College of Pharmacy, Dali University, Dali 671000, China
b.
College of Smart Energy, Shanghai Jiao Tong University, Shanghai 200240, China
c.
School of Materials Science and Engineering, Tianjin University, Tianjin 300350, China
d.
State Key Laboratory of Electrical Insulation and Power Equipment, Center of Nanomaterials for Renewable Energy, School of Electrical Engineering, Xi’an Jiaotong University, Xi’an 710049, China
e.
SINOPEC (Beijing) Research Institute of Chemical Industry Co., Ltd., Beijing 100013, China
f.
Guangzhou Key Laboratory for Surface Chemistry of Energy Materials, New Energy Institute, School of Environment and Energy, South China University of Technology, Guangzhou 510006, China
g.
Tsinghua University, Institute of Nuclear and New Energy Technology, Beijing 100084, China
h.
Institute of Nanoscience and Nanotechnology, College of Physical Science and Technology, Central China Normal University, Wuhan 430079, China
i.
Department of Chemical, Materials and Production Engineering, University of Naples Federico Ⅱ, Piazzale Tecchio 80, 80125 Naples, Italy
j.
Saint Petersburg University, St. Petersburg State University, 7/9 Universitetskaya Nab., St. Petersburg 199034, Russia
k.
Hydrogen Energy Laboratory, Ioffe Institute, St. Petersburg 194021, Russia
l.
College of Chemistry and Chemical Engineering, Shanxi Key Laboratory of Gas Energy Efficient and Clean Utilization, Taiyuan University of Technology, Taiyuan 030024, China
m.
Institute of Energy Materials Science, University of Shanghai for Science and Technology, Shanghai 200093, China
n.
Beijing Key Laboratory of Chemical Power Source and Green Catalysis, School of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing 100081, China
o.
School of Chemistry, Tiangong University, Tianjin 300387, China
Received Date:
12 December 2024 Accepted Date:
30 July 2025 Revised Date:
17 February 2025 Available Online:
15 January 2026
Abstract:
The catalytic transferred of small molecules into high-value chemical products in green methods are highly perused, and has obtained huge attention. In this field, great progress has been achieved during the past five years. Followed by the roadmap (Chinese Chemical Letters, 2019, 30, 2089–2109) written by us before five years, we think that it should be updated to give more insights in this field. Thus, we write the present roadmap based on the fast changed background. In this roadmap, oxygen and carbon dioxide reduction reactions (including at high temperature), photocatalytic hydrogen generation and carbon dioxide reduction reactions, (photo)electrocatalytic reduction of O2 to H2O2 and NH3 generated from N2 are discussed. The progress and challenges in above catalytic processes are given. We believe this manuscript will give the researchers more suggestions and help them to obtain useful information in this field.
Photocatalytic CO2 reduction reactions (CO2RR), which could directly utilize solar energy to achieve the synthesis of fuels and value-added chemicals, served as one of the most significant parts of the artificial photosynthesis technology [1]. Nevertheless, due to the thermodynamic requirements of the direct activation of CO2 (the CB position of the catalysts should be more negative than −1.5 eV, vs. RHE, pH 0), the CO2RR was more inclined towards the CO2 hydrogenation process with lower thermodynamic requirements (proton derived from the activation of H2O), which made the synergistic migration of "protons-carriers" as one of the crucial steps in photocatalytic CO2RR. Therefore, as schematically exhibited in Fig. 1 [2], some issues still need to be paid attention to (1) The low efficiency of carrier separation, which will not be favorable for the charge separation and the activation of protons in H2O effectively; (2) The competitive relationship between proton reduction for hydrogen production and CO2RR, significantly increasing the consumption of photogenerated electrons in this reaction; (3) The CO2 reduction in H2O or with H2O vapor reaction involves multiple tandem reaction processes, and the migration and transformation of carriers need to simultaneously span both temporal and spatial scales. Based on the abovementioned, the current research primarily concentrated on how to enhance carrier separation and thereby improve the rate and efficiency of proton dissociation and activation. Among these, the design of heterojunction catalysts, the loading of cocatalysts, the addition of hole scavengers, etc. have enabled the efficient conversion of CO2 to produce CO, CH4, C2H6, and even C3+ products [3–6]. In recent years, studies have also focused on the design of photocatalytic reactors, a 100 m2-scale photocatalytic mini-plant was equipped with the CO2 methanator for the production of CH4 by the sunlight-driven photocatalysis method [7].
Figure 1
Figure 1.
(a) Photocatalytic processes on semiconductor nanocrystals involving photoexcitation and formation of electron-hole pair in the nanocrystal. (b) Energy diagram of the same process for a semiconductor. (c) Energy loss mechanisms in solar fuel formation: ① below band gap photons, ② thermalization, ③ charge recombination, ④ overpotential, ⑤ back reaction, ⑥ separation and product extraction losses. (d) Semiconductor band alignment with possible levels of self-oxidation redox potential (left) and with two different hole scavengers (HS1 and HS2, shown on the right in blue and red), representing two potential energy balance scenarios. Item ⑦ represents a loss of stored energy due to the use of a sacrificial agent. Reproduced with permission [2]. Copyright 2018, American Chemical Society.
The rate-limiting step of photocatalytic CO2RR lies in the four-electron process of water oxidation, which is a sluggish kinetic process and will lead to the low efficiency of photocatalytic CO2RR and poor selectivity of products. The CO2 photoreduction involves multiple tandem catalytic processes, and the migration and transformation of carriers coupled with proton has emerged as one of the significant challenges: Firstly, at the mechanistic level, it could be separated into three aspects: the efficient separation of charge carriers in the bulk phase (or near the surface) and surface of the catalysts; the generation and activation of hydrogen species; the migration and transformation of charge-coupled hydrogen species. Current studies proposed that strategies, like S-scheme heterojunction construction, the built-in electric field regulation, and the control of microstructure distortion, could effectively enhance the separation efficiency of charge carriers, thereby achieving efficient activation and dissociation of protons from H2O [8–11]. Nevertheless, rare attention has been devoted to the migration and transformation process of carriers coupled with protons.
Due to the low conversion efficiency of photocatalytic CO2RR, it is necessary to eliminate interference factors that could affect the products during the experiment. Firstly, regarding the purity of the CO2 gas used for the reaction: we employed gases with a purity of 99.995% and discovered that the gas contains impurity gases such as CH4, C2H4, C2H6, and CO. Moreover, during the reaction, H2 produced from water splitting could react with CO to generate hydrocarbon products. Secondly, interference from the reactor sealing ring exists. Under illumination, the sealing ring could be oxidized and decomposited, and its products include CO, CH4, etc. Thirdly, the interference of the organic solvents from the catalyst preparation process. Furthermore, to enhance the separation efficiency of charge carriers, organic sacrificial agents are often added. During the reaction, the sacrificial agents will tend to decompose and generate hydrocarbon products as well, which will impact the evaluation of the true CO2 reduction performance and result in false positive outcomes. Although most of the studies have fulfilled the 13CO2 isotope tracing experiment to prove the products originated from the CO2 photoreduction, however, the ratio of hydrocarbon products generated by the sacrificial agents to those from photocatalytic reduction of CO2 is difficult to determine. Therefore, to evaluate the true CO2 reduction performance, it is necessary to eliminate possible false positive results in the experiment.
1.3
Advances in science and technology to meet challenges
To enhance the performance of CO2RR, novel, and effective photocatalysts have been developed for enhanced carrier separation, such as the utilization of "junction" engineering (including heterojunctions, p-n junctions, facet junctions, and homojunction), regulation of the built-in electric field, and construction of single-atom active sites [8,12]. The advancements in the above studies have improved the efficiency of CO2 reduction and expanded the generation system of CO2 products. CO, CH4, C2H6, and even C3+ products could be obtained via different pathways, as exhibited in Fig. 2 [13–15]. Additionally, the development of advanced instrumental techniques, such as the combined use of SEM-SKPFM, ultrafast transient absorption, and in situ infrared spectroscopy, has provided a supporting platform for revealing the mechanisms of photocatalytic CO2 reduction.
Figure 2
Figure 2.
(a) Mechanistic pathways of CO2 reduction to commonly observed C1 and C2 products. Reproduced with permission [13]. Copyright 2022, American Chemical Society. (b) Mechanism illustration for photocatalytic conversion CO2 to CH3CH2COOH on Cu-ZnS. Reproduced with permission [14]. Copyright 2024, Wiley. (c) Free-energy profile of the CO2RR pathway on CuN2O2 sites in Cu-MOF and Structural models of the Gibbs free energy calculations for CO2 reduction on Cu-MOF. Reproduced with permission [15]. Copyright 2024, Wiley-VCH.
Furthermore, the application of artificial intelligence in screening catalysts also offers the possibility of discovering highly efficient photocatalytic CO2 reduction materials in the future. The database of the catalysts with photocatalytic CO2 properties could be established and the AI will help researchers to find the possible catalysts with an efficient CO2 conversion rate quickly, even the CO2RR selectivity.
1.4
Concluding remarks and prospects
Photocatalytic CO2RR remains a major challenge in the interdisciplinary fields of catalytic chemistry, materials science, and even energy science, offering a direct energy utilization and cost-effective route for artificial photosynthesis as well as possessing vast potential for industrial and environmental applications. However, the underutilization of solar energy spectrum, low CO2 conversion rate and product selectivity have limited the development of solar CO2 reduction technology. In addition, the reaction path and mass transfer process of photocatalytic CO2 reduction are extremely complicated, and the future industrialization of photocatalytic CO2RR will face more scientific and engineering complexities, such as high product efficiency and high selectivity, product separation, and large-scale photocatalytic device design and optimization. For example, the reaction pathway and catalytic mechanism of photocatalytic CO2RR are still unclear, and in-situ analysis, numerical simulation, and density functional theory are needed to further understand the catalytic mechanism of photocatalytic CO2RR and the structural reorganization of intermediates/products in the future. Although the efficiency and selectivity of the photocatalytic CO2 reduction products have been improved, the current efficiency, selectivity, stability and product separation cost are still far from meeting the industrialization standard, and the improvement of the product selectivity can greatly reduce the cost of product separation in the future industrialization of photocatalytic CO2RR. In addition, the photocatalytic CO2RR efficiency has been improved but still relatively low, limited by the fact that most of the reported photocatalytic materials can only utilize the ultraviolet and visible light regions, and the utilization of infrared light and its photothermal synergistic catalytic mechanism have not been thoroughly studied. In the future, it is expected to construct a new type of photothermal coupling catalytic system without external heat source through solar energy concentrating technology, which can not only realize the photochemical conversion of high-energy photons, but also make full use of low-energy photons to increase the local temperature of solid-liquid reaction and thus reduce the activation energy of the reaction to promote the charge transfer and thermal catalytic activity, and realize the photon-phonon coupling matching and the graded utilization of energy in a real sense. Therefore, the future development of photocatalytic CO2RR not only needs to consider the high efficiency and high selectivity of CO2 reduction products, but also needs to optimize the coupling match between material conversion and energy transfer on the temporal and spatial scales, and reduce the energy consumption of the reaction, so as to pave the way for the future industrialized large-scale solar-powered CO2 reduction applications.
2.
Electrocatalytic CO2 reduction
2.1
Status
The dependence of fossil fuels has led to excessive emissions of CO2, causing irreversible damage to the climate and environment, including droughts and rising sea levels due to global warming, as well as an energy crisis [16]. Therefore, an ocean of efforts have been made to explore CO2 capture and utilization technologies. Electrocatalytic CO2RR offers a promising strategy to convert CO2 into chemicals and fuels, with advantages of economic feasibility, operability, and environmental friendliness.
Electrocatalytic CO2RR occurs through multiple pathways, with several electron-proton coupled transfer steps. Therefore, the reaction products of electrocatalytic CO2RR are diverse. The electrocatalytic CO2RR products could be classified into C1 products (CO, CH4, and CH3OH) and multi-carbon (C2+) products (C2H4, C2H5OH, CH3COOH, etc.) [17]. Multi-carbon products are more attractive due to their excellent energy density and high economic value. However, the referred multiple electron/proton transfer steps result in wide distribution of the products, the ECRR faces high energy cost and low selectivity. Therefore, it is challenging but significant to explore suitable method to decrease the reaction energy barrier and improve the selectivity towards specific products [18,19]. In the past decades, numerous efforts have been made from different aspects of the electrocatalysis, such as accelerating the activation of CO2, promoting the binding stability of key intermediates, as well as unrevealing the relationship between electrocatalytic CO2RR performance and the overpotentials [20,21]. With the deepening understanding to the electrocatalytic CO2RR process, peoples also demonstrated that the design of new type electrolytic cells can facilitate the transfer of CO2 to the active catalyst and decreasing the mass transfer resistance and Ohm resistance in the traditional H-type cell [22]. But it worth to notice that, all of the mentioned factors will be in low efficiency if the heart part of electrocatalytic CO2RR, electrode materials, themselves are improper.
2.2
Current and future challenges
In the past decades, Cu catalyst have attracted numerous studies because of the interesting product distributions including both hydrocarbons (methane, ethylene) and alcohols (ethanol and propanol) [23–25]. However, although progress has been made in laboratory scale, Cu-catalyzed CO2RR still face significant challenges for industrial applications due to the issues such as highly energy consumption, catalyst deactivation and uneven product distribution [26]. The core task is how to improve the selectivity towards single product of Cu catalyst to meet the economic viability of electrocatalytic CO2RR through reasonable catalyst design. In addition, towards C2+ products, the promotion of C–C coupling reaction is the key in electrocatalytic CO2RR, which asks for the modulation of catalyst electronic structure to activate CO2 and trigger the conversion of CO2 to deep reduction products. So far, the selectivity of C2+ products could reach over 80%, but it is still rare to have a single multi-carbon product selectivity exceeding 80% [27].
Current density and reaction stability are core indexes in evaluating the application potential of a material in industrial electrocatalytic CO2RR. The ideal industrial catalyst should have a partial current density of > 300 mA/cm2 for specific products and excellent stability [28]. Therefore, in order to meet the demand of industrial application, it is important to change the interfacial electron transfer and mass transfer resistance and thus improving the energy efficiency by adjusting the surface micro-environment of the catalyst.
2.3
Advances in science and technology to meet challenges
An important advancement made in the last decade is the multi-scale size and morphology control of Cu catalyst, including unsupported Cu (such as Cu nanoparticles and Cu single crystals) and supported Cu catalysts (such as Cu-MOF and single atomic Cu) (Fig. 3). The well-defined Cu nanocrystals provide an ideal model for revealing selectivity rules in electrocatalytic CO2RR. Based on experimental and theoretical studies, the products selectivity on Cu(100), Cu(111), Cu(110) facets and on the step sites have been revealed [29]. Nevertheless, only partial Cu nanocrystals (such as cubes, octahedra, spheres, and nanorods) can be obtained through the current synthetic methods, the fabrication of Cu catalysts exposing high index facets is still a challenge. Despite this, it is yet highly desirable to guide the regulation of product selectivity by using the results of theoretical calculations and studies on simple low-index crystal faces as a reference. Compare with unsupported Cu, supported Cu usually depicted highly dispersed active sites guarantees an ultra-high atomic utilization and outstanding reaction activity. For example, Cu-based MOFs have exhibited devisable textural, high dispersed reaction sites, and high adsorption capacity for reaction intermediates [30]. Moreover, carbon-based catalysts obtained by using Cu-MOFs or their derivatives as precursors can inherit the good catalytic activity of Cu-MOF [31]. These supported Cu catalysts have shown large potential in electrocatalytic CO2RR.
Figure 3
Figure 3.
Scheme of the design of Cu catalysts towards tuning the product selectivity of electrocatalytic CO2RR.
Moreover, centered on promoting the C–C coupling reaction, a crucial way is to reduce the *CO dimerization energy barrier by adjusting the adsorption strength and state of *CO intermediates on the catalyst surface. For example, by introducing a thin interface layer of Cu2O/CeO2, our group presented a metal/metal oxide synthetic strategy to enlarge the lattice spacing of Cu2O, which enhanced the adsorption of *CO intermediates and promoted *CO dimerization (Fig. 4a). A C2+ product faradaic efficiency of 76.4% was achieved at a high current density of 300 mA/cm2 [32]. In addition, Zhang et al. designed Cu nanosheets with nano-defects as electrocatalysts for electrocatalytic CO2RR to produce C2H4. The nano-defects can enhance the adsorption, enrichment, and confinement of *CO on the electrocatalyst, thereby promoting C–C coupling and obtaining an ethylene selectivity of 83.2% (Fig. 4b) [33]. Park et al. demonstrated that oxygen plasma-assisted nitrogen doping can improve the catalytic activity and durability of CuO for electrocatalytic CO2RR, which is because the highly active N radicals in the N2 plasma can increase the binding energy between Cu and CO intermediate (Fig. 4c) [34].
Figure 4
Figure 4.
(a) Schematic illustration of Cu-CeO2 promotes C2+ production. Reproduced with permission [32]. Copyright 2022, American Chemical Society. (b) Schematic illustration of nano-defects promotes C2+ production. Reproduced with permission [33]. Copyright 2020, American Chemical Society. (c) Schematic illustration of increased CO binding energy favorable for C–C coupling on ON-CuO. Reproduced with permission [34]. Copyright 2023, American Chemical Society.
Many modulation strategies, such as elemental doping and alloying, have also been developed in regulating the electrocatalytic CO2RR performance of Cu catalysts. All these factors can manipulate the adsorption energy of *H, *CO, as well as the C–C coupling process, and finally change the electrocatalytic CO2RR performance of Cu materials [35–37]. For example, the addition of second-phase elements can adjust the d-band center of Cu, thereby adjusting the interaction strength between Cu and intermediates, accelerating the kinetics of C–C coupling and further inducing the catalytic reaction to proceed in a specific direction. Qiao et al. classified the metals that affect the adsorption, hydrogenation or coupling process of the reaction intermediates when alloyed with Cu into four groups based on the affinity of the second-phase elements for H and O (Fig. 5) [35]. In addition, our group successfully induced the transformation of the Cu crystal face from (111) to (100) by introducing phosphate ligands, which changed the coordination environment of Cu atoms on the catalyst surface and promoted the production of ethylene. The development in fabrication and modification of Cu catalysts pave solid way for understanding the electrocatalytic CO2RR process from different scale and effectively guiding the design of catalysts targeting desired products.
Figure 5
Figure 5.
(a-d) Second phase metals are grouped according to various descriptors. Reproduced with permission [35]. Copyright 2018, Elsevier.
The precise and efficient catalyst design depends the accurate understanding of the underlying reaction mechanisms. Fortunately, researchers have realized the role of high-quality experiments in accurately revealing the reaction mechanism and the importance of reaction-mechanism-based catalyst design in promoting the generation of target product, which has also excited the development of DFT, operando electron microscopy method, as well as in-situ methods. The combination of operando electron microscopy and in-situ analytical tools enable the identification of the intermediates and the analysis to the reconstruction of catalysts. For example, in-situ X-ray photoelectron spectroscopy (XPS) and in-situ Raman are useful in probing the generation and evolution of key intermediates. Especially, in the first few minutes of reaction, the in-situ characterizations provide direct evidence for the change of surface chemical states of Cu species. In addition, in-situ Fourier transform infrared spectroscopy (FTIR) is another important method in analyzing the adsorption and activation of a reactant molecule and tracing reaction intermediates [38]. It worth noticing that, with the rapid advancement of computer technique, DFT will become another powerful tool in clarifying the reaction mechanism and revealing structure-property relationships.
2.4
Concluding remarks and prospects
Electrocatalytic CO2RR provides a new strategy to perform effective utilization of CO2 and produce high value-added chemicals. Although important progress has been made in this field, especially in catalyst design, reaction mechanism understanding and electrolyzer development, there are still many challenges. To achieve industrial-scale application, future research should focus on improving the activity and stability of catalysts, optimizing reaction conditions, as well as promoting reactor design and system integration [39]. In addition, it is also feasible to enhance the activity and long-term stability by adjusting the composition of the catalyst (e.g., increasing or decreasing the content of specific elements) or adopting new synthesis methods (e.g., sol-gel method instead of co-precipitation method), as well as optimizing the reaction conditions (e.g., reaction temperature, pressure, and reactant concentration) [40]. On that same note, exploration to the possibility of realizing efficient catalytic conversion under milder conditions (e.g., low-temperature or low-pressure processes with reduced energy consumption) is of significant economic and practical meaning. With the continuous development of materials science, characterization technology and theoretical calculation, CO2 reduction technology is expected to achieve major breakthroughs in the next decade. Especially driven by carbon neutrality policies and market demand, electrocatalytic CO2 reduction technology has the potential to become an important means to reduce CO2 emissions and achieve sustainable chemical production.
3.
High-temperature electrocatalytic reduction of CO2
3.1
Status
The selective conversion of CO2 to valuable chemicals provides a promising approach to realizing a sustainable, carbon-neutral economy. In recent years, significant progress has been made toward developing electrochemical techniques to achieve efficient and stable CO2RR. Among various electrocatalytic CO2RR devices, solid oxide electrolysis cells (SOECs) operating at high temperatures (> 600 ℃) have attracted increasing interest. Using SOECs, CO2 can be directly electrolyzed into CO (Eq. 1) or co-electrolyzed with steam (Eq. 2) to produce syngas (the mixture of CO and H2). The produced syngas can be further converted into high-value carbon-containing products, such as methane, methanol, and ethylene (Fig. 6). The high-temperature nature of SOEC allows for a portion of the reaction energy of CO2 electrolysis to be supplied by thermal energy, which lowers the requirement for electrical energy compared to low-temperature electrolysis. In addition, SOEC is a more reliable and effective option for large-scale carbon conversion and energy applications than liquid-phase catalysis, as it provides faster reaction kinetics on a more advantageous gas-solid interface for molecular processes. Therefore, high temperature electrocatalytic reduction CO2 using SOECs exhibited significant advantages in conversion rate, current density, and Faraday efficiency [41–43].
Despite the broad application prospects of high-temperature CO2 electrolysis or co-electrolysis, it still possesses a relatively high technical threshold. First, it is critical to develop highly active and stable electrodes materials for SOECs. The molecular structure of CO2 is stable due to its strong double bonds (C=O) and linear structure, which requires relatively higher activation energy to the fracture of two π34 bonds during the electrolysis process. Therefore, the electrode materials need to present high inherent activity for activating C=O bond to lower energy consumption for CO2RR. Meanwhile, the CO produced during CO2 electrolysis can be over-reduced to carbon, which could cover the electrode surface and lead to the blocking of active sites on the surface and the increase of electrode resistance. Therefore, in addition to high activity, coking resistance is another criterion for SOEC electrode materials to realize steady CO2 electrolysis. For the most commonly used Ni-YSZ electrodes, performance degradation was proved to originate by not only coking but also Ni migration and agglomeration at cathode/electrolyte interface, which leads to the formation of a dense Ni layer or a dense YSZ layer and increases the impedance [42]. At present, electrode materials that can perfectly meet the requirements of electrocatalytic activity and long-term operation stability are still lacking.
Another serious problem that influences the long-term stability of SOECs is interfacial delamination, which is fundamentally initiated by the structural defects of electrodes. SOEC electrodes typically possess sponge-like morphology with irregular/closed pores, which is obstructive to gas release. Under high electrolysis current, the sponge-like morphology may contribute to the accumulation of oxygen pressure at the electrode/electrolyte interface, especially under elevated current densities [44]. Finally, the large-scale application of high-temperature electrochemical CO2RR requires several technological achievements. These include large-scale CO2 capture and purification technology, heat management systems for high-temperature SOEC stacks, and highly efficient separation technology for obtaining single-component outlet gas [42]. In a word, the industrial applications of CO2RR rely on the comprehensive development of materials, stacks, and supporting systems.
3.3
Advances in science and technology to meet challenges
Perovskite-based oxides were considered as potential alternative electrode materials for the nickel-based materials. However, perovskite-based oxides normally encounter difficulties such as poor catalytic activity towards CO2 reduction. By annealing perovskite-based electrode in a reductive atmosphere or applying cathodic polarization, the transition metal cations at B-site (including Ni, Mn, Co, Fe, Cr, Cu, etc.) can be exsolved to the surface in the form of metallic nanoparticles or simple oxides [45,46]. These exsolved nanoparticles possess high electrochemical activity and show high stability under high temperatures. Meanwhile, the exsolved nanoparticles can promote the electrochemical reduction kinetics of CO2 and inhibit the carbon deposition process through rapid electron exchange. Another commonly used strategy to enhance the catalytic activity of existing electrodes is surface modification. By transformation between Ce3+ and Ce4+, for instance, CeO2-based material can participate in a carbon redox reaction to prevent solid carbon deposition, substantially modifying CO2 reduction pathways and preventing carbon deposition [47–49].
To enhance the structure stability of SOEC cells, electrodes with various micro-structures were developed to reduce gas diffusion resistance. For instance, a microchannel-structured Gd0.1Ce0.9O2-δ (GDC) framework was fabricated by means of a freezing-casting method and obtained a Sm0.5Sr0.5CoO3-Gd0.1Ce0.9O2–δ (SSC-GDC) porous anode via an infiltration-sintering method, leading to an extremely low polarization resistance of only 0.05 Ω cm2 at 600 ℃ [50]. Based on this, a honeycomb-structured LSC-YSZ microchannel anode with regular morphology was subsequently designed, which possessed only 0.0094 Ω cm2 polarization resistance at 800 ℃, and the corresponding single cell could operate at a large current density of 5.9 A/cm2 with no obvious performance degradation during 6-hour operation [44,51]. An LSF-GDC|GDC-YSZ|Ni-GDC electrolysis cell with a microchannel anode was also prepared and applied in pure CO2 electrolysis, which demonstrated good stability under a large current density of 2.5 A/cm2 at 800 ℃, and no performance degradation was observed within 124-h operation [52]. These results provide guidance for the rational design of electrodes with a fine-designed micro-structure for high-performance and sustainable CO2RR.
In addition to developing novel materials and structures, researchers have also expanded the application of SOEC technology for the conversion of other small molecules, such as alkane, N2, and NOx (Fig. 7). The produced molecules such as CO, NH3, and olefine, can further went through oxidation, polymerization, or other conversion to obtain high-value products such as fertilizers, synthetic resins, and synthetic rubbers [41]. From an energy-use standpoint, by integrating SOECs with exothermic reactions, the heat generated can be utilized for the SOEC, further optimizing energy utilization and having a significant synergistic effect. Developing novel reaction pathway-based SOECs can potentially provide an alternative approach for chemical products derived from the petroleum industry [43].
Figure 7
Figure 7.
Schematic diagram of potential chemical processes based on SOECs.
High-temperature electrocatalytic reduction of CO2 based on SOECs holds great importance for realizing a sustainable, carbon-neutral economy. While great progress has been made at the laboratory and pilot stages, the large-scale industrial application of this technology still needs further development. The fundamental challenges include how to improve both the activity and durability of the SOEC cathode materials. At the current stage, while Ni-YSZ is still one of the most extensively researched SOEC cathode materials for CO2RR, perovskite-based materials have been proven to be promising candidates. In the future, the application of state-of-the-art in situ characterization methods and theoretical tools is needed to provide guidance for the rational design of novel electrode materials and micro-structure to enable high-performance CO2 electrolysis. Meanwhile, the economic and technical feasibility of SOEC-based CO2RR technology needs to be systematically evaluated, thereby providing guidelines for the establishment of a complete CO2 conversion and utilization industrial chain.
4.
Photocatalytic hydrogen production
4.1
Status
Hydrogen has emerged as a versatile energy source to combat energy shortages and environmental crises due to its high superior gravimetric energy density, cleanliness, and zero emissions during combustion. Harvesting hydrogen using solar energy is an attractive strategy for reducing carbon footprint in terminal consumption [53]. Photocatalysis, photovoltaic electrochemistry (PV-EC), and photoelectrocatalysis (PEC) are the three main routes for solar-driven hydrogen production. Compared to PEC and PV-EC technologies, photocatalytic hydrogen generation eliminates the need for external sensors or circuitry, lowering system costs and environmental impact with thrifty and compact core construction. Photocatalytic hydrogen production relies solely on the supply of sunlight and a hydrogen source (water or organics) to sustain spontaneous reactions on the photocatalyst under ambient conditions. Therefore, photocatalytic hydrogen production offers three key advantages: (1) It avoids using fossil fuels, contributing to environmental protection; (2) Both solar energy and hydrogen energy are renewable, ensuring strong sustainability; (3) Reactors for photocatalytic hydrogen production can be decentrolized and be set up outdoors, providing broad applicability. As a result, photocatalytic hydrogen production, as an emerging green energy technology, holds promising prospects for application in the sectors of solar energy utilization, hydrogen energy generation, and environmental protection.
By harnessing solar energy to drive proton reduction for hydrogen evolution, the photocatalytic reaction comprises three main processes: the absorption of suitable light in the solar spectrum, the separation/transfer of photo-excited carriers to the interface, and the redox reactions at the interface to generate target products. Photon absorption efficiency, quantum efficiency, and solar-to-hydrogen (STH) efficiency are key indices for evaluating catalytic efficiency. Notably, STH efficiency is generally low for current photocatalysts, which is the main bottleneck hindering the commercialization of photocatalytic hydrogen production. Over the past few years, numerous efforts have been undertaken to enhance STH efficiency, for example, researchers have sought to minimize energy loss in the reaction by increasing the active surface area, adjusting the refractive index of the reaction medium, and designing solar concentrators. More than that, novel types of photocatalysts have been investigated, representatively, ZnIn2S4-based heterojunction photocatalysts show considerable improvement in photocatalytic performance. Both STH efficiency and reaction selectivity of these catalysts can be enhanced by optimizing their structures, compositions, and surface modifications. In addition, the understanding of reaction mechanisms and optimising reaction settings stemed from ZnIn2S4-based heterojunction photocatalysts are expected to inspire the design of photocatalysts, potentially leading to breakthrough of STH efficiency limiting. However, STH efficiency for photocatalytic solar hydrogen production is generally < 3% at present, a recent study reporting up to 9.2% under concentrated solar light and 6.2% in outdoor test, yet these levels of STH efficiency still fall short of what is needed for large-scale practical applications [54].
Overall, photocatalytic hydrogen production has made significant advancements in the laboratory, but it still has a considerable way to go before practical application. Photocatalysts, which play a vital role in this hydrogen production process, are being developed towards low cost, high efficiency, and long lifespan.
4.2
Current and future challenges
To enhance the STH efficiency, efforts must focus on addressing the substantial problems in three main processes of photocatalytic hydrogen production, including narrow solar light absorption range, the rapid recombination rate of photogenerated carriers, and the limited number of active sites. Additionally, innovative approaches should be pursued to reduce costs, thereby building a competitive advantage of this hydrogen generation technology for future commercialization.
The primary challenge lies in the construction of efficient photocatalysts. The standard free energy for the decomposition of water into H2 and O2 is 0 and 1.23 V, respectively. For the photocatalytic overall water splitting to occur, the conduction band (CB) potential of a semiconductor should be < 0 V, while the valence band (VB) potential of a semiconductor should exceed 1.23 V (Fig. 8) [55]. For note, these potentials of the energy band may differ for the decomposition of other hydrogen sources. The prerequisite is in accordance with following principles of energy bands for logical design and construction of photocatalysts: (1) A wide bandgap (Eg) of semiconductors results in weak light absorption capability, meaning that only a small portion of photons with shorter wavelength in solar spectrum can be effectively absorbed. (2) The separation and transfer of photogenerated carriers was facilitated though a built-in field to achieve high quantum efficiency, which relies on the appropriate construction of semiconductors with varying electrical characteristics. (3) Carriers in semiconductor construction require extra overpotential to overcome the energy barrier of redox reactions at interface, thereby sustaining a considerable rate of hydrogen production.
Figure 8
Figure 8.
Schematic energy diagrams of water-splitting photocatalysts. Reproduced with permission [55]. Copyright 2023, Springer Nature.
In addition, developing simple and effective synthetic approaches for producing high-quality photocatalysts on a wide scale while keeping costs low is essential for building a competitive advantage. Numerous synthetic techniques, such as hydrothermal synthesis, sol-gel synthesis, precipitation, and impregnation methods, have been developed. However, these methods often fall short of meeting the demands of mass production. For example, the hydrothermal method can effectively control reactions and crystal development by altering the reaction temperature, pressure, and time, making it suitable for the synthesizing a wide range of materials. Nonetheless, it faces challenges such as the need for high-temperature and high-pressure resistance, as well as corrosion issues in pilot-scale equipment. Additionally, the size of the produced crystals is limited by the dimension of the high-pressure vessel and the feeding mass of raw materials. The inability to directly observe the reaction process complicates timely identification and resolution of issues. As a result, mass production of photocatalysts with optimal structures and high catalytic efficiency through low-cost and environment-friendly synthesis processes remains a significant long-term challenge.
Using widely used and easily available raw materials for photocatalytic hydrogen production is another crucial factor in building competitive advantage. Currently, the raw materials of hydrogen source for photocatalytic hydrogen production primarily include freshwater, seawater, and biomass-based organic materials. Biomass-based raw materials encompass lignocellulosic components (cellulose, hemicellulose, lignin), monosaccharides, and biomass-based liquid hydrogen carriers (methanol, formic acid, ethanol, glycerol), etc. Unfortunately, current studies reveal that the insolubility and recalcitrant molecular structure of raw biomass, particularly lignocellulose, provide significant obstacles to hydrogen production due to sluggish reaction kinetics and unsatisfactory selectivity. On the other hand, because of the unfavourable physical qualities and complicated chemical structure, there still exist a knowledge gap to understand the reaction pathways and molecular mechanisms in the photodriven biomass conversion process. From this perspective, using water, particularly seawater (over 97% of the Earth's water), for photocatalytic hydrogen production is noteworthy for the benefits of simpler systems, rapid reaction kinetics, and more abundant natural sources.
4.3
Advances in science and technology to meet challenges
According to the three main steps of photocatalytic process, investigating novel catalysts and constructing suitable hetero/homo-junctions in semiconductors represent the effective strategies to alleviate the efficiency limiting of photocatalytic hydrogen production. Advanced materials or appropriate structures can not only enhance the generation of photogenerated carriers at the desired energy but also improve the migration of these carriers, while facilitating redox reactions at the interfaces. Many advanced photocatalysts have been deliberated for the adoption this strategy: such as transition metal sulphide semiconductor catalysts (NiPS3 ultrathin nanosheets [56], ZnCdS [57]), graphitic carbon nitride (g-C3N4)-based catalysts (Pd/DN-UCN [58]), Bi-based catalysts (BiOBr/C [59], ZnIn2S4/BiVO4 [60]), layered double hydroxides (LDHs)-based catalysts [61], metal oxide-based catalysts [62]. Enhancement of photocatalytic performance on corresponding photocatalysts proved the feasibility of this strategy. However, low activity and durability of photocatalysts significantly hinder the development of these semiconductors, indicating ongoing efforts are necessary to better understand the relationship between photocatalytic performance and photocatalysts.
Modification of photocatalysts also demonstrates significant improvements in optimizing photocatalytic performance through various strategies, including morphology and structural regulation, surface crystal plane engineering, co-catalyst loading, element doping, and defect engineering. Among these strategies, passivation engineering helps to mitigate the effects of photogenerated holes on the catalyst surface and to offer additional protection for sensitive catalyst surfaces. This results in a relatively stable and mild environment conducive to the efficient separation and utilization of photogenerated carriers. In recent years, passivation engineering has gained popularity as an effective approach to enhance both the activity and longevity of photocatalysts. For example, Huang et al. designed an Sx-In-P1-x passivation monolayer structure, wherein the Sx-In-P1-x buffer layer could minimize the lattice mismatch between the ZnS and InP to passivate the interface defects. This strategy inhibits undesirable recombination of photogenerated charges at trap sites, thereby yielding beneficial outcomes for photocatalytic H2 evolution [63]. Very recently, numerous studies have found that organic semiconductors and perovskite materials possess excellent absorption, tunable band gaps, and good stability, positioning them as promising candidates for photocatalytic water splitting [64]. For instance, Zhou et al. reported a hydrophilic one-dimensional microporous organic framework, 1, 3, 6, 8-tetrakis (p-benzoic acid) pyrene (HOF-H4TBAPy), for achieving high-performance photocatalytic hydrogen production. Experimental results show that under light irradiation, the photocatalytic H2 yield of HOF-H4TBAPy can reach 358 mmol h-1 g-1, with an apparent quantum yield of 28.6% at 420 nm (Fig. 9) [65].
Figure 9
Figure 9.
Photocatalytic H2 evolution performance of HOF-H4TBAPy. Reproduced with permission [65]. Copyright 2023, Nature Publishing Group.
A novel approach to enhancing photocatalytic performance is to improve the sluggish reaction kinetics by accelerating the desorption and separation of the product H2. Metal organic frameworks and two-dimensional multi-layer catalysts have been discovered to be successively applied to photocatalytic generation of hydrogen coupled with in-situ hydrogen gas storage. This advanced strategy eliminates the need for additional considerations regarding hydrogen separation and transportation [60,66]. However, these catalysts are often limited by stability and hydrogen storage rates, making them temporarily unsuitable for industrial production.
As mentioned above, utilizing seawater as a hydrogen source instead of pure water in photocatalytic hydrogen production is highly advantageous for enhancing the competitiveness of this technology. Seawater is nearly unlimited and widely available on Earth, and the longer sunlight duration along with higher radiation intensity contributes to techno-economics for offshore photocatalytic application. Consequently, the direct use of seawater in photocatalysis has attracted widespread attention as a promising avenue for sustainable research. Over the last decade, significant progress has been made in the use of seawater for photocatalytic hydrogen production. Inorganic-based photocatalysts, organic-based photocatalysts, and hybrid organic-inorganic heterojunction photocatalysts as prospective materials for seawater splitting have been explored. As an example, Zhang et al. built a novel ZnIn2S4/polymer (ZIS/PAN) nanofiber membrane by electrospinning technology. Its photoactivity performance was tested in seawater splitting without cocatalyst, and the HER could be measured to be 1836 µmol h-1 g-1. The excellent catalytic performance is due to the cyanide groups on PAN fibres, which could capture a part of photogenerated electrons. These captured electrons subsequently adsorb ions from seawater, thereby mitigating their adverse effects on the photocatalyst and displaying superior seawater hydrogen generation capability [67]. Although the current STH is below 5% and the ionic environment in seawater is complex, the potential for using seawater in photocatalytic hydrogen production is substantial when compared to the scarcity of freshwater resources and the high cost of desalination.
4.4
Concluding remarks and prospects
Photocatalytic hydrogen production, as an environment-friendly energy production strategy, holds significant potential in addressing pressing environmental and energy crisis. As scientific and technological advancements continue to unfold, photocatalytic hydrogen production is poised to assume an increasingly vital role within the realm of hydrogen energy. Despite substantial research and development efforts, there are still many challenges to be addressed before practical implementation, including unclear theoretical frameworks, low photocatalytic stability and efficiency, and unidentified reactive mechanisms. Furthermore, there is an urgent need to reconcile the disparities between laboratory-scale innovations and their large-scale operational viability, along with the difficulties in achieving high selectivity and yield. Each of these challenges is unique and requires careful analysis and creative solutions. In summary, further efforts are essential to advance the technology of photocatalytic hydrogen production for effective and sustainable hydrogen energy conversion.
5.
Photocatalytic total splitting of water
5.1
Status
The undelayable transition toward cleaner and renewable energy production pushes forward the research and development of efficient and sustainable technologies to convert solar energy into energy vectors (solar fuels such as H2 or hydrocarbons) through processes that resemble artificial photosynthesis. Hydrogen is expected to play a key role in this scenario, indeed, its demand approached 100 Mt in 2023 and is quickly rising. However, despite the growth of electrolysis, whose capacity has reached 20 GW, most of hydrogen is still produced from fossil fuels [68]. To be regarded as a clean fuel, "green" hydrogen must be produced from abundant sources and renewable energy, ideally water and solar radiation. Photocatalytic water splitting is a strongly endoergonic (uphill) reaction:
In acidic environment, H2 and O2 are produced through the following half-reactions, exploiting the electrons and holes provided by an irradiated semiconductor with suitable potentials of conduction and valence band edges:
The oxygen evolution reaction (OER) implies the transfer of 4 electrons, which makes it rather sluggish, with the consequence of slowing down the kinetics of hydrogen evolution reaction (HER) [69]. Therefore, cocatalysts are frequently deposited onto the photocatalyst, for promoting either the oxidation or reduction step or both (Fig. 10a). Typical reduction cocatalysts are noble metals, particularly Pt, Pd, Rh, Au, while oxides such as RuO2 and IrO2 have been recognized as efficient cocatalysts for OER. To counter the depletion of natural resources, many research works have recently explored alternative cocatalysts based on less precious transition metals; among them, oxides, nitrides, sulfides and phosphides containing Ni, Co and Cu have given promising results [70]. To increase the H2 production rates, sacrificial agents are often employed: such compounds have more advantageous electrochemical potentials than water, so their oxidation or reduction is thermodynamically favoured. Alcohols and triethanolamine are common electron donors, while sodium persulfate and ferric nitrate are examples of electron acceptors. Though sacrificial agents may be useful to gain insight into the reactivity of semiconductors, the consumption of such compounds, particularly in case of industrial products, lowers the overall energy efficiency and environmental benefits of the process. The preferable option is the direct conversion of water, namely total (or overall) water splitting, avoiding the concurrent consumption of other chemicals, also resulting in the depletion of valuable resources.
Figure 10
Figure 10.
(a) Scheme of photocatalytic total water splitting and its three main steps, excitation (ⅰ), charge carrier migration (ⅱ) and surface reactions (ⅲ). Reproduced with permission [70]. Copyright 2022, Wiley. (b) Absorption spectrum of g-C3N4/Co3O4 photocatalyst and AQE in overall water splitting under simulated solar light. Reproduced with permission [77]. Copyright 2023, Elsevier. (c) Illustration of the charge carriers transfer and H2 and O2 evolution by piezo-photocatalysis on BiFeO3/COF. Reproduced with permission [81]. Copyright 2023, Springer Nature.
Since the famous pioneering work by Honda and Fujishima (1972), decades of research efforts have brought great advances in the photocatalytic splitting of water. Along with classical photocatalysts, like titanium dioxide and strontium titanate, a vast number of materials have been proposed to this aim, including single and mixed metal oxides, chalcogenides, nitrides. In the last years, hybrid semiconductors, such as metal-organic frameworks (MOFs), as well as metal-free ones, chiefly carbon nitrides and covalent organic frameworks (COFs), have emerged as promising candidates for total water splitting [71,72]. Here some significant recent results are briefly summarised. A breakthrough was reported in 2020 by Domen and coworkers, who achieved an impressive apparent (external) quantum efficiency (AQE) of 96% in overall water splitting under 350–360 nm irradiation wavelength, using an Al-doped strontium titanate (SrTiO3: Al) photocatalyst with precisely photodeposited cocatalysts Rh/Cr2O3 and CoOOH for H2 and O2 evolution reactions, respectively [73]. They also shed light on the mechanism, ascribing the outcome to the separation of reduction and oxidation sites, multiple charge transfers and suppressed recombination. More recently, SrTiO3: Al coupled with CoOx was modified with nanoparticles obtained from a Ni-MOF, so that the Ni-based cocatalyst increased the electrical conductivity of the photocatalyst and promoted HER, leading to AQE of 10.3% at 365 nm for overall water splitting, a remarkable value considering the absence of noble metals [74].
Metal chalcogenides are attracting increasing attention as solar-active photocatalysts, due to their suitable band energy levels and optoelectronic properties. Doping can positively affect the behaviour of metal sulfides. In 2023, a V-doped indium sulfide heterostructure with cadmium telluride quantum dots (CdTe/V-In2S3) was reported to achieve 114% internal quantum efficiency at 350 nm, which was explained as an effect of the built-in electric field that allowed multiple exciton generation [75]. The corresponding STH conversion efficiency was 1.31%. ZnIn2S4 was subjected to cation-site O doping through a structural distortion, and the large electronegativity differences between adjacent atoms prompted notable STH efficiency (0.57%) and good stability in prolonged testing up to 120 h [76]. Photothermal catalysis offered optimal performances for InGaN/GaN nanowires with Rh/Cr2O3 and Co3O4 nanoparticles as cocatalysts, which worked around 70 ℃ under concentrated simulated solar light reaching an outstanding STH efficiency of 9.2% with pure water and about 7% with tap water and seawate. Also, g-C3N4, modified through N-vacancy formation and decorated with Co3O4 nanorods and Pt as cocatalysts, was tested under simulated sunlight and achieved AQE of 11.94% at 400 nm and STH efficiency of 1.48% in the half-reaction experiments (Fig. 10b) [77]. To give an idea, the H2 evolution rates by pure water splitting in the best performing systems are typically in the order of 100 µmol/h.
5.2
Current and future challenges
The application of heterogeneous catalysts for the direct solar-to-hydrogen conversion using pure water remains full of hurdles in terms of commercial scalability. The first challenge in the development of photocatalytic technology for total water splitting is related to the performances, which are still unsatisfactory because of thermodynamic and kinetic limitations, associated with a severe overpotential, and the occurrence of backwards and side reactions. The slower O2 diffusion compared to H2 and its generally higher affinity for the photocatalyst's surface, which hinders the desorption of O2 molecules, contribute to the slow overall kinetics. An appropriate photocatalyst for water splitting has the typical basic requirements, namely: (1) Proper potentials of conduction and valence band edges; (2) a broad light absorption range to maximize solar radiation harvesting; (3) effective separation and migration of photogenerated electron/hole pairs. In overall splitting, reduction and oxidation cocatalysts play a central role. Besides offering efficient adsorption and active sites, they may have additional effects, such as improving light capture, accelerating charge carriers transfer or lowering the overpotential [70]. The ideal cocatalysts might depend on the photocatalyst, so their choice, design, and deposition procedure at optimal load should be carefully performed. The synthesis routes of nanostructured photocatalytic materials should give high yields, ensure reproducibility, possibly also on a larger scale, and follow the principles of green and sustainable chemistry. As far as possible, the use of Earth-abundant and inexpensive elements in the design of novel photocatalytic systems is encouraged.
The photoreactor configuration and reaction conditions are crucial. In a water splitting photoreactor, both temperature and pressure need to be carefully monitored, since they will affect the water vapour pressure, the solubility of the gas products, besides the effect of temperature on reaction kinetics. The effective transport of the products and separation of evolved hydrogen must be assured to avoid potential hazards due to the explosive mixture. As total H2O splitting is attained, H2 and O2 are produced in 2:1 molar ratio, which should be detected in the outgoing gas stream. It is very important to test long-term stability under operating conditions and scale-up feasibility. The design of photoreactors at an industrially relevant scale is a general challenge in photocatalysis. This includes the immobilization of photocatalytic nanomaterials on suitable supports avoiding dramatic losses of activity, because the use of suspended nanoparticles, common in laboratory experiments, is hardly transferrable to a real plant.
Last but not least, the correct interpretation, reporting and comparison of experimental results should be pursued. Especially in a field like photocatalytic H2 production, expanding and advancing at a fast pace, it is essential for researchers to stay updated on the developments reported in the literature and to be able to compare own results with those of others, which becomes tricky when dealing with photocatalysis because relevant variables are sometimes overlooked or not reported in detail. Nevertheless, standardized procedures to measure the H2 evolution efficiency of photocatalytic systems are still lacking. The H2 evolution rate, usually normalized by photocatalyst mass (µmol h-1 g-1), is the general figure of merit for the photocatalytic activity. However, this value alone is not sufficient to compare the intrinsic activity of different materials, since other variables are involved, primarily the intensity and emission wavelength range of the light source. The apparent (or external) quantum efficiency normalizes the reaction yield with respect to the incident radiation energy, although it neglects the actual absorption by the solid catalyst (which would give the internal quantum efficiency). The specific wavelength at which AQE is measured must be reported and ideally a photoaction spectrum should be provided (see for example Fig. 10b). Additionally, in simulated of real sunlight tests, the STH energy conversion efficiency should be estimated. It can be noted that few photocatalytic systems reported to date enable STH efficiencies close to 10%, which is as an indicative target for large scale application.
5.3
Advances in science and technology to meet challenges
The progress in photocatalysis research is led by the development of advanced photocatalytic materials, including both the deeper understanding and optimization of traditional photocatalysts, and the discovery of new semiconductors with promising properties. Besides well-known modification methods (doping, sensitization, noble metal deposition), the engineering of morphology and structural defects and the construction of heterojunctions are among the strategies that reveal the greatest potential to improve solar energy conversion performances. Heterojunctions, especially those exploiting the advantage of a charge transfer that follows a Z-scheme or the novel concept of S-scheme, can effectively separate the sites of reduction and oxidation on two suitable semiconductors. The variety of materials and composites developed for photocatalytic water splitting is testified by recent reviews on the topic [74,78].
On the other hand, as innovative photocatalysts and cocatalysts are proposed, often with increasing complexity (mixed phases, heterostructures, etc.), a detailed understanding of the relationship between their synthetic route, structure, physicochemical features and activity is needed. In the fabrication of multicomponent nanomaterials, the particle shape and dimensionality (0D, 1D, 2D) play a relevant role, for example by enhancing the surface-to-volume ratio, exposing more active sites or exploiting quantum effects. Accurately controlled synthesis protocols allow to define the desired crystalline structure, morphology, exposed facets and realize good interfacial contact with other components. Nevertheless, the connection between the procedure and the resulting features is not always well clarified. The spatial separation of HER and OER cocatalysts and the tuning of their surfaces can help inhibiting the reverse reactions of H2 and O2. The impact of a properly designed cocatalyst was recently demonstrated by decorating the Rh HER cocatalyst of the benchmark photocatalyst GaN-ZnO with Al2O3 nanostructures through atomic layer deposition [79]. This modification raised the AQE of overall water splitting from 0.3% to 7.1% at 420 nm, showing that the partial coverage of noble metal surface sites with inert oxides can effectively suppress the reverse reactions that lower HER rates. Fundamental insight into the electron transfer mechanisms in organic photocatalysts was lately reported. A study on molecular crystal nanobelts, obtained by self-assembly of spin-one open-shell perylene diimide diradical anions (:PDI2-) and their tautomeric spin-zero closed-shell quinoid isomers (PDI2-), stated that such :PDI2-/PDI2- structure altered the spin-dependent excitation pathways and induced H+ reduction through a hydrophilic diradical-mediated electron transfer, with AQE over 1.5% under visible-near IR solar spectrum [80].
While photocatalytic processes are usually conducted at or near room temperature, the effect of heating is increasingly investigated and an optimal temperature that maximizes the kinetics can be found, as cited above [53]. The energy balance must be taken into account, however moderate temperatures could be easily reached in a solar plant using light concentrators. Novel concepts in reactor setups include the introduction of additional energy sources, such as mechanical strain, ultrasounds, magnetic fields or external electric fields. For example, ultrasonication was applied with a core-shell composite piezo-photocatalyst obtained by covalent linking piezoelectric BiFeO3 nanosheets and a COF (TpPa-1-COF) [81]. The Z-scheme charge transfer and the combination of ultrasounds and simulated sunlight irradiation provided exceptional H2 and O2 production rates of 1416 and 708 µmol h−1 g−1 by the BiFeO3@TpPa-1-COF nanocatalyst. The polarization deriving from the piezoelectric potential difference could drive the migration of electron and holes (Fig. 10c). Photothermal catalysis was also improved by a weak magnetic field, testing a Fe3O4/N-TiO2 superparamagnetic photocatalyst with Au cocatalyst under simulated solar illumination at high temperature (270 ℃) [82]. The induced local magnetic flux prolonged charge carriers lifetime and boosted H2 evolution, affording 11.9% STH efficiency.
The possible coupling of H2 generation with the oxidation of organic compounds, which can greatly enhance HER rates, could be evaluated in case the electron donors are represented by waste-derived substances or organic contaminants in wastewater, if actual energy and economic benefits are attainable. The splitting of water in vapour phase has been proposed to solve some shortcoming of the liquid phase reaction, however in the gas phase lower activities have been recorded to date [69]. The scale-up of the process has been seldom investigated so far. A valuable report concerned the construction of a 100 m2 array of panel reactors, containing a film of the reference SrTiO3: Al-based photocatalyst and equipped with a polyimide membrane as gas separator [83]. The system was optimized for safety and durability and operated under natural sunlight for several months, reaching a maximum STH of 0.76%.
5.4
Concluding remarks and prospects
In order to give a significant contribution to the achievement of ambitious goals concerning renewable energy, such as European Union's program to accomplish carbon neutrality with net zero emissions by 2050, photocatalysis still needs to make a big leap forward. The emergence of innovative photoactive semiconductors and relative cocatalysts, the development of nanostructures (e.g., quantum dots, 2D nanosheets) as well as the deepening of the comprehension of all mechanisms involved in water splitting process will lead the improvement of the performances. The identification of standard testing procedures or at least best practices to evaluate the photocatalytic water splitting activity would guide and inspire researchers in the elaboration of rigorous and accurate reports and facilitate the comparison of results obtained in different laboratories. Nowadays, a fast evolution of computational methods is witnessed, including the simulation of materials properties and reactivity as well as artificial intelligence approaches based on machine learning. Rationally designed models and predictions may support the selection of the most promising compositions to be investigated experimentally, even in case of missing experimental data through the use of generative AI tools [84].
The transfer from the lab scale to the industrial scale needs further studies, considering potential prospects such as the combination of photocatalysis with other techniques (e.g., photoelectrocatalysis), its integration in solar energy capture and conversion systems (e.g., solar cells), or the splitting of seawater, which could also be integrated with desalination plants and during the reengineering of existing technologies. In summary, the solutions to boost total water splitting technology involve advanced materials, effective photoreactors and integrated processes. It is envisaged that the crossing of fundamental and applied research, with cooperative efforts by researchers from chemistry, physics, materials science and chemical engineering fields, can pave feasible ways toward clean hydrogen production by solar energy conversion.
6.
(Photo)electrocatalytic reduction of O2 to H2O2
6.1
Status
The oxygen reduction reaction (ORR) is a critical process in electrochemistry, playing a central role in various applications such as energy storage and conversion systems, particularly in fuel cells. In these systems, the four-electron reduction of oxygen to water is highly beneficial due to its high energy efficiency. However, the two-electron oxygen reduction reaction (2e− ORR) has gained significant interest for its potential in the chemical industry and wastewater treatment, owing to its ability to produce hydrogen peroxide (H2O2), a green and powerful oxidant. This partial reduction pathway is often in competition with the complete four-electron reduction, necessitating careful selection of cathode materials to selectively promote the 2e− ORR. While platinum group metals are known for their efficacy in facilitating the four-electron reduction, a broader range of materials, including tungsten (W) and mercury (Hg) compounds, have shown promise for the electrosynthesis of H2O2 from O2 [85]. The obvious problem associated with the use of these catalysts is their high price. Additionally, they suffer from poor stability, and, in the case of Hg, high toxicity. Earth abundant transition metal-based compounds (e.g., Fe, Co, Ni, Mn, Cu and their oxides, chalcogenides, phosphides) can be considered as advanced low-cost 2e− ORR electrocatalysts [86]. However, in acidic electrolytes, metal catalysts are unstable, which enforces the development of carbon materials as cheap and abundant electrocatalysts. These materials can be based either on doped elemental carbon in the form of graphite, graphene or carbon nanotubes or on composites of carbon nanomaterials with specifically tailored polymers.
The advance in H2O2 generation may be enhanced by photoactivation of the cathode within the framework of photoelectrocat-alytic (PEC) ORR. Photoactivation can either enhance the reaction rate, thereby increasing the current density, or affect the thermodynamics of the redox process, leading to a reduction in the applied bias voltage to as low as 0 V [87,88]. Over the past five years, several novel materials for the PEC 2e− ORR have been reported, including metal phthalocyaninates, nanostructured transition metal oxides and sulfides, 2D graphene-like materials and conductive polymers. The best of reported PEC 2e− ORR systems have demonstrated the ability to deliver 1 mol/L H2O2, which represents a potential breakthrough in industrial H2O2 production, surpassing the efficiency of existing industrial processes [89].
6.2
Current and future challenges
The efficiency of (photo)electrochemical H2O2 production is primarily governed by the rate of the 2e− ORR, which competes with the 4e− ORR and hydrogen evolution reactions. These competing processes are quantified by the cathode current of the ORR and the Faradaic efficiency (FE) of 2e− ORR over 4e− ORR. Achieving fast redox kinetics can shift the limiting factor to oxygen diffusion, where the concentration of O2 dissolved in the electrolyte constrains the ORR currents. So far, this limits the ORR (photo)current [90] by several mA/cm2. Typical stable currents of the 2e− ORR do not exceed 0.5 mA/cm2 in the dark [85] and 0.9 mA/cm2 for photocathodes. For practical applications of electrochemical H2O2 production, achieving a stable current density above 0.5 mA/cm2 and a Faradaic efficiency above 90% for the 2e− ORR are critical milestones. However, most current ORR electrocatalysts fall short of these targets, typically exhibiting a current density of < 0.3 mA/cm2 and an efficiency below 80%.
In the context of industrial H2O2 production, the maximum accumulated concentration of H2O2, [H2O2]max, achievable in the system, is critically important. To date, reported [H2O2]max values are 100 mg/L (3 mmol/L) for EC and 3.4% (1 mol/L) for PEC production [91]. These limitations originate from both the decomposition of the produced H2O2 the degradation of the catalyst [92]. A key challenge in this area is the higher reactivity of H2O2 compared to O2, both for the kinetic and the diffusion reasons.
To bridge the gap between the currently obtained and desired H2O2 production rates and achievable concentrations, it is essential to focus on innovative electrochemical reactor designs. Enhancing gas diffusion electrodes (GDE) and developing advanced membrane-based systems are critical steps in this direction. These improvements can facilitate better oxygen transport and distribution, overcoming the limitations imposed by oxygen diffusion in the electrolyte. Additionally, optimizing the activity, selectivity, and durability of catalysts through advanced phase engineering is necessary to enhance their performance for effective H2O2 electrosynthesis. For PEC processes, another significant limitation is the low light utilization efficiency, primarily due to the light absorbance being restricted to the blue-to-near UV range [93]. By refining these aspects, it is possible to achieve higher current densities, improved Faradaic efficiencies, and greater stability in the production of H2O2, making the process more viable for industrial applications [86,93].
6.3
Advances in science and technology to meet challenges
Efforts have been made to develop new and effective electrocatalysts for the 2e− ORR aimed at H2O2 production. Due to their abundance, low cost, and tunability, carbon materials doped with heteroatoms were extensively studied in recent years, demonstrating high H2O2 selectivity, but low overall ORR activity. In addition to tuning the electronic structure of carbon, the doped heteroatoms can act as coordination sites to anchor metal atoms, forming single atom metal catalysts (SACs) that overcome the low 2e− ORR activity of bare carbon materials. The active site configurations of SACs can be rationally regulated to alter electronic structures by tailoring coordination environments, introducing surface functional groups, and optimizing geometric structures. This approach shows great potential for controlling the ORR process with a specific reaction pathway towards the control of the dioxygen binding mode (Fig. 11). More importantly, reducing metal materials to the atomic scale can maximize atomic utilization and reduce costs in large-scale H2O2 electrosynthesis [86]. For example, on SAC selectivity of 2e− ORR increases to 95% [94].
Figure 11
Figure 11.
Advance of single atom catalysis for the 2e− ORR illustrated on oxygen binding modes. Reproduced with permission [94]. Copyright 2024, Springer Nature.
The development of materials for the PEC 2e− ORR has focused on the exploitation of the heterojunction structures, which enable more efficient charge separation and light utilization through fine band engineering. Fig. 12 illustrates various heterojunction photocatalyst strategies that can be applied in photoelectrochemical oxygen reduction reaction (PEC–ORR) systems [95]. Heterojunction-based composite materials based on different junction architectures such as Z- and S-schemes, typical for internal photocatalysts, are now widely implemented in the PEC catalysts. Much attention is paid to morphology optimization and mass transfer control. For example, recent studies have shown that hydrophobic PEC materials enhance the mass transfer of the dissolved dioxygen thereby increasing the H2O2 generation rate. Additionally, heating the cathode with incident light has been employed to achieve operando concentration of the obtained H2O2 [96].
Figure 12
Figure 12.
Various heterojunction schemes for photocatalysts, which may be employed in PEC ORR systems. Reproduced with permission [95]. Copyright 2023, Elsevier.
Over the last five years, the role of the mechanistic studies and computational support significantly grew in the field of PEC 2e− ORR. Advanced techniques such as transient absorption and emission spectroscopy, electron paramagnetic resonance (EPR), electrochemical impedance spectroscopy and chemical superoxide scavenging, are widely used. This enhanced understanding of the electron transport, photodynamics in the catalysts and phase boundaries and involved intermediates.
6.4
Concluding remarks and prospects
The pursuit of efficient and scalable methods for H2O2 production through electrochemical and photoelectrochemical processes remains a significant challenge in the fields of electrochemistry and material science. The two-electron oxygen reduction reaction offers a promising pathway for H2O2 generation, with substantial potential for industrial and environmental applications. However, the current limitations in catalyst performance, reactor design, and light utilization efficiency must be addressed to realize this potential fully. Advancements in electrocatalyst development, particularly with heteroatom-doped carbon materials and single atom metal catalysts, have shown promising results in enhancing the activity and selectivity of the 2e− ORR. These innovations, coupled with sophisticated band and phase engineering, have the potential to significantly improve the efficiency and cost-effectiveness of H2O2 production. For photoelectrochemical (PEC) processes, the development of heterojunction structures, Z- and S-schemes, and morphology optimization are crucial steps forward. Furthermore, the application of hydrophobic materials and the strategic use of light-induced heating have demonstrated enhancements in H2O2 generation rates. Considering the progress in the H2O2 production efficiency, the increase of the light efficiency, striving for the whole visible spectrum utilization, is expected to be the future trend in the (photo)electrochemical 2e− ORR.
7.
Photocatalytic synthesis of ammonia
7.1
Status
Ammonia (NH3) is an essential and highly versatile chemical that plays a critical role in agriculture, food production, and medicine [97]. Its applications range from manufacturing fertilizers, cosmetics, and dyes to producing soda ash, manganese, and ferroalloys. Additionally, ammonia holds promise as a sustainable fuel for internal combustion engines or as a source of hydrogen, which can be utilized in proton-exchange membrane fuel cells, alkaline fuel cells, and other energy systems [98].
Today, most ammonia is produced using the Haber-Bosch process, which involves converting atmospheric nitrogen (N2) into ammonia by reacting it with hydrogen in the presence of a metal catalyst, usually iron:
This process requires high temperatures (over 400 ℃) and pressures around 200 atm to break the strong N≡N bonds in nitrogen molecules, resulting in significant energy consumption for both heating and cooling. Moreover, the Haber-Bosch process contributes to the emission of over 500 million tons of CO2 annually [99]. Given these challenges, developing sustainable methods to optimize ammonia production while reducing greenhouse gas emissions is a crucial objective. One promising avenue for addressing these challenges is exploring alternative methods for NH3 synthesis, such as biological, electrochemical, and photocatalytic processes [100]. Biological approaches, which use microorganisms capable of fixing atmospheric nitrogen and converting it into ammonia, offer considerable potential for more environmentally friendly production. However, the cost of biologically synthesized ammonia is still far from competitive with that produced through the Haber-Bosch process. Electrochemical synthesis offers another route, where nitrogen is reduced to ammonia via an electric current. Although this process operates at lower temperatures and pressures, which could substantially reduce the carbon footprint, it remains economically challenging due to the need for expensive electrode materials. Photocatalytic nitrogen reduction, utilizing solar energy, has recently emerged as a particularly promising approach for producing ammonia under milder conditions [101]. While current photocatalytic ammonia production rates are not yet viable for industrial-scale use, promising materials have already been identified that could accelerate research progress, which is especially important in the global effort to address climate change.
7.2
Current and future challenges
Nitrogen naturally exists as diatomic molecules (N2), held together by a strong triple bond. As a result, nitrogen molecules are incredibly stable, and dissociating them under normal conditions is nearly impossible. To activate N2 for its reduction to NH3, the high dissociation energy of the triple bond must be overcome. Traditionally, this can only be achieved at high temperatures and pressures, as used in the Haber-Bosch process. Photocatalytic technologies offer an alternative, allowing nitrogen activation under milder conditions, but with relatively slow reaction rates. The photocatalytic ammonia synthesis process begins with the chemisorption of nitrogen molecules on the catalyst's active site. Following this, the N≡N triple bond is broken, and continuous hydrogenation occurs through protons derived from water. Nitrogen activation is typically regarded as the rate-limiting step, making it a crucial factor in evaluating the efficiency of nitrogen photocatalytic fixation. Therefore, the selection of a highly active photocatalytic material and optimization of the pH environment in which the reaction occurs are essential.
Photocatalytic ammonia synthesis can be broken down into a sequence of processes involving water splitting followed by nitrogen reduction [101]. Absorption of photons by the semiconductor photocatalyst initiates the generation of electron-hole pairs (Fig. 13a). Electrons excited in the photocatalyst cross the bandgap and enter the conduction band, leaving behind holes in the valence band. These electron-hole pairs migrate to the surface of the photocatalyst, where they participate in redox reactions with adsorbed N2 and H2O molecules [102]. The holes in the valence band oxidize water, while the electrons in the conduction band reduce nitrogen:
Figure 13.
(a) Schematic representation of the photocatalytic production of ammonia. Reproduced with permission [102]. Copyright 2024, Elsevier. (b) Schematic representation of possible mechanisms of nitrogen reduction to ammonia. Reproduced with permission [103]. Copyright 2022, Frontiers.
Fig. 13b illustrates two possible mechanisms for nitrogen reduction: dissociative and associative. In the dissociative mechanism, the N≡N bonds break before hydrogenation begins, leaving individual nitrogen atoms on the catalyst surface, which are then converted into ammonia [103]. It is believed that the Haber-Bosch process follows this dissociative pathway. On the other hand, the associative mechanism allows nitrogen reduction without breaking the N≡N bond before hydrogenation, and the reaction can proceed through either a distal or alternative route. In the distal pathway, the nitrogen atom farthest from the adsorption site is protonated first, producing the first ammonia molecule, while the second nitrogen atom undergoes hydrogenation to form the second ammonia molecule. The alternative pathway involves alternating hydrogenation of both nitrogen atoms until one is converted into ammonia and the N≡N bond breaks.
Key challenges in improving the efficiency of ammonia production through photocatalysis include the development of highly active photocatalytic materials, optimizing nitrogen fixation and activation methods, and enhancing selectivity for NH3 over H2. Additionally, optimizing reaction conditions-such as temperature, pH, and the composition of the reaction medium can significantly improve process efficiency. External factors like light irradiation, spectrum, and the presence of impurities in the reaction system, which may negatively affect yield, must also be considered. Further, new approaches to photocatalyst regeneration and degradation reduction are critical areas of research.
Overall, a multidisciplinary approach, combining advancements in materials science, chemistry, and engineering, will be key to developing efficient and sustainable ammonia production technologies through photocatalysis.
7.3
Advances in science and technology to meet challenges
One of the central challenges in optimizing photocatalytic ammonia production lies in identifying an effective and stable photocatalytic material that is highly sensitive to visible light and has a sufficient number of active sites per surface area. To date, numerous attempts have been made to design photocatalytic materials with optimal properties for nitrogen photoreduction. These materials can be broadly categorized into oxide- and oxyhalide-based photocatalysts [104,105], sulfide-based photocatalysts [106,107], carbon-based photocatalysts [108,109], and others. However, the use of these photocatalysts is still limited by the insufficient number of adsorption and activation sites for nitrogen on their surfaces, as well as poor charge transfer at the interface, resulting in suboptimal efficiency. These issues have led to new directions in the development of highly functional catalysts, including defect engineering, morphology modification, co-catalyst integration, and heterojunction formation.
Introducing defects in the surface of photocatalytic materials has emerged as a promising strategy for increasing the number of active sites, which enhances light absorption efficiency. By controlling the formation of vacancies, it is possible to fine-tune the optoelectronic properties of the material for specific photocatalytic applications. For instance [110], the influence of oxygen vacancies on the surface of commercial TiO2 powders was studied in relation to the photocatalytic ammonia synthesis under UV light. The study revealed that Ti3+ particles, created by surface defects in titanium dioxide, serve as highly active centers for N₂ reduction, achieving a solar-to-chemical energy conversion efficiency of 0.02%–one of the highest reported for powder photocatalysts. Zhang et al. [111] proposed a strategy to enhance the surface defect states of nitrogen photoreduction catalysts through doping, ultrathin wires of substoichiometric tungsten oxide (W18O49) were doped with molybdenum (Mo), where coordinatively unsaturated metal atoms with oxygen defects served as sites for N₂ chemisorption and electron transfer. As a result, the Mo-doped W18O49 sample (1 mol% Mo) exhibited an ammonia production rate under 400 nm light of 195.5 µmol g-1 h-1, which is seven times higher than the undoped W18O49 sample. In pure water, the catalyst exhibits an apparent quantum efficiency of 0.33% when illuminated at a wavelength of 400 nm. Additionally, it achieves a solar-to-ammonia conversion efficiency of 0.028% under simulated AM1.5 G light conditions. Fei et al. [112] introduced a synthesis method for a heterojunction based on bismuth tungstate (Bi2WO6) and graphene quantum dots (GQDs). By creating a tight contact between co-catalysts, the energy band alignment within the system improved, leading to a significant enhancement in the material's photocatalytic activity. Photocatalytic tests showed that the Bi2WO6 sample containing 10 wt% GQDs exhibited the highest ammonia synthesis rate under visible light, which was 8.9 times higher than pure Bi2WO6 and 33.8 times higher than GQDs alone. Another method of improving catalytic performance is through morphology control, specifically by increasing the surface area of the samples. While g-C3N4 has already demonstrated promise for various photocatalytic processes under visible light, its performance is still limited by its relatively low surface area [113,114]. To address this [115], proposed enhancing the textural properties of g-C3N4 by forming a heterojunction with reduced graphene oxide (rGO). The g-C3N4/rGO composite, with a mass ratio of rGO to g-C3N4 at 0.2, showed the highest NH3 production rate, achieving values 8.3 times higher than those of the original g-C3N4.
7.4
Concluding remarks and prospects
In conclusion, developing environmentally sustainable and efficient methods for ammonia production is a multifaceted challenge that requires an interdisciplinary approach. Photocatalytic nitrogen reduction holds significant promise for future scaling and industrial application. However, to fully realize this potential, several limitations must be addressed-namely, the discovery and development of efficient and cost-effective photocatalytic materials, as well as the optimization of process parameters. Ongoing research continues to focus on creating and refining stable, eco-friendly photocatalysts. Materials based on oxides, oxyhalides, metal sulfides, and carbon composites have demonstrated significant potential. In particular, surface modification strategies, defect engineering, and heterostructure formation have shown promise in enhancing the photocatalytic process. As advances in photocatalytic nitrogen reduction progress, they could lead to the development of a more sustainable and environmentally friendly ammonia production technology. Such advancements may offer a viable alternative to the traditional Haber-Bosch process, aligning with global efforts to reduce greenhouse gas emissions and mitigate climate change.
8.
Electrocatalytic reduction of N2 to NH3
8.1
Status
The electrochemical nitrogen reduction reaction (NRR) is considered a promising alternative to the Haber-Bosch process, as it enables the clean synthesis of ammonia with reduced energy consumption under ambient conditions. The NRR process involves proton-coupled electron transfer reactions, requiring the transfer of six electrons for the reduction of one N2 molecule. NRR mechanisms are generally classified into two pathways: dissociative and associative. In the dissociative pathway, the N≡N bond is cleaved into ∗N atoms, which then undergo protonation, a process analogous to the well-documented industrial production of NH3. The associative pathway is further divided into distal and alternating pathways. In the distal pathway, the more remote nitrogen atom is initially attacked by proton/electron pairs and subsequently protonated to release NH3 [116]. Conversely, in the alternating pathway, the two nitrogen atoms are hydrogenated alternately (Fig. 14). Numerous nanomaterials have been successfully engineered for NRR systems, with transition metal catalysts such as iron (Fe), ruthenium (Ru) [117], and molybdenum (Mo) demonstrating superior NRR activity [118]. This enhanced activity is attributed to the donation-backdonation effects, which facilitate N2 activation and hydrogenation. Both experimental and theoretical studies have confirmed the efficacy of these transition metal catalysts in NRR applications.
Figure 14
Figure 14.
Schematic diagram of electrocatalytic NRR process. Reproduced with permission [116]. Copyright 2018, Elsevier.
A highly intensive triple bond (N≡N) is formed by the hybridization of atomic orbitals (Fig. 15a). As an inert molecule, N2 possesses a quite high N≡N triple bond enegy of 941 kJ/mol, where its first bond cleavage energy is up to 410 kJ/mol. Moreover, the wide energy gap is up to 10.82 eV between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of N2 resulting in unfavored electron transfer processes. Additionally, the non-polar nature and high ionization potential (15.84 eV) further hinders electron transfer. All the above arguments answer for inertness of N2 (Fig. 15b) [119]. Thus, the design and synthesis of efficient electrocatalysts achieving high activity and selectivity at the same time is a great challenge. Up to now, various strategies to improve catalytic behavior of electrocatalysts have been proposed, i.e., structural optimization, heteroatom doping, vacancy engineering, surface/interface engineering, and synergistic effects. Due to the highly disperse active sites, single atom catalysts exhibit great performance toward the NRR. Geng and coworkers reported a Ru single atom supported on nitrogen-doped carbon (Ru SAs/N–C), which achieved high production rate of 120.9 µg h-1 mg-1 and a FE of 29.6% at −0.2 V vs. RHE [120]. In comparison, Ru nanoparticles (NPs) possessed significantly lower catalytic activity. The improved NRR activity of Ru SAs/N–C was attributed to the stronger adsorption of N2 and more negative free energy in the steps of *N2 to *NNH compared to Ru NPs. However, recent breakthroughs suggested that the reconstitution of catalysts may occur during nitrogen-transforming reaction processes, leading to inconsistencies between the genuine active site and the original catalytic configuration [121]. Investigating the dynamic reconstitution of catalysts to accurately identify the real active sites under working conditions is necessary for the design of an efficient electrocatalyst [122].
Figure 15
Figure 15.
(a) Molecular orbitals and (b) structure characteristics of N2 molecule. Reproduced with permission [119]. Copyright 2023, Wiley-VCH.
By contrast, the competitive hydrogen evolution reaction (HER) is a significant issue for the NRR in aqueous electrolytes, which is a kinetically favored two-electron process. Hydrophobic layer coating on the electrocatalyst to suppress HER is regarded as a direct method to enhance the faradaic efficiency [123]. Recently, Ling et al. fabricated dual-atom catalysts (X/Fe-N-C) and provided evidence that local hydrogen radical generated on the X site of the X/Fe-N-C can activate and reduce N2 molecule adsorbed on the Fe site [124]. It was found that the local hydrogen radical comes from the water dissociation. Accordingly, it is imperative to design an electrocatalyst that could balance the competition between H and N2 on the active site. The extremely low solubility and slow diffusion of N2 in aqueous solution are also major constraint for the NRR process [125]. Since N2 is supplied from the bulk electrolyte, which affects mass transport. Therefore, high current densities (< 1 mA/cm2) cannot be achieved, limiting practical applications [126]. Lastly, NH3 contamination from the gas feedstock, catalyst and electrolyte must be checked and determined. N2 isotope labeling experiments are crucial for elucidating the source of ammonia generated from NRR rather than contamination.
8.3
Advances in science and technology to meet challenges
Efforts have been made to develop effective electrocatalysts for improving catalyst performance, it is still a difficult task for NRR to satisfy the commercial demands. In contrast to the extensive works of HER, a clear mechanism of NRR on catalysts is still imperfect, which hinders the development of NRR. In situ characterization techniques are essential for providing insights into the electrode/electrolyte interface throughout the NRR process, thereby assisting in the design of novel catalysts. XRD is a nondestructive characterization technique that can analyze crystalline structures, strain, as well as crystal defects of bulk catalysts during the NRR process. X-ray absorption spectroscopy (XAS) can be used to in NRR to examine the coordination structure of bulk materials [127]. FTIR spectroscopy and Raman spectroscopy are the surface analytical technique for material to provide molecular level fingerprint information on the surface of material and intermediates [128].
With the fast development of computational methods, facilitating the exploration of efficient catalysts and trend prediction of activity during NRR process has been achieved from first-principles calculations. The Sabatier principle has suggested a qualitative framework for the optimum catalyst, which binds atoms and molecules neither too weakly nor too strongly. The scaling relations further describe correlations between the adsorption energies of intermediates and catalysts. To overcome the mass transport issues, Gas diffusion electrodes (GDEs) are proposed that maintain a high N2 concentration and modulate the concentration of H2O, thereby resulting in higher current densities.
8.4
Concluding remarks and prospects
Though the remarkable progress made in the field, designing effective NRR catalysts and achieving catalytic NH3 production at practical levels remain significant challenges. The parameter space for advanced catalytic molecules and materials remains largely unexplored. In this regard, the selection of catalysts can be achieved through theoretical calculations and machine learning, thereby reducing the costs associated with experimental exploration. However, further improvements of the computational methods, model accuracy and computational strategy are highly demanded. Moreover, it is insufficient to indentify the catalytic mechanism only based on the theoretical calculations. Therefore, the reconstruction of the surface structure, the changes in element valence state, and the exposed active centers should be analyzed through the high spatial temporal resolution in situ characterization techniques. The integration of theoretical calculations and experimental results helps us gain deeper understanding of the correlations between structures and performance and design various catalysts. Additionally, strategies to suitably suppress HER are requested. The p-block elements are influential in suppressing HER, including B, Bi, and Sn. Despite the grand challenges ahead, we believed that coupled with reliable theoretical and experimental investigations and advanced in situ/operando characterization techniques will further push the development of more active, more selective electrocatalysts for N2 reduction under ambient conditions soon.
9.
Electrocatalytic oxygen reduction used in fuel cells and metal-air batteries
9.1
Status
Fossil fuels pose energy and environmental challenges, driving the development of clean solutions like fuel cells and metal-air batteries, which are efficient and pollution-free for electric vehicles and global energy security [129]. However, their commercialization is limited by the catalysts required for the oxygen reduction reaction [130]. ORR is kinetically slow, involving multiple steps and diverse pathways. As shown in Fig. 16, it can be categorized into two main types based on electron transfer: the four-electron (4e-) pathway, which directly reduces oxygen to water (H2O) in acidic environments or to hydroxide (OH-) in alkaline environments, and the two-electron (2e-) pathway, which partially reduces oxygen to hydrogen peroxide (H2O2) in acidic environments or to superoxide (HO2-) in alkaline environments [131]. The 4e- pathway is more efficient, while the 2e- pathway produces reactive oxygen species that can harm catalysts and system performance. Currently, state-of-the-art platinum (Pt) catalysts for ORR are hindered by high costs and poor durability. Consequently, finding alternatives to Pt catalysts is crucial for fuel cell commercialization. There are three primary strategies for developing new ORR catalysts: low-Pt catalysts, which include Pt-transition metal (TM) alloys or Pt nanostructures, TM-based Pt-free catalysts, and metal-free catalysts. Among these, TM-based Pt-free catalysts are particularly notable due to their cost-effectiveness, abundant precursors, and high catalytic activity [132,133].
Figure 16
Figure 16.
Schematic presentation of the unified 2e- and 4e- pathways in electrochemical ORR. Reproduced with permission [131]. Copyright 2024, Wiley.
The development and application of ORR catalysts face several key challenges, including scaling up from lab to industry, improving mechanistic understanding, stabilizing single atom catalysts, exploring sustainable water sources, and aligning evaluation standards between rotating disk electrode (RDE) and membrane electrode assembly (MEA) tests. Overcoming these issues is crucial for advancing sustainable and efficient fuel cell and metal-air batteries technologies.
(1) In laboratory-scale research, scientists can precisely control reaction conditions, material ratios, and synthesis steps to produce catalysts with excellent performance. However, when these catalysts move to industrial-scale production, it is often challenging to maintain the same performance due to the larger production scale, differences in equipment, and changes in process parameters. Therefore, a key challenge is how to preserve the catalyst's activity, selectivity, and stability during large-scale production. To address this issue, researchers need to develop new synthesis methods and processes that enable precise control in industrial-scale manufacturing. This may involve optimizing reaction conditions, improving equipment design, and increasing automation. Additionally, real-time monitoring and evaluation of the catalyst's performance is necessary to allow for timely adjustments to the production process, ensuring consistent product quality. Reducing production costs is another crucial concern in the large-scale preparation of ORR catalysts. In laboratory studies, researchers often focus more on the catalyst's performance rather than its cost. However, in industrial production, cost is a key factor. To reduce costs, researchers must look for cheaper raw materials, simplify synthesis steps, and improve production efficiency. Moreover, by enhancing the design and structure of the catalyst, its activity and durability can be improved, reducing the amount needed and extending its lifespan, which further lowers overall costs [134].
(2) Despite significant advancements in modeling and simulation of electrocatalyst, our understanding of their mechanisms during ORR remains limited. To unravel complex ORR catalytic mechanisms, machine learning combined with advanced in situ techniques like in situ X-ray absorption spectroscopy, in situ Raman spectroscopy and in situ X-ray diffraction is essential. This integration allows researchers to comprehensively understand the mechanisms and dynamic processes at the electrocatalyst surface.
(3) Single atom catalysts (SACs) have shown great potential for achieving maximum atomic efficiency, high ORR activity, and stability [135]. These properties make them highly attractive candidates in recent years. However, SACs are susceptible to agglomeration due to their large surface free energy. This aggregation can occur during the electrocatalytic process, resulting in a mixed phase that includes single atoms, metal nanoparticles, and nanoclusters. Such a mixture can significantly impact the catalytic activity and reaction mechanism, posing challenges in maintaining the desired performance of SACs.
(4) ORR and its application in fuel cells offer a promising clean technology, but large-scale implementation raises concerns about strain on freshwater resources. Sustainable alternatives like seawater-based fuel cells are gaining interest to mitigate this issue. Seawater's unique characteristics–abundant, no freshwater consumption, and near-neutral pH–offer advantages for ORR applications. Expanding ORR to non-traditional water sources can help address water scarcity and promote sustainable energy conversion.
(5) The difference in catalyst evaluation standards between half-cell RDE tests and full-cell MEA assessments lead to a mismatch in observed activities. As the core component of fuel cells, the MEA plays a crucial role in determining their overall performance. RDE testing, though rapid and sensitive to layer quality, provides data at low current densities that often do not correlate with higher-density real-cell conditions. Conversely, MEA evaluations, which are more aligned with fuel cell environments, face challenges such as catalyst degradation, mass transport issues, and water management. This gap highlights the need for bridging the differences in testing standards to ensure high-performance ORR catalysts identified in RDE can translate to MEA efficacy.
9.3
Advances in science and technology to meet challenges
This section discusses ways to improve ORR efficiency. It highlights the effectiveness of transition metals like Fe and Co, especially in acidic conditions. SACs with modified ligands, such as iodine in FePc, enhance ORR by altering electronic properties. vacancy engineering and catalysts' structural morphology optimization also play crucial roles in enhancing ORR performance. The choice of carrier material, with its surface area, pore size, and interaction with active sites, further influences catalytic efficiency and durability.
(1) The use of transition metals like Fe and Co in 4e- catalysts enhance oxygen and intermediate adsorption, promoting efficient 4e- processes. Notably, in acidic conditions, Fe-N-C catalysts exhibit superior activity, highlighting their enhanced performance in such environments [136,137].
(2) SACs show superior ORR activity due to their unique structure. As an important component of SACs, ligands have a significant impact on ORR performance. For instance, when axial iodine ligands are modified, the ORR electrocatalytic activity and stability of FePc are significantly enhanced. This enhancement is attributed to the fact that iodine can generate high-spin electron states, which regulate the electronic structure. Additionally, the presence of iodine ligands decreases the interaction strength between reactive oxygen species and catalytic active sites, making the reaction thermodynamically favorable.
(3) Creating vacancies on the surface of catalytic active sites can optimize the ORR performance. Vacancy engineering influences the 4e- activity of ORR in multiple ways, including enhancing the conductivity of the active sites, influencing the filling of the Eg states and spin states, and activating adjacent atoms. Yang et al. reported a novel strategy to boost reversible oxygen electrocatalytic performance by a synergistic effect between electronically correlated Co-N-C and oxygen vacancy at the interface of a non-stoichiometry Co3O4-x/N-doped graphene composite catalyst. As shown in Fig. 17a, the Co-N-C cooperates with its neighboring oxygen vacancy to effectively modulate the charge density of oxygen-vacant Co active site, thus optimizing its adsorption energies and in turn remarkably enhancing intrinsic ORR/OER activities [138].
Figure 17
Figure 17.
The mechanism of enhancing catalyst ORR performance through vacancy engineering. Reproduced with permission [138,139]. Copyright 2020, Elsevier; Copyright 2022, American Chemical Society.
(4) The regulation of catalysts' structural morphology by carefully selecting the active center metals is an effective strategy for optimization. For instance, catalysts engineered with a conical geometry are conducive to facilitating both product desorption and adsorption processes, while leveraging the sharp-tip effect can further improve catalytic performance. Fig. 17b demonstrates a special ultrathin N-doped graphene nanomesh (NGM) as a robust scaffold for highly exposed Fe–N4 active sites. The pore sizes of the NGM can be elaborately regulated by adjusting the thermal exfoliation conditions to simultaneously disperse and anchor Fe–N4 moieties, ultimately leading to highly loaded Fe single-atom catalysts (SA-Fe-NGM) and a highly exposed morphology [139].
(5) The shape, size, and pore structure of a carrier strongly affect the performance of catalyst active sites. Carriers with larger surface areas provide more active sites, improving catalytic efficiency, while specific pore sizes help selectively trap reactant molecules, enhancing selectivity and improving mass transfer [140]. The interaction between the carrier and active sites, through chemical bonds or van der Waals forces, affects stability and durability. Additionally, the carrier's impact on the dispersion and aggregation state of active sites further modulates their catalytic efficacy.
9.4
Concluding remarks and prospects
The pursuit of efficient and sustainable ORR catalysts is a critical effort in the advancement of clean energy technologies, particularly for fuel cells and metal-air batteries. The current status of ORR catalysts, dominated by platinum-based materials, is fraught with challenges such as high cost and limited durability. This has spurred extensive research into alternative catalysts, with a focus on transition metal-based, metal-free, and SACs. Despite the significant strides made in the laboratory, translating these advances to industrial-scale production remains a challenge. The preservation of catalyst performance at scale requires innovative synthesis methods, precise control over production parameters, and the development of cost-effective processes. Additionally, the aggregation of SACs during electrocatalysis poses a significant obstacle, necessitating strategies to maintain their dispersed state and thereby their high activity and stability. The integration of machine learning with advanced in situ techniques offers a promising avenue for unraveling the complex mechanisms of ORR catalysts. This approach could lead to a more understanding of the dynamic processes at the electrocatalyst surface, potentially revealing new pathways for catalyst optimization. Looking ahead, the development of ORR catalysts must address not only performance metrics but also the broader implications of their implementation. The exploration of non-traditional water sources, such as seawater, for fuel cell applications could mitigate the strain on freshwater resources and promote sustainable energy conversion. In conclusion, while the path to commercializing ORR catalysts for widespread use in clean energy technologies is complex, it is not insurmountable. Continued research into novel catalyst materials, synthesis methods, and a deeper understanding of catalytic mechanisms will be essential. The future holds the promise of more efficient, durable, and cost-effective ORR catalysts that can drive the transition to a cleaner and more sustainable energy future.
10.
Oxygen reduction reaction at high temperature
10.1
Status
High-temperature oxygen reduction reaction (HTOR) is a crucial electrochemical process in solid oxide fuel cells (SOFCs), which play a vital role in the energy conversion process of the fuel cells. As shown in Fig. 18, solid oxide fuel cells (SOFCs) are generally categorized into two main types. Oxygen-ion-conducting SOFCs (O-SOFCs), operates by reducing oxygen at the cathode to form oxygen ions, which subsequently drive the electrochemical reaction. Another type is proton-conducting SOFCs (H-SOFCs), where O2 is electrochemically reduced with protons to produce water molecules at the cathode. Notably, oxygen activation poses a greater challenge compared to hydrogen activation, necessitating a higher activation energy for the SOFC cathode. Consequently, the advancement of novel cathodes exhibiting superior activity and durability is imperative for the commercial viability of SOFCs.
Figure 18
Figure 18.
High-temperature oxygen reduction reaction in solid oxide fuel cell with different electrolytes.
Given the variations in cathodic reaction mechanisms across SOFCs with different electrolytes, it is crucial to emphasize the importance of designing cathode materials specifically tailored to each type of electrolyte [141].
In O-SOFC, the oxygen reduction reaction primarily encompasses the diffusion of oxygen and the surface exchange process of oxygen, such as the adsorption, dissociation, and charge transfer of O atoms, the diffusion of O2− ions on the oxide surface or bulk and the transfer of O2− ions at the electrode/electrolyte interface. A number of researchers have embarked on optimizing the electronic conductivity and oxygen ion transport characteristics of materials, with the ultimate goal of elevating the electrochemical performance of cathodes [142].
During the nascent phase of H-SOFC development, the majority of cathodes were adapted from those employed in SOFCs, which utilized typical mixed oxygen-ionic and electronic conductors as electrocatalysts. However, a notable limitation of most MOEC cathodes is their inherent proton insulation. The electrochemically active sites on MOEC cathodes are confined predominantly within triple-phase boundaries (TPBs), resulting in suboptimal performance in H-SOFCs. Recent research has revealed that cathode materials possessing intrinsic proton mobility can expand the active sites beyond TPBs to the entire cathode surface, enhancing the vitality of the oxygen reduction reaction process [143]. Nonetheless, integrating simultaneous conduction of electrons, oxide ions, and protons within the cathode remains a formidable challenge. Hence, incorporating intrinsic proton conduction properties into mixed oxide ion-electron conductors stands as a pivotal challenge for the advancement of H-SOFC cathode technology in the future.
Despite exhibiting excellent catalytic activity at high temperatures, perovskite-type materials like La0.6Sr0.4Co0.2Fe0.8O3 undergo structural modifications and chemical degradation, leading to a decline in reactivity. The degradation of material undermines the electrochemistry performance, drastically reducing its lifespan and compromising its durability [144]. Additionally, the increased resistance and the instability of chemical reactions at the electrode-electrolyte boundary critically undermine the long-term stability of the system. Enhancing the stability of oxygen electrode materials is crucial technical challenge that needs to be addressed to improve overall system performance and reliability.
10.3
Advances in science and technology to meet challenges
Currently, perovskite oxides stand out as a promising option for electrocatalysts in high-temperature oxygen reduction reactions, attributed to their distinctive physical and chemical attributes, encompassing a modifiable crystal structure and non-stoichiometric composition. As shown in Fig. 19, various structural forms of perovskite oxides, including ABO3-type single perovskite, double perovskites, and Ruddlesden-Popper (RP) perovskites, are employed for high-temperature oxygen reduction. Over the past few years, researchers have devised diverse optimization strategies to tailor the conductive properties and bolster the stability of perovskite cathodes.
Figure 19
Figure 19.
Materials and optimization strategies for high-temperature oxygen reduction reaction.
The first, and most widely adopted method, is doping elements into perovskite to create a single-phase cathode with mixed conductors. Specifically, Zn2+ doping within the double perovskite oxide PrBaFe2O5+δ increases the concentration of oxygen vacancies and holes, thereby improving its mixed oxygen ionic and electronic conduction. As a result, an oxygen-based solid oxide fuel cell (O-SOFC) equipped with a Zn-doped PrBaFe2O5+δ cathode achieves a peak power density of 1.06 W/cm2 at 750 ℃ and exhibits remarkable stability over 240 h at 700 ℃ [145]. In addition to cation doping, anion doping can also effectively regulate ion transport in perovskite. For instance, F- doping in BaCo0.4Fe0.4Zr0.1Y0.1O3-δ perovskite oxide leads to an inductive effect, which increases protonic defects and lowers the migration energy barrier for protons on lattice oxygen. Consequently, a H-SOFC with an F- doped BaCo0.4Fe0.4Zr0.1Y0.1O3-δ perovskite cathode demonstrates a peak power density of 0.921 W/cm2 at 650 ℃ [146].
Another effective method is building heterojunctions, which has shown substantial potential in reducing interfacial resistance of SOFC cathode and enhancing electrochemical performance. For example, a SraCebFecNidO3-δ (SCFN) nanocomposite has been reported as the oxygen electrode of reversible H-SOFC. This nanocomposite consists of RP perovskite, NiO and CeO2 phases. The RP phase within the SCFN nanocomposite is proposed to bolster hydration capacity and expedite proton transfer, while NiO and CeO2 nanoparticles facilitate O2 surface exchange and O2- transfer, collectively enhancing the ORR activity [147].
In addition to the two strategies mentioned above, surface reconstruction is also proposed to modify the perovskite cathode. For example, the structure of PSFC is optimized by the exsolution of nanoparticles, creating B-site ion vacancies and oxygen vacancies that enhance the adsorption and migration of oxygen species [148]. A novel composite cathode with surface reconfiguration, which BaCoO3-δ (BCO) nanoparticles formed by water-induced precipitation on a Ba0.9Pr0.1Co0.7Fe0.2Y0.1O3-δ (BPCFY) substrate with e−/O2−/H+ triple conductivity demonstrates high catalytic activity for the oxygen reduction reaction and excellent stability under high steam concentrations [149].
10.4
Concluding remarks and prospects
Despite the diversification and advancement of materials research aimed at HTOR performance, numerous challenges and limitations persist. To gain a deeper understanding of the HTOR process, the following approaches can be pursued:
(1) Develop innovative material optimization strategies. Identifying novel materials and structures that augment both ORR activity and stability remains a pivotal objective. Addressing the synergistic effects of multiple conductive mechanisms through meticulous material design will be crucial for future material advancements.
(2) Investigate the underlying mechanisms of HTOR in greater depth. Current research largely focuses on material synthesis, yet the ion and electron transport mechanisms, as well as their reaction mechanisms during the electrochemical oxygen reduction process, necessitate further elucidation. For instance, in H-SOFC, the formation and transport of intrinsic protons, the concentration of oxygen vacancies, and their specific impacts on the HTOR process are currently limiting factors in material design.
(3) Establish high-throughput material screening methodologies. By integrating artificial intelligence and high-throughput technologies, material screening, structural optimization, and process parameter prediction can be accelerated, thereby expediting the development of new materials and reducing the number of experimental cycles.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Kezhen Qi: Writing – original draft. Zhidong Wei: Writing – original draft. Haibin Wang: Writing – original draft. Hongyan Liang: Writing – original draft. Dandan Ma: Writing – original draft. Jian-Wen Shi: Writing – original draft. Yifeng Li: Writing – original draft. Xuepeng Xiang: Writing – original draft. Yan Chen: Writing – original draft. Bo Yu: Writing – original draft. Chunchun Wang: Writing – original draft. Zhuo Xing: Writing – original draft. Claudio Imparato: Writing – original draft. Aurelio Bifulco: Writing – original draft. Daniil A. Lukyanov: Writing – original draft. Elena V. Alekseeva: Writing – original draft. Oleg V. Levin: Writing – original draft. M.I. Chebanenko: Writing – original draft. V.I. Popkov: Writing – original draft. Tan Zhang: Writing – original draft. Jinping Li: Writing – review & editing. Guang Liu: Writing – original draft. Wei Li: Writing – original draft. Linlin Song: Writing – original draft. Rongzheng Ren: Writing – original draft. Zhenhua Wang: Writing – original draft. Jianmin Ma: Writing – review & editing.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos. 22268003, 22102095, 52204320, U20A20246 and 12275199, U22A20418, 22075196, 21972110, 52202208), National Key Research and Development Program of China (Nos. 2023YFA1507903, 2022YFB3803600, 2022YFB4002501), SINOPEC (Beijing) Research Institute of Chemical Industry Co., Ltd. (No. 223239), the Fundamental Research Funds for the Central Universities (No. CCNU22JC017), the Postdoctoral Science Foundation of China (No. 2021M692535), the Natural Science Foundation of Shaanxi Province (No. 2022JQ-095), the Basic Research Project Foundation of Xi’an Jiaotong University (No. xzy012024012), the Youth Foundation of State Key Laboratory of Electrical Insulation and Power Equipment (No. EIPE2131), the Russian Science Foundation (No. 22-13-00035), the Ministry of Science and Higher Education within the framework of a State Assignment of the Ioffe Institute, Russian Academy of Sciences (No. FFUG-2024-0036), Yunnan Fundamental Research Projects (No. 202305AF150116), and the Research Project Supported by Shanxi Scholarship Council of China (No. 2022-050).
Figure 1
(a) Photocatalytic processes on semiconductor nanocrystals involving photoexcitation and formation of electron-hole pair in the nanocrystal. (b) Energy diagram of the same process for a semiconductor. (c) Energy loss mechanisms in solar fuel formation: ① below band gap photons, ② thermalization, ③ charge recombination, ④ overpotential, ⑤ back reaction, ⑥ separation and product extraction losses. (d) Semiconductor band alignment with possible levels of self-oxidation redox potential (left) and with two different hole scavengers (HS1 and HS2, shown on the right in blue and red), representing two potential energy balance scenarios. Item ⑦ represents a loss of stored energy due to the use of a sacrificial agent. Reproduced with permission [2]. Copyright 2018, American Chemical Society.
Figure 2
(a) Mechanistic pathways of CO2 reduction to commonly observed C1 and C2 products. Reproduced with permission [13]. Copyright 2022, American Chemical Society. (b) Mechanism illustration for photocatalytic conversion CO2 to CH3CH2COOH on Cu-ZnS. Reproduced with permission [14]. Copyright 2024, Wiley. (c) Free-energy profile of the CO2RR pathway on CuN2O2 sites in Cu-MOF and Structural models of the Gibbs free energy calculations for CO2 reduction on Cu-MOF. Reproduced with permission [15]. Copyright 2024, Wiley-VCH.
Figure 4
(a) Schematic illustration of Cu-CeO2 promotes C2+ production. Reproduced with permission [32]. Copyright 2022, American Chemical Society. (b) Schematic illustration of nano-defects promotes C2+ production. Reproduced with permission [33]. Copyright 2020, American Chemical Society. (c) Schematic illustration of increased CO binding energy favorable for C–C coupling on ON-CuO. Reproduced with permission [34]. Copyright 2023, American Chemical Society.
Figure 10
(a) Scheme of photocatalytic total water splitting and its three main steps, excitation (ⅰ), charge carrier migration (ⅱ) and surface reactions (ⅲ). Reproduced with permission [70]. Copyright 2022, Wiley. (b) Absorption spectrum of g-C3N4/Co3O4 photocatalyst and AQE in overall water splitting under simulated solar light. Reproduced with permission [77]. Copyright 2023, Elsevier. (c) Illustration of the charge carriers transfer and H2 and O2 evolution by piezo-photocatalysis on BiFeO3/COF. Reproduced with permission [81]. Copyright 2023, Springer Nature.
Figure 11
Advance of single atom catalysis for the 2e− ORR illustrated on oxygen binding modes. Reproduced with permission [94]. Copyright 2024, Springer Nature.
Figure 12
Various heterojunction schemes for photocatalysts, which may be employed in PEC ORR systems. Reproduced with permission [95]. Copyright 2023, Elsevier.
Figure 13
(a) Schematic representation of the photocatalytic production of ammonia. Reproduced with permission [102]. Copyright 2024, Elsevier. (b) Schematic representation of possible mechanisms of nitrogen reduction to ammonia. Reproduced with permission [103]. Copyright 2022, Frontiers.
Figure 17
The mechanism of enhancing catalyst ORR performance through vacancy engineering. Reproduced with permission [138,139]. Copyright 2020, Elsevier; Copyright 2022, American Chemical Society.

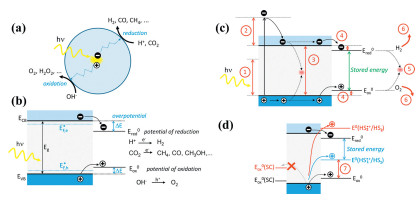
 DownLoad:
DownLoad:
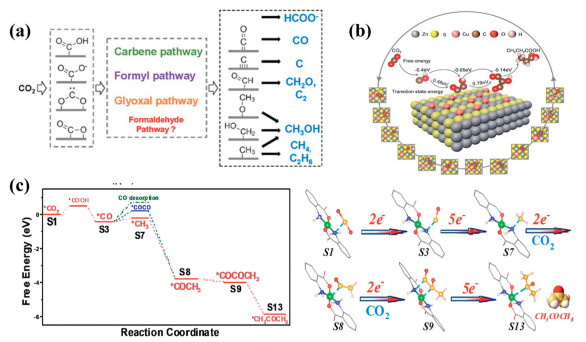
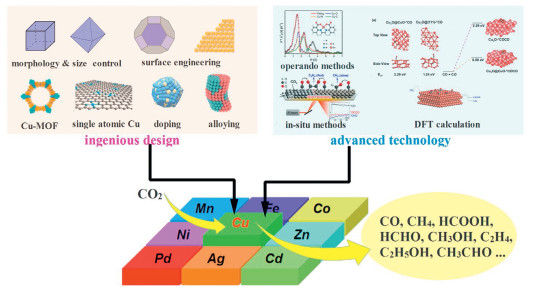
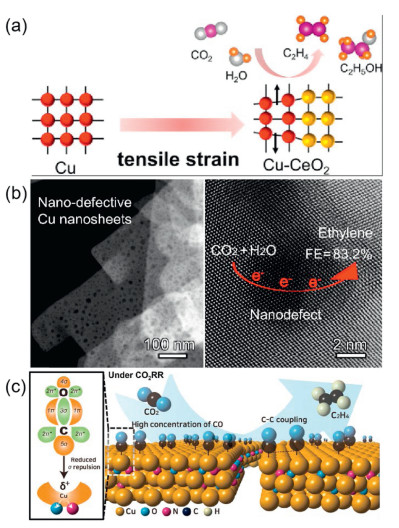
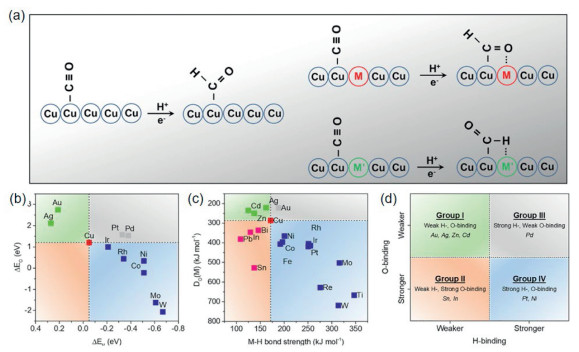
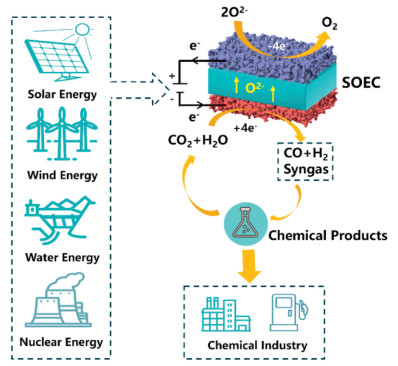
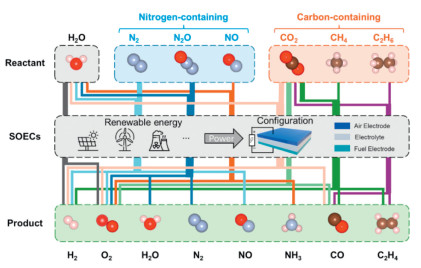
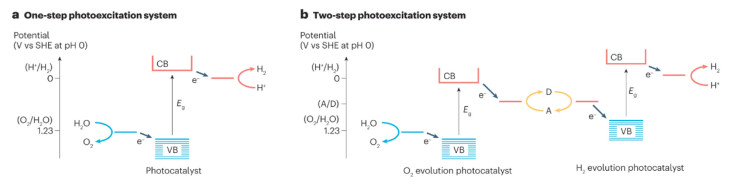
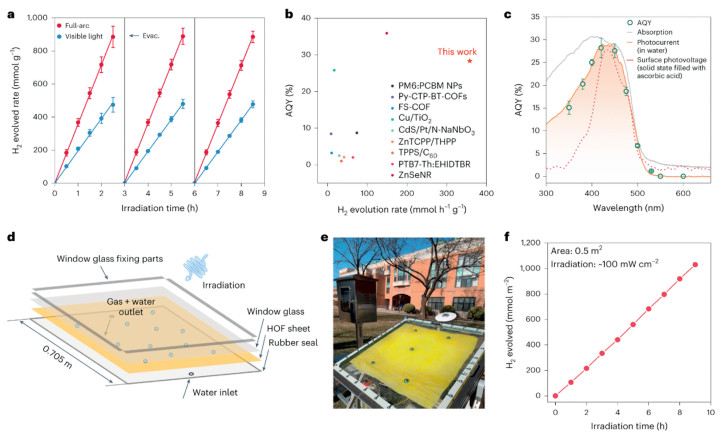
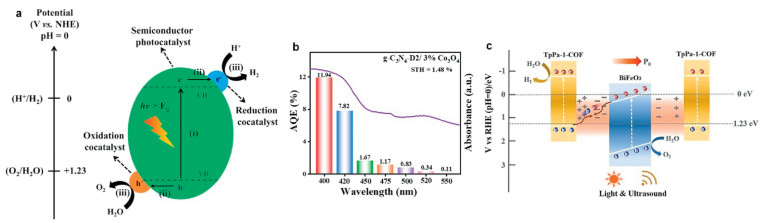
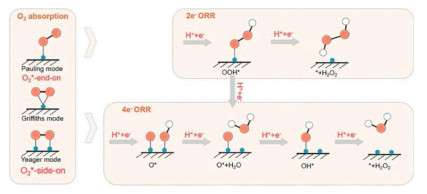
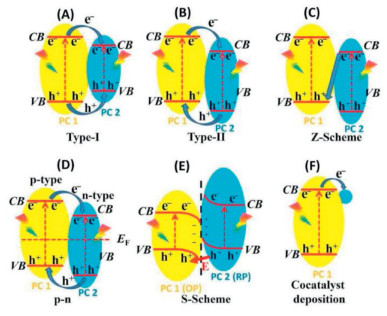


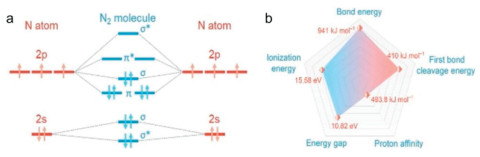
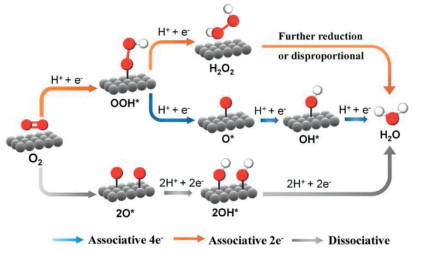



 下载:
下载:




















 下载:
下载:
