Received Date:
15 April 2025 Accepted Date:
31 July 2025 Revised Date:
30 July 2025 Available Online:
15 July 2026
Abstract:
Excessive and prolonged inflammatory monocyte activity accelerates left ventricular remodeling after myocardial infarction (MI). After myocardial reperfusion, the CCL2/CCR2 axis is considered to be a key chemokine axis that initiates and modulates tissue inflammatory damage. Effectively blocking the CCL2/CCR2 axis can reduce the chemotaxis of inflammatory monocytes to the infarct area, which has important therapeutic value for the repair of myocardium after reperfusion injury (MI/R). Here, we propose a nanoparticle carrying CCL2 silencing short interfering RNA (siRNA), which is prepared by coating a lipid hybrid CCR2-overexprssion macrophage cell membrane on a siRNA-loaded mesoporous silica nanoparticle. The overexpression of CCR2 and adhesion factors on the macrophage membrane improves its targeting ability, directing nanoparticle to the myocardial infarction area of MI/R-induced mice, the overexpress CCR2 in the macrophage cell membrane also adsorb more CCL2. Then by fusing with the cell membrane, siRNA is released into the injured endothelial cells cytoplasm, silencing the expression of CCL2. In vivo administration of the formula blocked CCL2/CCR2 axis, reduce the chemotaxis of inflammatory cells, regulate the immune microenvironment, further decreasing myocardial infarction area and improving cardiac function. Targeted block of CCL2/CCR2 axis represent a new therapeutic intervention for inflammatory disease.
Myocardial infarction (MI), a form of ischemic heart disease, is characterized by high global rates of morbidity and mortality [1]. Timely reperfusion therapy significantly reduces the acute mortality of MI, but it can also trigger a series of events that accelerate and prolong ischemic injury, putting survivors at risk for heart failure [2]. We refer to this as MI-reperfusion injury (MI/R). During the process of MI repair, inflammation is initiated. While timely and appropriate activation of inflammation is essential for effective cardiac repair, excessive, prolonged, or dysregulated inflammation is closely linked to adverse outcomes and complications [3–5]. Clinical studies have also proven that anti-inflammatory treatment is beneficial for the prognosis of patients after MI [6,7]. However, systemic anti-inflammatory treatment has serious side effects, hindering its clinical application [8]. We continue to need to search for new therapeutic targets and targeted delivery strategies.
CC chemokine ligand 2 (CCL2) plays a pivotal role in the regulation of inflammatory processes by binding to its receptor, CC chemokine receptor 2 (CCR2). This interaction is instrumental in drawing in and activating immune cells involved in inflammation [9,10]. Research has shown that CCL2/CCR2 signaling pathway is responsible for mobilizing inflammatory monocytes to the site of injury or infection [11,12]. Monocytes/macrophages would continuously chemotaxis to areas where CCL2 is overexpressed due to their surface CCR2 [11,13]. Further research indicates that CCL2 triggers the secretion of other chemokines during the acute phase of injury, promotes the release of inflammatory factors, and induces the activation and differentiation of more immune cells [14]. Over the long term, CCL2 continuously mediates the infiltration of immune cells, exacerbating damage [15,16]. And the expression and functional role of CCL2 have been extensively reported in various inflammatory diseases, including MI, atherosclerosis, multiple sclerosis, rheumatoid arthritis, stroke, and nephritis [17,18]. Therefore, blocking the CCL2/CCR2 axis maybe a therapeutic target for treating MI and improving post-MI healing. For example, the ablation of CCL2 has demonstrated efficacy in impeding macrophage infiltration into the healing myocardial infarct, diminishing the expression levels of inflammatory cytokines, and mitigating ventricular remodeling processes subsequent to MI [19]. And in other cardiovascular and inflammatory diseases, the genetic deficiency of either CCL2 or CCR2 leads to a significant reduction in inflammation [18–21]. So blocking the CCL2/CCR2 axis maybe a new therapeutic target for the treatment of MI/R. Research efforts are currently focused on small molecules and neutralizing antibodies that target the CCL2/CCR2 signaling axis [22]; however, these agents frequently suffer from limitations in terms of specificity and in vivo therapeutic efficacy [23].
Small interfering RNA, also known as siRNA, hold great potential to selectively inhibit specific genes [24,25]. However, clinical applications of siRNA is limited by the lack of safe delivery technologies, especially for specific tissue needs [26]. Naked RNA has a short half-life in vivo and is easily internalized by endocytosis after entering cells, resulting in most of the siRNA payload being expelled outside the cells, with the remaining siRNA being degraded by lysosomes [27,28]. Moreover, there is a lack of targeting in tissue delivery, and when systemically administered siRNA, the limited target retention necessitates higher siRNA concentrations to maintain efficacy. Additionally, siRNA requires a certain amount of time to take effect, which is not conducive to the alleviation of acute inflammation [29]. Here, we aim to design a targeted delivery platform that uses engineered macrophage membrane-modified fusogenic liposomes encapsulating mesoporous silica nanoparticles (MSNs) to deliver siRNA, thereby blocking the CCL2/CCR2 axis, reducing the infiltration of inflammatory monocytes, modulating the immune microenvironment (IME), and thus improving the repair of MI/R (Fig. S1 in Supporting information). First, we loaded siRNA via calcium silicate sealing using MSNs because of the biological protection and carrying effectiveness of siRNA [30]. In vivo, it can successfully shield siRNAs against deterioration. Second, macrophage migration to infarct area depends on membrane adhesion molecules and chemokine receptors such as macrophage receptor 1 (Mac1), lymphocyte function associated antigen 1 (LFA1), P-selectin glycoprotein ligand 1 (PSGL1), very late antigen 4 (VLA4) and CCR2 [31]. Therefore, by upregulating the expression of CCR2 on the macrophage membrane through viral transfection, nanoparticles (NPs) can not only serve as a “nanonavigator” to chemotaxis to the inflammatory site of MI but also act as a “nanodecoys” to adsorb a large amount of CCL2. Then, the adhesion molecules on the macrophage membrane can also precisely target the MI area. Furthermore, we used a previously documented technique to make fusogenic liposomes [32], the direct transport of liposome inclusions into the cytoplasm through membrane fusion circumvents their endocytosis by recipient cells [33]. This mechanism facilitates the delivery of liposome contents straight into the cytoplasm, thereby bypassing the endocytic pathway of the recipient cell and ensuring efficient membrane fusion-mediated internalization. After the NPs are targeted to the MI injury area, the overexpress CCR2 plays an adhesive role, providing time for the siRNA to take effect. Finally, siRNA carried by the NPs is directly released into the cytoplasm of endothelial cells in the injured area through membrane fusion to silence the production of CCL2. The two work synergistically and are spatially and temporally controllable, blocking the CCL2/CCR2 axis and reducing the chemotaxis of inflammatory monocytes. Overall, this work demonstrated that the spatiotemporal regulation of NP inhibits the CCL2/CCR2 axis, reduces inflammatory cell chemotaxis, and improves MI repair, offering a comprehensive therapeutic approach for the prognosis of MI reperfusion.
As described in Result 1.1 (Supporting information), we successfully constructed the MACCCR2-LM-siRNA. Next, we investigated whether macrophage membranes enhance the targeting of NPs [34], which are then internalized by inflammatory endothelial cells and cardiomyocytes. Inspired by the inherent CCR2-CCL2 signal axis, MACCCR2-LM-siRNA was expected to serve as a “nanonavigator”, chemotaxing to the MI site through the binding of CCR2 to CCL2. The experimental procedure is shown in Result 1.2 (Supporting information). The data showed that under inflammatory conditions, macrophage membrane decoration increased the transport of NPs through the endothelial monolayer and their tendency to reach injured endothelial cells and cardiomyocytes. Additionally, CCR2 overexpression further enhanced chemotaxis to the MI area by binding to CCL2.
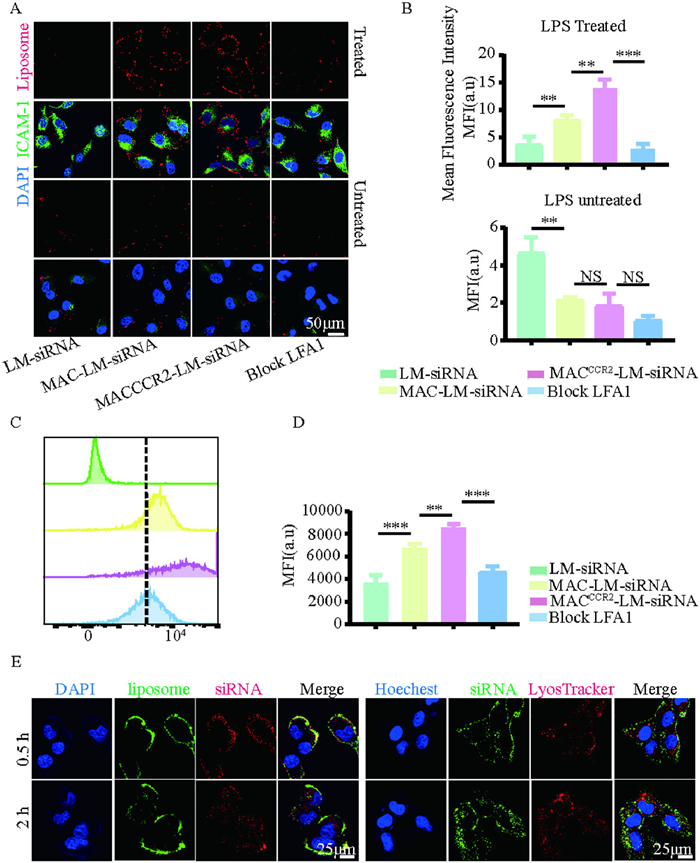
Besides, macrophage influx to inflamed sites largely depends on integrin-dependent adhesion. Mac-1 and LFA-1 binding to intercellular adhesion molecule 1 (ICAM-1) is particularly vital. Notably, ICAM-1 was found to be significantly upregulated in pre-infarcted regions, coinciding with increased leukocyte infiltration [35,36]. Therefore, we chose to validate the adhesion factor LFA1 guide MACCCR2-LM-siRNA to the damaged heart by binding to ICAM-1. We investigated the interaction of MACCCR2-LM-siRNA with both inflamed and non-inflamed endothelial cells using flow cytometry and immunofluorescence analysis in vitro. Both non-inflamed and inflamed human umbilical vein endothelial cells (HUVECs) were exposed to 1,19-dioctadecyl-3,3,39,39-tetramethylindodicarbocyanine perchlorate (DiD)-labeled (on liposome) LM-siRNA, MAC-LM-siRNA or MACCCR2-LM-siRNA for 30 min, respectively. As expected, the binding of MACCCR2-LM-siRNA to the inflamed HUVECs cells were significantly increased (Figs. 1A and B). Similar results were obtained by flow cytometry (Figs. 1C and D). Furthermore, pre-treatment with an anti-LFA-1 antibody markedly diminished the interaction between MACCCR2-LM-siRNA and inflamed HUVECs, while the binding of MACCCR2-LM-siRNA to non-inflammatory HUVECs was not affected (Figs. 1A–D). These results indicated that macrophage membranes on NP can mediate their adhesion to damaged endothelial cells, which in turn facilitates their subsequent migration to injured myocardial tissue.
Figure 1
Figure 1.In vitro targeting of MACCCR2-LM-siRNA. (A) The binding of LM-siRNA, MAC-LM-siRNA, MACCCR2-LM-siRNA, or MACCCR2-LM-siRNA treated with anti-LFA1 antibody to inflamed (or not) HUVECs were imaged after staining with ICAM-1 (nuclei shown in blue, ICAM-1 in green, and DiD-labeled lipids in red). (B) Measurement of the average fluorescence intensity of DiD-labeled lipids across various groups. (C) The interaction of DiD-labeled PBS, LM-siRNA, MACCCR2-LM-siRNA, or LFA1-blocked MACCCR2-LM-siRNA with LPS-pretreated HUVECs was analyzed using flow cytometry and further quantified in (D). (E) Confocal laser scanning microscope (CLSM) images were acquired from HUVECs undergoing inflammation, which were incubated with double-labeled MACCCR2-LM-siRNA for durations of 0.5 and 2 h. The siRNA was tagged with Cy5 (red) and encapsulated within liposomes tagged with DiD (green) on the left panel, whereas on the right panel, the siRNA was labeled with FAM (green). The nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (right), while lysosomes were visualized using Lyso-Tracker Red, and the nuclei were counterstained with Hoechst (left). Results are presented as mean ± SD (n = 3). NS, not significant. **P < 0.01, ***P < 0.001.
Efficient delivery of siRNA into the cytoplasm is a prerequisite for their effectiveness. Therefore, we investigated the cellular internalization pathway of MAC CCR2-LM-siRNA after selective binding to injured myocardium. To monitor MACCCR2-LM-siRNA incorporating dynamic process, we used DiD to label lipids and Cy5 labeled siRNA. When incubated with the HUVECs and the MACCCR2-LM-siRNA 0.5 h, DiD and Cy5 fluorescent signal are mainly distributed in the cell surface, showing distinct localization (Fig. 1E). After incubation 2 h, lipid DiD red fluorescence signal mainly appear in the HUVECs surface, while siRNA Cy5 fluorescent signal evenly dispersed in the cytoplasm (Fig. 1E, left). Next, we determine whether the internalization of siRNA can evade degradation by endosomes/lysosomes, which is crucial for the effective function of siRNA in the cytoplasm later on. We employed FAM to tag the siRNA carried by MACCCR2-LM-siRNA and stained the lysosomes of the recipient cells with the Lyso-Tracker Red. Confocal images revealed that scattered siRNA fluorescence signals were observed in the cytoplasm after 2 h, and lysosome tracing red staining confirmed that there was little endosome/lysosome colocalization (Fig. 1E, right). In conclusion, after binding to recipient cells, the siRNA encapsulated within MACCCR2-LM-siRNA is delivered directly into the intracellular environment through membrane fusion, thereby preventing siRNA degradation by the lysosomal phagocytic system.
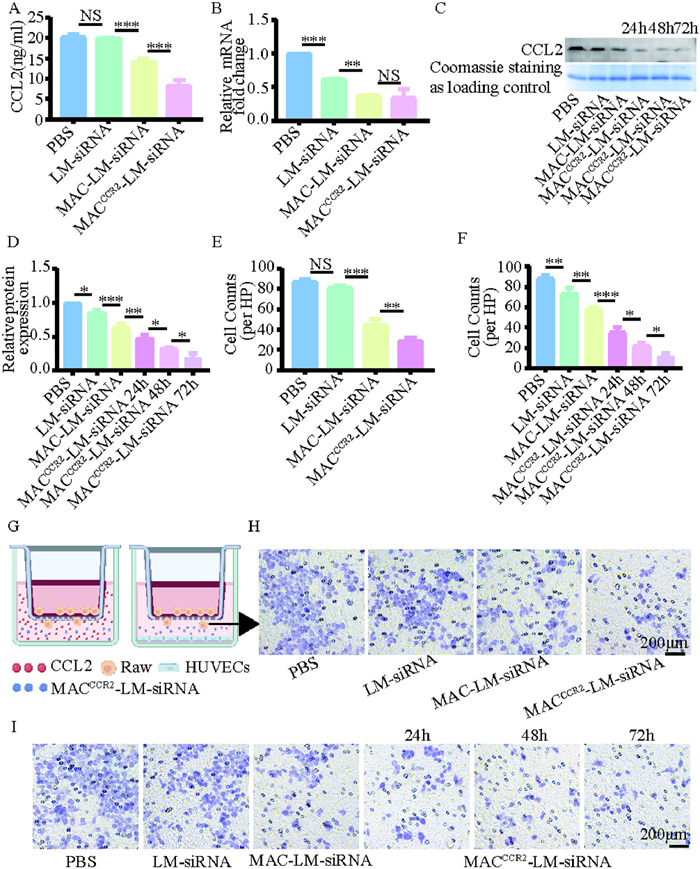
Following verification of MACCCR2-LM-siRNA cytosolic delivery and targeting capability, we evaluated whether NP was able to block the CCL2/CCR2 axis. In the area of MI injury, endothelial cells continuously secrete CCL2 [14], which attracts a large number of inflammatory monocytes expressing CCR2 to the center of inflammation. MACCCR2-LM-siRNA was expected to serve as a “nanodecoys”, alleviating the inflammatory response by adsorbing CCL2. A key requirement for competition with inflammatory cells to adsorb CCL2 is the effective binding of CCR2 to CCL2. Therefore, the binding capacity of MACCCR2-LM-siRNA was investigated to detect the residual content of CCL2 in the supernatant using the enzyme-linked immunosorbent assay (ELISA) kit. We added a medium containing CCL2 factor to the petri dish, then added phosphate-buffered saline (PBS), LM-siRNA, MAC-LM-siRNA, and MACCCR2-LM-siRNA, respectively. The results showed that the absorption of CCL2 in MAC-LM-siRNA and MACCCR2-LM-siRNA groups was significantly increased, and the absorption of CCL2 in MACCCR2-LM-siRNA group was more (Fig. 2A). Next, we assessed the ability of siRNA to silence CCL2. Inflamed HUVECs were treated with PBS, LM-siRNA, MAC-LM-siRNA, MACCCR2-LM-siRNA, separately for 24 h. Then, the RNA was collected to analyze the relative expression of CCL2 by reverse transcription quantitative polymerase chain reaction (RT-qPCR). As shown in Fig. 2B, the level of CCL2 in MAC-LM-siRNA group was significantly lower than that in LM-siRNA group, but there was no significant difference between MAC-LM-siRNA group and MACCCR2-LM-siRNA group. This suggests that siRNA can silence CCL2 production, while overexpression CCR2 has no effect on CCL2 production. In addition, we cultured inflammatory HUVECs with PBS, LM-siRNA, MAC-LM-siRNA, and MACCCR2-LM-siRNA for 24, 48, and 72 h, respectively. Afterward, we collected the media, extracted the proteins, and confirmed the expression level of CCL2 using Western blot (WB). The LM-siRNA, MAC-LM-siRNA group and MACCCR2-LM-siRNA group all can reduce the expression of CCL2, with the MACCCR2-LM-siRNA group showing the most significant reduction (Figs. 2C and D). As well, CCL2 levels decreased notably after 48 and 72 h of treatment in the MACCCR2-LM-siRNA group, indicating that the effect of siRNA was sustained over time (Figs. 2C and D). The results showed that MACCCR2-LM-siRNA not only adsorb CCL2, but also silence the production of CCL2. Together, these mechanisms effectively block the CCL2/CCR2 axis, providing a sustained effect in vitro.
Figure 2
Figure 2.
MACCCR2-LM-siRNA absorbed and silenced CCL2 and reduced the chemotaxis of inflammatory monocytes in vitro. (A) Concentration of CCL2 in the supernatant after CCL2-contianed medium incubated with PBS, LM-siRNA, MAC-LM-siRNA, or MACCCR2-LM-siRNA. (B) RT-qPCR was used to evaluate CCL2 expression levels in HUVECs following various treatments. (C) WB was conducted to assess the protein levels of CCL2, with further quantification in (D) using ImageJ. (E) Chemotaxis was quantitatively analyzed from the images in (H) using ImageJ. (G) Schematic diagram of the established transwell system. (H) A Transwell assay was conducted to assess the migration of inflamed monocytes in response to 20 ng/mL of CCL2, following exposure to various combinations of NPs. (I) Transwell assay of inflamed monocyte towards LPS- pretreated HUVECs produce CCL2 following different treatments and the chemotactic quantitative analysis was further quantified in (F) by ImageJ. Results are presented as mean ± SD (n = 3). P < 0.05, **P < 0.01, ***P < 0.001.
Following cardiac ischemic injury, an excessive systemic influx of inflammatory monocytes can contribute to heart failure and subsequent ischemic complications [37]. The migration of Ly6Chigh inflammatory monocytes during MI/R was mediated through the CCL2/CCR2 axis [38]. To replicate the IME observed in vivo after cardiac ischemic injury, we developed a Transwell model. In this model, LPS-stimulated monocytes were seeded in the upper chamber to simulate inflammatory chemotaxis in vitro (Fig. 2G, left). Added the culture medium containing the CCL2 factor to the lower chamber, then added PBS, LM-siRNA, MAC-LM-siRNA, and MACCCR2-LM-siRNA for 24 h (Fig. 2G, left). Observe the migration of monocytes under a microscope and record the number of cells at the bottom of the upper chamber. As shown in Figs. 2E and H, treatment with LM-siRNA not reduced monocyte migration, but both MAC-LM-siRNA and MACCCR2-LM-siRNA significantly reduced monocyte migration, particularly in the CCR2 overexpressing group. These results demonstrated that MACCCR2-LM-siRNA with high CCR2 expression can effectively adsorb CCL2, thereby inhibiting the invasion of inflammatory monocytes. Then, similarly, the upper chamber was plated with LPS-stimulated monocytes, while inflammatory stimulated HUVECs in the bottom chamber (Fig. 2G, right), PBS, LM-siRNA, MAC-LM-siRNA and MACCCR2-LM-siRNA were added to the bottom chamber and incubated for 24, 48 and 72 h, respectively. As predicted, incubation with LM-siRNA, MAC-LM-siRNA and MACCCR2-LM-siRNA all inhibited monocyte migration, with MACCCR2-LM-siRNA showing the most effective suppression (Figs. 2F and I). Moreover, the number of monocytes continued to decrease after 48 and 72 h of MACCCR2-LM-siRNA treatment (Figs. 2F and I). This implied that MACCCR2-LM-siRNA may reduce recruitment of inflammatory monocytes by cutting off the CCL2/CCR2 axis through adsorbing and silencing the CCL2.
For the subsequent in vivo experiments, mice were used, purchased from Shanghai SLAC Laboratory Animal Co., Ltd. All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of Zhongshan Hospital, Fudan University, and complied with the relevant guidelines outlined in the "Guide for the Care and Use of Laboratory Animals" published by the National Research Council of the Institute for Laboratory Animal Research.
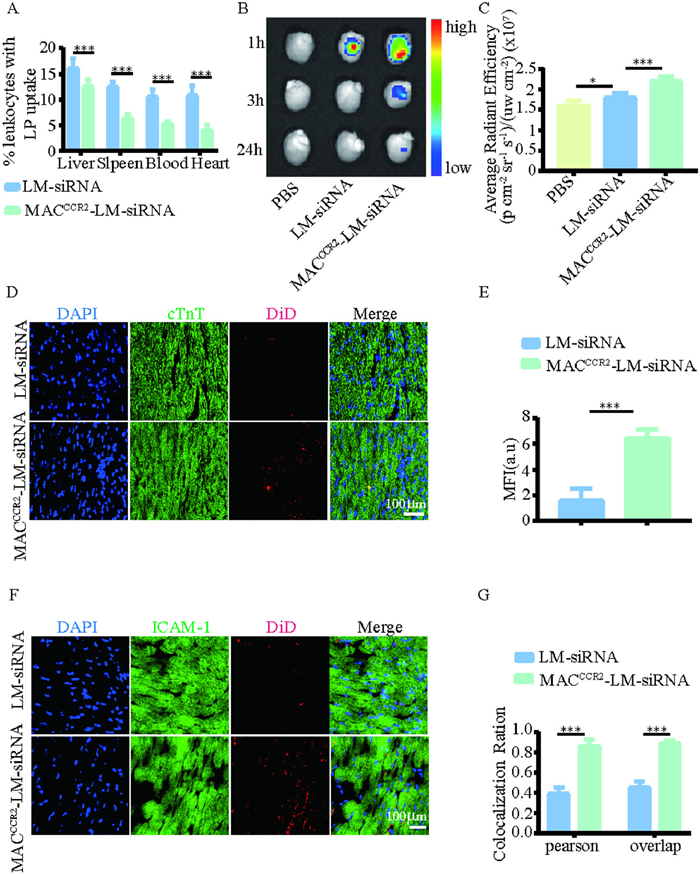
The prolonged circulation of MACCCR2-LM-siRNA is a necessary condition for its targeted capability and clinical potential in vivo. First, the blood circulation profile of MACCCR2-LM-siRNA was studied. Cy5 (on siRNA) labeled LM-siRNA, or MACCCR2-LM-siRNA were administered intravenously to naive mice via tail vein. As shown in Fig. S9 (Supporting information), MACCCR2-LM-siRNA exhibits a longer circulation time than LM-siRNA, which is important for targeting the injured heart (Fig. S9A). In addition, flow cytometry assessed leukocyte uptake of NPs. Six h following intravenous injection of LM-siRNA or MACCCR2-LM-siRNA, mice were euthanized, and samples of the heart, blood, liver, and spleen were harvested and processed for analysis via flow cytometry. The findings revealed that the incorporation of macrophage membranes notably diminished the internalization of NPs by leukocytes, implying that the presence of these membranes could potentially attenuate the clearance of NPs by the mononuclear-phagocyte system (Fig. 3A and Fig. S8 in Supporting information). After that, we constructed a MI/R mice model, and one day later, injected PBS, LM-siRNA, or MACCCR2-LM-siRNA (DiD labeled liposomes), then the heart was extracted at different time points (1, 3, 24 h) and observed through ex vivo macroscopic fluorescence imaging using the in vivo spectral imaging system (IVIS) (Fig. 3B). The findings revealed that the accumulation of MACCCR2-LM-siRNA in damaged cardiac tissue significantly exceeded that of all other groups (Fig. 3C), IVIS imaging was further employed to analyze the distribution of DiD-labeled (on liposome) NPs in other major organs of MI/R-induced mice (Fig. S9B), showing that MACCCR2-LM-siRNA exhibited lower liver uptake compared to LM-siRNA (Fig. S9C). Their distribution in other organs was not significantly different (Fig. S9C). Then, the heart was sectioned into 5 µm sections and stained with cardiac troponin T (cTnT), and it was found that DiD-labeled (on liposome) MACCCR2-LM-siRNA was predominantly localized at the interface of the infarcted and healthy myocardium 3 h after intravenous injection, whereas LM-siRNA showed minimal localization (Figs. 3D and E). In addition, the heart tissue underwent staining for ICAM-1 (Fig. 3F), and subsequent analysis demonstrated a statistically significant colocalization of ICAM-1 with DiD-labeled MACCCR2-LM-siRNA in the MACCCR2-LM-siRNA group, but not in the LM-siRNA group (Fig. 3G, Figs. S9D and E). In summary, these results indicated that MACCCR2-LM-siRNA has a considerable circulation time in the blood circulation of mice, and this NP exhibited preferential targeting potential to the injured heart.
Figure 3
Figure 3.In vivo targeting specificity of MACCCR2-LM-siRNA. (A) Flow cytometry analysis of LM-siRNA or MACCCR2-LM-siRNA uptake by CD45+ leukocytes in the liver, spleen, blood, and heart. (B) IVIS images showing the accumulation of LM- siRNA or MACCCR2-LM-siRNA in the hearts of MI/R mice at three time points (1, 3, and 24 h) after intravenous injection, with data from the first time point further quantified in (C). (D) Fluorescent images and quantitative data of heart sections presents in (E), which have been stained with cTnT to visualize the targeted accumulation of both LM-siRNA and MACCCR2-LM-siRNA (blue, nuclei; green, cTnT; red, DiD-labeled lipid). (F) Fluorescent micrographs of heart tissue sections, following immunostaining with ICAM-1, exhibit the colocalization of ICAM-1 with the respective NPs (red, DiD-labeled lipid; green, ICAM-1). (G) Pearson's correlation and overlap coefficient were calculated following colocalization analysis of the images in (F) using ImageJ. Results are presented as mean ± SD (n = 6). P < 0.05, ***P < 0.001.
After MACCCR2-LM-siRNA was recruited to the area of cardiac injury, their effects on local CCL2 in the infarcted heart were evaluated. We constructed a MI/R mice model and injected PBS, LM-siRNA,MAC-LM-siRNA and MACCCR2-LM-siRNA one day later. Then, we measured serum CCL2 concentrations. As shown in Fig. S10 (Supporting information), serum levels of CCL2 decreased in LM-siRNA, MAC-LM-siRNA and MACCCR2-LM-siRNA groups, the most pronounced reduction was detected in the MACCCR2-LM-siRNA treatment cohort (Fig. S10A).
Additionally, one day after established the MI/R mouse model, PBS, LM-siRNA, MAC-LM-siRNA and MACCCR2-LM-siRNA were injected, and after 24, 48, and 72 h, the damaged heart tissues were collected to determine CCL2 expression using WB. The LM-siRNA, MAC-LM-siRNA and MACCCR2-LM-siRNA groups all reduced CCL2 expression, while the MACCCR2-LM-siRNA groups showed the greatest reduction, and CCL2 levels continued to decrease after 48 and 72 h of treatment with MACCCR2-LM-siRNA (Figs. S10B and C), which is consistent with our in vitro results. Next, we evaluated whether MACCCR2-LM-siRNA could reduce the number of inflammatory Ly6Chigh monocytes in the infarcted heart in MI/R mice. Flow cytometry showed that the lowest number of inflammatory Ly6Chigh monocytes accumulated in the infarcted heart of mice receiving MACCCR2-LM-siRNA treatment, while LM-siRNA and MAC-LM-siRNA also reduced their recruitment, but not as effectively as MACCCR2-LM-siRNA. In addition, Ly6Chigh monocytes continued to decrease 48 and 72 h after MACCCR2-LM-siRNA injection (Figs. S10D and E). These data indicated that MACCCR2-LM-siRNA treatment markedly decreases the infiltration of inflammatory monocytes into the infarcted area. To further demonstrate this, we performed immunohistochemistry and flow cytometry experiments to evaluate the changes in macrophage infiltration at the infarcted myocardium following different NP treatments. As shown in Result 1.3 (Supporting information), the conclusions of both are consistent with those previously obtained. Furthermore, the levels of inflammatory cytokines (IL-1β and TNF-α) in heart homogenates of MI /R mice after 3 days of MACCCR2-LM-siRNA treatment was measured by ELISA to verify the efficiency of MACCCR2-LM-siRNA immuneomodulation. The results showed that MACCCR2-LM-siRNA could significantly reduce the concentrations of IL-1β and TNF-α (Fig. S10F). Moreover, MACCCR2-LM-siRNA could also significantly inhibit the production of IL-1β and TNF-α in serum compared with LM-siRNA and PBS (Fig. S12 in Supporting information). Overall, these findings have effectively proved that MACCCR2-LM-siRNA can by cutting off the CCL2/CCR2 axis to reduce inflammatory monocytes recruited, thereby regulating the IME and alleviating the inflammatory response. To further analyze the effect of NPs on systemic inflammation in MI/R mice, we performed whole-blood transcriptomic profiling of mice treated with MACCCR2-LM-siRNA and untreated MI/R negative control mice. The RNA sequencing results are presented in Result 1.4 (Supporting information).
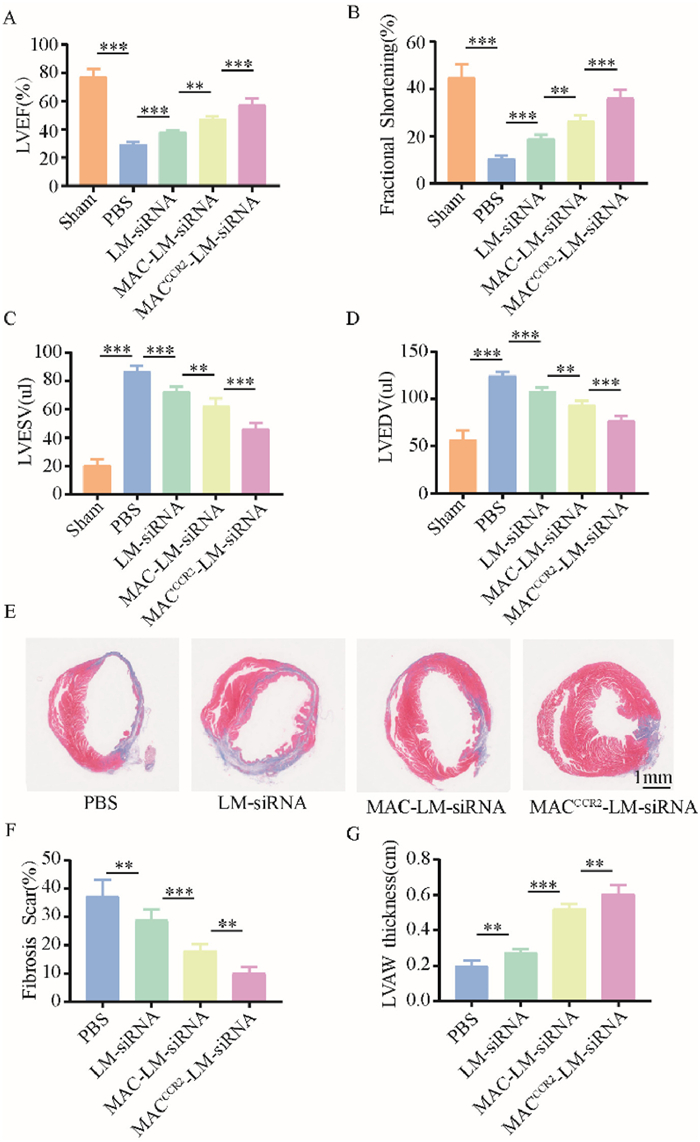
Finally, to evaluate the cardioprotective efficacy of MACCCR2-LM-siRNA, echocardiographic evaluations were conducted on mice subjected to MI/R four weeks post-administration of PBS, LM-siRNA, MAC-LM-siRNA or MACCCR2-LM-siRNA. Echocardiography results revealed an improvement in left ventricular ejection fractions (LVEF) and fraction shortening (FS) in the MACCCR2-LM-siRNA group (Figs. 4A and B). Moreover, the LV end-systolic volume (LVEDV) and LV end-diastolic volume (LVESV) decreased most in the MACCCR2-LM-siRNA group compared to the other groups (Figs. 4C and D). The findings indicated that MACCCR2-LM-siRNA significantly mitigates the decline in cardiac function in mice following MI/R. Subsequently, to evaluate pathological remodeling, cardiac tissue sections were collected, and collagen volume as well as infarct area were analyzed using Masson staining (Fig. 4E). Consistent with expectations, the MACCCR2-LM-siRNA group exhibited the greatest preservation of myocardium and the least fibrosis compared to the control groups (Figs. 4F and G). Overall, these findings confirmed that MACCCR2-LM-siRNA enhanced cardiac function and alleviated fibrosis in MI/R mice. In addition, the evaluation of NP safety is described in Result 1.5 (Supporting information).
Figure 4
Figure 4.
Cardiac Protection Efficiency of MACCCR2-LM-siRNA. (A–D) Four weeks post MI/R injury, LVEF, FS, LVESV and LVEDV were assessed via echocardiography in animals that had been treated with PBS, LM-siRNA, MAC-LM-siRNA or MACCCR2-LM-siRNA. (E) Paraffin sections of hearts subjected to MI/R were stained with Masson's trichrome at four weeks post-treatment to evaluate the histological changes associated with different therapeutic interventions. (F, G) Quantitative evaluation of fibrosis remodeling (F) and left ventricular anterior wall (LVAW) thickness (G) in the Masson’s trichrome staining images. Results are presented as mean ± SD (n = 6). **P < 0.01, ***P < 0.001.
In this study, we developed a CCR2-overexpressing macrophage membrane-coated lipid NP delivery system (MACCCR2-LM-siRNA) that achieves precise active targeting of siRNA to the MI/R injury site. The targeting capability stems mainly from two key features of the macrophage membrane coating. First, adhesion molecules naturally present on the macrophage membrane enable the NP to preferentially home to the inflamed myocardial tissue. Second, the overexpressed CCR2 receptors on the macrophage membrane specifically interact with the chemokine CCL2, which is highly expressed in the infarcted region, facilitating chemotaxis and accumulation of the NPs at the injury site. Importantly, CCR2 overexpression not only enhances targeting but can also adsorb excess CCL2 locally, providing an early blockade of the CCL2/CCR2 axis before siRNA-mediated gene silencing takes effect, thereby better controlling acute inflammation. Following NP accumulation, MACCCR2-LM-siRNA fuses with injured endothelial cell membranes to efficiently deliver siRNA into the cytoplasm, where it silences CCL2 expression. This suppression prevents recruitment of inflammatory monocytes to the damaged myocardial tissue, exerting a potent anti-inflammatory effect. Our results demonstrate that the MACCCR2-LM-siRNA platform integrates precise cell membrane-based targeting, local inflammation clearance, and IME remodeling to promote myocardial repair.
The significance of our work lies in the novel combination of biomimetic targeting via macrophage membrane adhesion factors and CCR2/CCL2 axis modulation with siRNA therapy. The CCR2/CCL2 interaction is well recognized as a pivotal chemokine pathway regulating monocyte recruitment in various inflammatory contexts, including MI/R injury. Recent studies have further elucidated the critical role of this axis in orchestrating inflammatory cell infiltration and tissue remodeling after cardiac injury [39,40]. Our dual-targeting approach enhances delivery specificity and therapeutic efficacy by simultaneously utilizing macrophage membrane adhesion molecules and CCR2-mediated chemotaxis, thereby effectively intercepting this key inflammatory signaling. This strategy minimizes systemic immunosuppression risks by confining the intervention locally to the infarct zone. Additionally, leveraging a natural cell membrane coating improves biocompatibility and immune evasion, offering a promising therapeutic platform for nucleic acid delivery in the highly complex and dynamic inflammatory milieu.
However, there are limitations to consider. The long-term safety and pharmacokinetics of these membrane-coated NPs require rigorous evaluation. Our current findings are based on murine MI/R models, which may not fully capture the complexity of human pathophysiology, potentially affecting clinical translation. Moreover, while targeting the CCL2/CCR2 axis is effective, myocardial reperfusion injury involves multiple inflammatory pathways, suggesting that combining multi-target strategies could yield improved therapeutic outcomes.
In conclusion, our MACCCR2-LM-siRNA platform exploits macrophage membrane adhesion molecules and CCR2 overexpression to actively and specifically target inflamed myocardial tissue, facilitating efficient siRNA delivery to silence CCL2 and reduce inflammation. This study provides important insights into spatiotemporal regulation of inflammation via biomimetic nanomedicine, offering a promising avenue for treating MI/R injury and other inflammatory diseases.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by National Natural Science Foundation of China (Nos. 82170254 and 82370257 to Y. Song, 82070281 to Z. Huang) and Shanghai Rising-Star Program (No. 23QA1401300 to Y. Song). The authors are grateful to Genekinder Medicaltech (Shanghai) Co., Ltd., China for their technical support in bioinformatics analysis.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.111666.
Figure 1In vitro targeting of MACCCR2-LM-siRNA. (A) The binding of LM-siRNA, MAC-LM-siRNA, MACCCR2-LM-siRNA, or MACCCR2-LM-siRNA treated with anti-LFA1 antibody to inflamed (or not) HUVECs were imaged after staining with ICAM-1 (nuclei shown in blue, ICAM-1 in green, and DiD-labeled lipids in red). (B) Measurement of the average fluorescence intensity of DiD-labeled lipids across various groups. (C) The interaction of DiD-labeled PBS, LM-siRNA, MACCCR2-LM-siRNA, or LFA1-blocked MACCCR2-LM-siRNA with LPS-pretreated HUVECs was analyzed using flow cytometry and further quantified in (D). (E) Confocal laser scanning microscope (CLSM) images were acquired from HUVECs undergoing inflammation, which were incubated with double-labeled MACCCR2-LM-siRNA for durations of 0.5 and 2 h. The siRNA was tagged with Cy5 (red) and encapsulated within liposomes tagged with DiD (green) on the left panel, whereas on the right panel, the siRNA was labeled with FAM (green). The nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (right), while lysosomes were visualized using Lyso-Tracker Red, and the nuclei were counterstained with Hoechst (left). Results are presented as mean ± SD (n = 3). NS, not significant. **P < 0.01, ***P < 0.001.
Figure 2
MACCCR2-LM-siRNA absorbed and silenced CCL2 and reduced the chemotaxis of inflammatory monocytes in vitro. (A) Concentration of CCL2 in the supernatant after CCL2-contianed medium incubated with PBS, LM-siRNA, MAC-LM-siRNA, or MACCCR2-LM-siRNA. (B) RT-qPCR was used to evaluate CCL2 expression levels in HUVECs following various treatments. (C) WB was conducted to assess the protein levels of CCL2, with further quantification in (D) using ImageJ. (E) Chemotaxis was quantitatively analyzed from the images in (H) using ImageJ. (G) Schematic diagram of the established transwell system. (H) A Transwell assay was conducted to assess the migration of inflamed monocytes in response to 20 ng/mL of CCL2, following exposure to various combinations of NPs. (I) Transwell assay of inflamed monocyte towards LPS- pretreated HUVECs produce CCL2 following different treatments and the chemotactic quantitative analysis was further quantified in (F) by ImageJ. Results are presented as mean ± SD (n = 3). P < 0.05, **P < 0.01, ***P < 0.001.
Figure 3In vivo targeting specificity of MACCCR2-LM-siRNA. (A) Flow cytometry analysis of LM-siRNA or MACCCR2-LM-siRNA uptake by CD45+ leukocytes in the liver, spleen, blood, and heart. (B) IVIS images showing the accumulation of LM- siRNA or MACCCR2-LM-siRNA in the hearts of MI/R mice at three time points (1, 3, and 24 h) after intravenous injection, with data from the first time point further quantified in (C). (D) Fluorescent images and quantitative data of heart sections presents in (E), which have been stained with cTnT to visualize the targeted accumulation of both LM-siRNA and MACCCR2-LM-siRNA (blue, nuclei; green, cTnT; red, DiD-labeled lipid). (F) Fluorescent micrographs of heart tissue sections, following immunostaining with ICAM-1, exhibit the colocalization of ICAM-1 with the respective NPs (red, DiD-labeled lipid; green, ICAM-1). (G) Pearson's correlation and overlap coefficient were calculated following colocalization analysis of the images in (F) using ImageJ. Results are presented as mean ± SD (n = 6). P < 0.05, ***P < 0.001.
Figure 4
Cardiac Protection Efficiency of MACCCR2-LM-siRNA. (A–D) Four weeks post MI/R injury, LVEF, FS, LVESV and LVEDV were assessed via echocardiography in animals that had been treated with PBS, LM-siRNA, MAC-LM-siRNA or MACCCR2-LM-siRNA. (E) Paraffin sections of hearts subjected to MI/R were stained with Masson's trichrome at four weeks post-treatment to evaluate the histological changes associated with different therapeutic interventions. (F, G) Quantitative evaluation of fibrosis remodeling (F) and left ventricular anterior wall (LVAW) thickness (G) in the Masson’s trichrome staining images. Results are presented as mean ± SD (n = 6). **P < 0.01, ***P < 0.001.

 DownLoad:
DownLoad:

 下载:
下载:





 下载:
下载:
