were in good agreement with the single-crystal XRD results, with small mean deviations for the bond lengths in the core coordination geometry, Mo1-C1 for 1.800 Å. The orbital diagram and corresponding energy level are shown in Fig. 2. HOMO orbital was stabilized due to the overlap of the dxz orbital of Mo with the antibonding orbital of the CO fragment (Fig. 2a), and HOMO-1 and HOMO-2 orbitals participated in the π bond of the Mo≡C bond (Figs. 2c and d). LOL [40] was also used to determine the bonding characterization (Fig. 3a), and it is worth noting that the orbital localization component between Mo and C was polarized toward C atom of carbyne, and calculated Fuzzy bond order was 2.247, which showed a typical Mo≡C strength [41]. Electrophilic strength analysis using Fukui function (Fig. 3b) [42] showed that the fw+s was mainly located on Mo and C of carbyne, indicating that these two atoms were electrophilic sites.
With the successful synthesis of PNP-pincer molybdenum and tungsten carbynes in hand, we commenced to investigate their reduction reactions with hydride reagents. It turned out that treatment of these metal carbyne complexes with excess amounts of LiAlH4 in toluene at 80–90 ℃ yielded a series of unexpected PP-bidentate η6-arene molybdenum and tungsten complexes 7a-d, 8 (Scheme 4a). It is noteworthy that the substituents on the carbyne ligands were observed to appear on the η6-arene moiety of the products, suggesting that the η6-arene moiety was derived from the carbyne ligand and did not exchange with the toluene solvent in the reactions. Meanwhile, complex 7a was also obtained in 79% NMR yield by treatment of 2a with NaBHEt3 in a THF solution (Fig. S55 in Supporting information). The 1H NMR spectrum of 7a in C6D6 showed that the benzene ring originated from the carbyne ligand changed to 4.08 ppm (2H), 3.86 ppm (1H), and 3.54 ppm (2H), while the signal of hydrogenated carbyne carbon was at 1.98 ppm (3H) (Fig. S52 in Supporting information). The single-crystal structure of complex 7a revealed that the Mo-N distance was elongated, and the π-bond of η6-benzene molybdenum complex was nearly equal to six benzene carbons (Fig. 4). The Mo-C(O) and Mo-P bond lengths remained unchanged and slight elongation of C1-C2 [1.510(3) Å] was observed. The deuterium-labelling experiments further confirmed that the carbyne carbons in complexes 2 were completely reduced by hydrides (Scheme 4b). Specifically, after treatment of 2a with LiAlD4, the 1H NMR spectrum of product 7a-D showed that the hydrogenated methyl peak of complex 7a at 1.98 ppm almost disappeared. The integration ratio of this peak to the methyl group on the PN(CH3)P ligand was 0.07/1 indicating a 93% deuterium ratio, which was consistent with the 95% deuterium ratio of LiAlD4 (Fig. S70 in Supporting information). Similar deuterium-labelling result was obtained for 7b. The η6-arene ring of complex 7b corresponded to the signals at 3.71 ppm (4H) and 1.75 ppm (6H) for the two methyl groups in 1H NMR (Fig. S56 in Supporting information). After the deuterium-labelling experiments, the integration of the two methyl signals at 1.75 ppm changed from 6 H to 3 H in 7b-D (Fig. S72 in Supporting information).
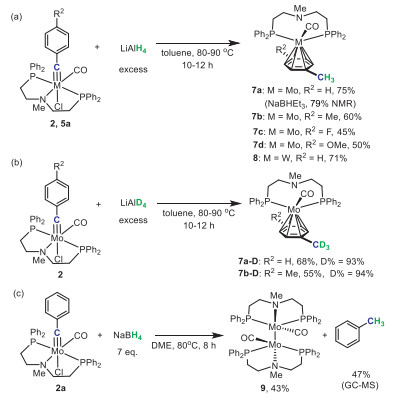
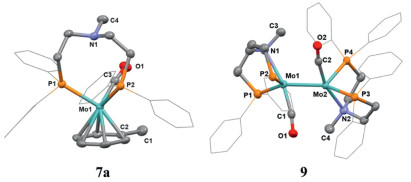
To our surprise, when NaBH4 was used as the reductant instead of LiAlH4 to react with PNP-pincer molybdenum carbyne complex 2a, a reduction pathway distinct from the previously observed reactions occurred and a dimolybdenum biscarbonyl complex [(PNP)Mo(CO)]2 9 was isolated after recrystallization in 43% yield (Scheme 4c). The single crystal X-ray diffraction analysis on the structure of 9 revealed complete detachment of the original carbyne ligand in 2a (Fig. 4). Further analysis of the reaction mixture using GC–MS detected the formation of toluene with a yield of 47%, in a good agreement with the fact that the carbyne carbon was fully reduced with hydrides.
In summary, a series of PNP-pincer molybdenum and tungsten carbyne complexes were successfully synthesized and fully characterized for the first time. Furthermore, it was showed that PP-bidentate η6-arene molybdenum and tungsten complexes were easily accessed from the reduction reactions of these PNP-pincer molybdenum and tungsten carbyne complexes with LiAlH4. Meanwhile, toluene and dimolybdenum biscarbonyl complex [(PNP)Mo(CO)]2 with the carbyne ligand fully reduced and detached, were generated when employing NaBH4 as the hydride reductant. Such reactivity on the carbyne carbons of PNP-pincer molybdenum and tungsten carbyne complexes with hydride reagents are in sharp contrast to previous reactivity on the metal centers of monodentate and bidentate molybdenum and tungsten carbyne complexes with hydrides. Further investigations on new reactions and catalytic reactivity of these metal carbyne complexes are underway in our lab.
We herein declare that all the authors have no conflict of interest.
Xiwen Yu: Writing – original draft, Validation, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Haiqi Xu: Validation, Investigation. Tao Xu: Investigation, Data curation. Tongling Liang: Resources, Data curation. Yunhui Yang: Writing – review & editing, Supervision, Investigation. Congyang Wang: Writing – review & editing, Writing – original draft, Validation, Supervision, Resources, Project administration, Methodology, Investigation, Funding acquisition, Formal analysis.
Financial supports from the National Key R&D Program of China (No. 2023YFA1507902), the National Natural Science Foundation of China (Nos. 22025109, 22371283), CAS Project for Young Scientists in Basic Research (No. YSBR-050), and the State Key Laboratory of Fine Chemicals, Dalian University of Technology (No. KF2102) are gratefully acknowledged.
Supplementary material associated with this article can be found, in the online version, at doi:
E.O. Fischer, G. Kreis, C.G. Kreiter, et al., Angew. Chem. Int. Ed. 12 (1973) 564–565. doi: 10.1002/anie.197305641
S.J. McLain, C.J. Wood, L.W. Messerle, et al., J. Am. Chem. Soc. 100 (1978) 5962–5964. doi: 10.1021/ja00486a069
A. Mayr, H. Hoffmeister, Recent advances in the chemistry of metal-carbon triple bonds, in: F.G.A. Stone, R. West (Eds.), Advances in Organometallic Chemistry, Academic Press, 1991, pp. 227–324.
R.R. Schrock, Chem. Rev. 102 (2002) 145–180. doi: 10.1021/cr0103726
M. Cui, G. Jia, J. Am. Chem. Soc. 144 (2022) 12546–12566. doi: 10.1021/jacs.2c01192
A. Reinholdt, J. Bendix, Chem. Rev. 122 (2022) 830–902. doi: 10.1021/acs.chemrev.1c00404
Y. Yang, C. Wang, Chin. J. Chem. 39(2021) 3481–3484. doi: 10.1002/cjoc.202100461
X. Wang, W.Y. Tong, B. Huang, et al., J. Am. Chem. Soc. 144 (2022) 4952–4965. doi: 10.1021/jacs.1c12874
K. Weiss, Angew. Chem. Int. Ed. 25 (1986) 359–360. doi: 10.1002/anie.198603591
C. Sergey, C.M. Alvaro, M. Logesh, K.V. Bukhryakov, Organometallics 39 (2020) 3453–3457. doi: 10.1021/acs.organomet.0c00491
F. Blanc, J. Thivolle-Cazat, J.M. Basset, C. Copéret, Chem. Eur. J. 14 (2008) 9030–9037. doi: 10.1002/chem.200800864
T.J. Katz, T.H. Ho, N.Y. Shih, Y. Ying, V.I.W. Stuart, J. Am. Chem. Soc. 106 (1984) 2659–2668. doi: 10.1021/ja00321a029
A. Mortreux, M. Blanchard, J. Chem. Soc., Chem. Commun. (1974) 786–787.
J.H. Wengrovius, J. Sancho, R.R. Schrock, J. Am. Chem. Soc. 103 (1981) 3932. doi: 10.1021/ja00403a058
R.R. Schrock, C. Czekelius, Adv. Synth. Catal. 349 (2007) 55–77. doi: 10.1002/adsc.200600459
A. Fürstner, Angew. Chem. Int. Ed. 52 (2013) 2794–2819. doi: 10.1002/anie.201204513
A. Mayr, A.M. Dorries, G.A. McDermott, J. Am. Chem. Soc. 107 (1985) 7775–7776. doi: 10.1021/ja00311a099
A.C. Filippou, P. Portius, C. Jankowski, J. Organomet. Chem. 617-618 (2001) 656–670. doi: 10.1016/S0022-328X(00)00701-4
J. Heppekausen, R. Stade, A. Kondoh, et al., Chem. Eur. J. 18 (2012) 10281–10299. doi: 10.1002/chem.201200621
M. Green, A.G. Orpen, I.D. Williams, J. Chem. Soc., Chem. Commun. (1982) 493–495.
I.J. Hart, J.C. Jeffery, R.M. Lowry, F.G.A. Stone, Angew. Chem. Int. Ed. 27 (1988) 1703–1705. doi: 10.1002/anie.198817031
A.P. James, F.G.A. Stone, J. Organomet. Chem. 310 (1986) 47–54. doi: 10.1016/S0022-328X(00)99659-1
N.H.T. Huy, J. Fischer, F. Mathey, Organometallics 7 (1988) 240–242. doi: 10.1021/om00091a040
O.R. Inderwildi, S.J. Jenkins, D.A. King, J. Phys. Chem. C 112 (2008) 1305–1307. doi: 10.1021/jp710674q
R.A. van Santen, A.J. Markvoort, I.A.W. Filot, M.M. Ghouri, E.J.M. Hensen, Phys. Chem. Chem. Phys. 15 (2013) 17038–17063. doi: 10.1039/c3cp52506f
J. Buss, T. Agapie, Nature 529 (2016) 72–75. doi: 10.1038/nature16154
J.H. Wengrovius, R.R. Schrock, M.R. Churchill, H.J. Wasserman, J. Am. Chem. Soc. 104 (1982) 1739–1740. doi: 10.1021/ja00370a051
E. Bannwart, H. Jacobsen, F. Furno, H. Berke, Organometallics 19 (2000) 3605–3619. doi: 10.1021/om000129h
F. Furno, T. Fox, H.W. Schmalle, H. Berke, Organometallics 19 (2000) 3620–3630. doi: 10.1021/om000332c
N. Aristov, P.B. Armentrout, J. Am. Chem. Soc. 108 (1986) 1806–1819. doi: 10.1021/ja00268a017
R.L. Hettich, B.S. Freiser, J. Am. Chem. Soc. 108 (1986) 2537–2540. doi: 10.1021/ja00270a008
R.L. Hettich, B.S. Freiser, J. Am. Chem. Soc. 109 (1987) 3543–3548. doi: 10.1021/ja00246a009
G.A. McDermott, A.M. Dorries, A. Mayr, Organometallics 6 (1987) 925–931. doi: 10.1021/om00148a005
H. Huang, R.P. Hughes, A.L. Rheingold, Dalton Trans. 40 (2011) 47–55.
M.V. Baker, D.H. Brown, Tungsten compounds with CO or isocyanide, in: D.M.P. Mingos, R.H. Crabtree (Eds.), Comprehensive Organometallic Chemistry III, Comprehensive Organometallic Chemistry III, 5, Elsevier, Oxford, 2007, pp. 597–722.
A.F. Hill, J.M. Malget, A.J.P. White, D.J. Williams, Chem. Commun. (1996) 721–722.
A.F. Hill, J.M. Malget, Chem. Commun. (1996) 1177–1178.
M.J. Frisch, G.W. Trucks, H.B. Schlegel, et al., Gaussian 16, Revision C.01, Wallingford CT, 2019.
T. Lu, F. Chen, J. Comput. Chem. 33 (2012) 580–592. doi: 10.1002/jcc.22885
H.L. Schmider, A.D. Becke, J. Mol. Struct. 527 (2000) 51–61. doi: 10.1016/S0166-1280(00)00477-2
I. Mayer, P. Salvador, Chem. Phys. Lett. 383 (2004) 368–375. doi: 10.1016/j.cplett.2003.11.048
R. Pino-Rios, D. Inostroza, G. Cárdenas-Jirón, W. Tiznado, J. Phys. Chem. A 123 (2019) 10556–10562. doi: 10.1021/acs.jpca.9b07516

Figure 1 Thermal ellipsoid drawing of the molecular structures of 2a, 2a′, 4a, 5a, 6a at 50% probability. H atoms, some of the phenyl substituents, disordered parts, and solvent molecules are omitted for clarity.

Scheme 4 (a) Reaction of PNP-pincer Mo/W-carbyne complexes with LiAlH4. (b) D-Labelling experiments with LiAlD4. (c) Reaction of PNP-pincer Mo-carbyne complex with NaBH4.

Figure 4 Thermal ellipsoid drawing of the molecular structures of 7a, 9 at 50% probability. H atoms, some of the phenyl substituents, disordered parts, and solvent molecules are omitted for clarity. Selected bond lengths (Å) and angles (deg): 7a, Mo-C2 2.301(2) Å, Mo-C3 1.950(2) Å, C1-C2 1.510(3) Å, Mo1-C2-C1 129.17(17)o, C2-Mo1-C3 88.60(9)°. 9, Mo1-Mo2 2.5795(3), Mo1-C1 1.922(3), Mo1-C2 1.910(3), P1- Mo1-P2, 94.81(2)°, P3- Mo1-P4, 122.42(2)°.
Table 1. Selected core bond lengths (Å) and bond angles (deg) of molybdenum and tungsten carbyne complexes.
| Complex | M1-C1 (Å) | M1-C3 (Å) | M1-Cl (Å) | M1-N1 (Å) | C1-C2 (Å) | C1-M1-X1 (deg) | C1-M1-C3 (deg) |
| 2a | 1.808(4) | 1.978(4) | 2.5698(9) | 2.355(3) | 1.432(6) | 173.14(11) | 85.19(18) |
| 2a′ | 1.796(3) | 1.964(3) | 2.5269(7) | 2.464(2) | 1.443(3) | 101.20(8) | 85.02(11) |
| 4a 5a 6a |
1.821(3) 1.817(3) 2.007(3) |
2.023(3) 1.963(3) 2.138(3) |
2.5504(7) 2.5445(7) 2.5482(8) |
- 2.346(2) 2.313(3) |
1.433(4) 1.443(4) 1.455(5) |
169.33(9) 172.55(9) 90.29(10) |
85.37(13) 85.27(12) 38.48(13) |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们





 下载:
下载: