Department of Chemistry and Chemical Engineering, Henan Institute of Science and Technology, Xinxiang 453000, China
b.
Nanophotonics and Biophotonics Key Laboratory of Jilin Province, School of Physics, Changchun University of Science and Technology, Changchun 130022, China
c.
Key Laboratory of Applied Surface and Colloid Chemistry, Ministry of Education, School of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi'an 710119, China
* Corresponding authors. E-mail addresses: xugr70@163.com (G. Xu)
Received Date:
30 May 2025 Accepted Date:
30 July 2025 Revised Date:
27 July 2025 Available Online:
15 January 2026
Abstract:
Structural instability and sluggish lithium-ion (Li+) kinetics of spinel NiCo2O4 anodes severely hinder their applications in high-energy-density lithium-ion batteries. Mesocrystalline structures exhibit promising potential in balancing structural stability and enhancing reaction kinetics. However, their controlled synthesis mechanisms remain elusive. Herein, a substrate interface engineering strategy is developed to achieve controllable synthesis of mesocrystalline and polycrystalline NiCo2O4 nanorods. Remarkably, mesocrystalline NiCo2O4 exhibits a high capacity retention rate of 85.7% after 500 cycles at 2 A/g, attributed to its porous structure facilitating Li+ transport kinetics and unique stress-buffering effect validated by ex-situ TEM. Theoretical calculations and interfacial chemical analysis reveal that substrate-crystal surface engineering regulates the nucleation-growth pathways: Acid-treated nickel foam enables epitaxial growth via lattice matching, acting as a low-interfacial-energy template to reduce nucleation barriers and promote low-temperature oriented crystallization. In contrast, carbon cloth requires high-temperature thermal activation to overcome surface diffusion barriers induced by elevated interfacial energy. This substrate-driven crystallization kinetic modulation overcomes the limitations of random nucleation in conventional hydrothermal synthesis. The established substrate-crystal interfacial interaction model not only clarifies the kinetic essence of crystal orientation regulation but also provides a universal theoretical framework for lattice-matching design and mesostructural optimization of advanced electrode materials.
High-energy-density lithium-ion batteries (LIBs) urgently require the synergistic optimization of lithium-ion (Li+) transport kinetics and structural stability [1,2]. Transition metal oxide anodes (e.g., NiCo2O4) demonstrate significant potential owing to their multi-electron redox characteristics and natural abundance [3–7]. Compared to lithium metal batteries and solid-state batteries, they reduce the risk of dendrite formation while maintaining compatibility with existing electrolytes. However, the inherent rigidity of traditional polycrystalline NiCo2O4 induces to severe volumetric expansion (> 200%) during cycling, leading to structural pulverization and rapid capacity decay [4,6–8]. Although nanostructuring strategies can mitigate volumetric strain and enhance reaction kinetics, the disordered stacking of nanoparticles introduces abundant inactive grain boundaries, disrupting the structural continuity of conductive networks and creating an inherent conflict between kinetics and stability [4,6,7].
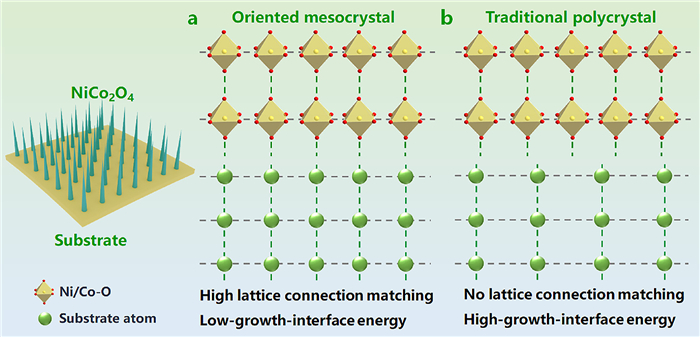
Mesocrystals composed of ordered nanoparticles offer novel strategies to address these challenges [9–16]. They maintain structural stability through 3D interconnected networks while preserving high surface area advantages [11–17]. Furthermore, intergranular stress redistribution mechanisms suppress structural collapse during cycling [1,12,14]. Nevertheless, the metastable nature of mesocrystals inevitably triggers uncontrolled recrystallization into thermodynamically stable singlecrystals or polycrystals [11,14]. Existing synthesis methods (e.g., template-induced and external-field-assisted syntheses) [13–24] lack universal control mechanisms for spinel oxide mesocrystals. Notably, the controlled epitaxial growth technique of nanostructures on low-dimensional substrates utilizing lattice-matched interfacial engineering [25], provides a promising direction for the fabrication of oriented mesocrystalline NiCo2O4 (Scheme 1). Particularly, the formation of highly interconnected and lattice-coherent interfacial can significantly reduce the interfacial energy during crystal growth, thus effectively promoting oriented crystallization.
Scheme 1
Scheme 1.
The epitaxial growth of (a) oriented mesocrystalline NiCo2O4 with highly connected and matched interfacial lattices (present work) and (b) traditional polycrystalline NiCo2O4 without highly connected and matched interfacial lattices.
Previous studies have demonstrated that mesocrystalline NiCo2O4 with distinct morphologies can be synthesized on nickel foam (NF) substrates through hydrothermal temperature tuning. Their growth mechanism follow a topological transformation process controlled by edge-to-core corrosive etching [1]. Based on this, we innovatively proposed a grain orientation regulation strategy based on interfacial engineering between NF and carbon cloth (CC) substrates, successfully realizing controllable synthesis of NiCo2O4 mesocrystalline and polycrystalline nanorods. The orientationally assembled mesocrystalline structure significantly optimized Li+ transport kinetics through constructing interconnected grain boundary networks and a unique grain-boundary synergistic deformation mechanism, with a high delithiation specific capacity of 1892 mAh/g (0.1 A/g) and 85.7% capacity retention rate (2 A/g, 500 cycles). By combining substrate-crystal interface analysis and density functional theory (DFT) calculations, we have identified the regulatory mechanism of substrate-crystal interfacial interactions on nucleation kinetics and growth modes: Acid-etched NF substrate establishes an epitaxial interface with merely 0.3% lattice mismatch NiCo2O4 (400) and Ni (110) planes, effectively reducing the nucleation activation energy, thereby enabling oriented nucleation and growth at low temperatures (30–60 ℃). In contrast, the chemically inert CC substrates required a thermodynamic driving force at 120 ℃ to overcome surface diffusion barriers to form polycrystals. This study elucidates the nature of substrate-induced crystal orientation in hydrothermal synthesis. The established substrate-crystal interaction model not only deepens fundamental understanding of heterogeneous nucleation dynamics, but also provides a universal theoretical framework for lattice-matched design and mesostructure controlled synthesis of advanced energy materials.
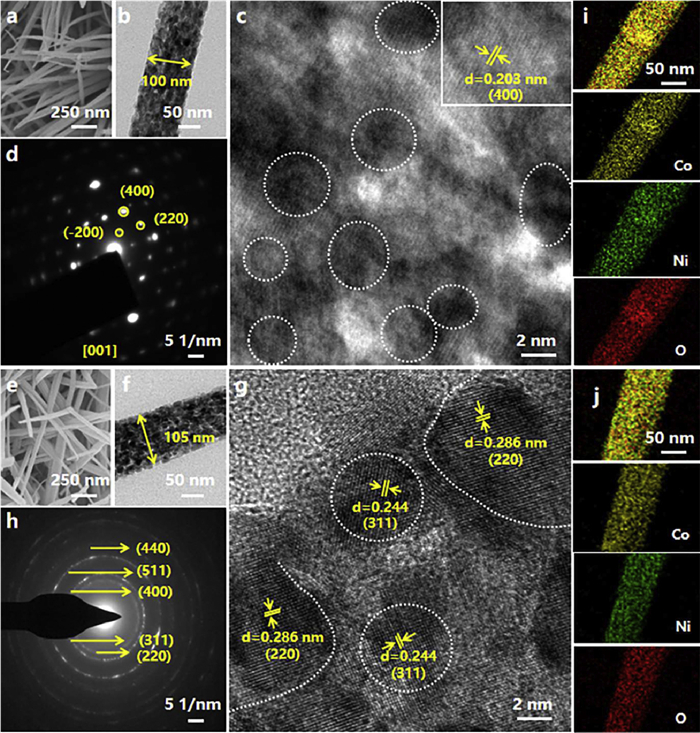
Scanning electron microscopy (SEM) and low-resolution transmission electron microscopy (TEM) analyses (Figs. 1a and b, Fig. S1a in Supporting information) revealed that the NiCo2O4 nanorods (approximately 100 nm in diameter) synthesized on the NF substrate constructed a porous network through the self-assembly of primary nanoparticles (2–5 nm). Additionally, uniform lattice fringes with a spacing of 0.203 nm (marked by white circles), corresponding to the (400) planes of spinel NiCo2O4, were observed through high-resolution TEM (HR-TEM) within the individual nanorods in Fig. 1c. Remarkably, the (400) lattice fringes maintain strict parallel alignment along the nanorod axis despite the existence of distinct grain boundaries between nanoparticles, indicating unified crystallographic orientation among primary particles during self-assembly. More importantly, the selected-area electron diffraction (SAED) in Fig. 1d displayed periodic single-crystal diffraction spots along the [001] zone axis of the nanorods, further confirming their single-crystalline diffraction characteristics. Notably, this single-crystal pattern formed through oriented self-assembly of crystallographically aligned nanoparticles is a typical example of mesocrystalline structures [1,11,18]. In contrast, NiCo2O4 nanorods (with a diameter of about 105 nm, Fig. S1b in Supporting information) synthesized on CC substrates consisted of primary particles sized 5–10 nm in Figs. 1e and f. In addition, (220) and (311) planes corresponding to lattice fringes of 0.286 nm and 0.244 nm, respectively, exhibiting disordered orientation characteristics in Fig. 1g, suggesting a lack of preferred crystallographic alignment. More importantly, further evidence of the polycrystalline nature was provided by the SAED pattern, showing distinct diffraction rings (Fig. 1h). In summary, these observations have confirmed the successful synthesis of polycrystalline nanorods on CC substrates. Elemental mapping further demonstrated the homogeneous distribution of Ni, Co, and O across the nanorods from both substrates (Figs. 1i and j). In addition, X-ray diffraction (XRD) analysis showed that NiCo2O4 nanorods synthesized on different substrates maintained high crystallinity and phase purity (Fig. S2 in Supporting information) [26–28], indicating that substrate variations mainly influence crystallographic growth modes rather than chemical composition. Brunauer-Emmett-Teller (BET) measurements quantitatively highlighted the structural advantages of mesocrystalline NiCo2O4 (Fig. S3 in Supporting information).
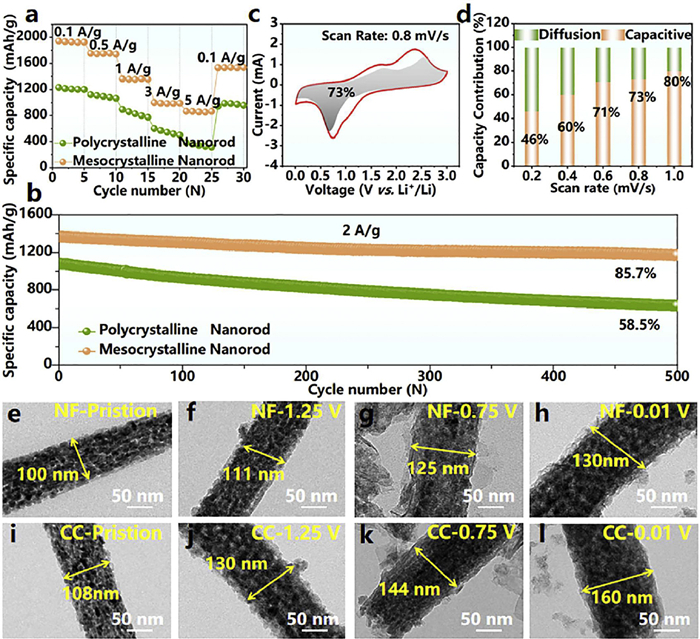
Mesocrystalline structure provided a delithiation specific capacity of 1892 mAh/g at a current density of 0.1 A/g, exhibiting a remarkable 64.5% enhancement over polycrystalline nanorods (1150 mAh/g) (Fig. S4 in Supporting information). Furthermore, the mesocrystalline nanorods demonstrated exceptional rate capability, retaining a reversible specific capacity of 1440 mAh/g even when the current density was increased to 5 A/g, with a capacity recovery rate of 78% upon returning to 0.1 A/g (Fig. 2a). In addition, long-term cycling stability tests further highlighted the structural advantages of mesocrystalline NiCo2O4 nanorods (Fig. 2b), where the mesocrystalline electrode maintained a stable reversible specific capacity of 1173 mAh/g after 500 cycles at 2 A/g, corresponding to an outstanding capacity retention rate of 85.7%. This exceptional cyclability significantly surpasses previously reported NiCo2O4-based electrodes (Table S1 in Supporting information). Different Li+ transport behaviors between mesocrystalline and polycrystalline NiCo2O4 nanorods were systematically investigated through scan-rate-dependent voltammetric kinetic analysis [29–35]. The redox peak currents of both materials within the scan rate range of 0.1–1 mV/s followed a power-law relationship as shown in Fig. S5 (Supporting information), where the kinetic exponent b quantitatively distinguishes capacitive-dominated (b = 1) and diffusion-controlled (b = 0.5) charge storage mechanisms [1,32,33]. Notably, the b values of mesocrystalline nanorods closer to 1, highlighting their superior capacitive-dominated behavior (Fig. S6 in Supporting information). These structural advantages were further confirmed by the capacity contribution analysis at 0.8 mV/s (Fig. 2c and Fig. S7 in Supporting information), where the mesocrystalline electrode delivered a 73% capacitive contribution, representing an 8.9% enhancement over polycrystalline nanorods (67%). Scan-rate-dependent quantitative analysis (Fig. 2d and Fig. S8 in Supporting information) consistently validated this advantage [31,32]. Complementary evidence from electrochemical impedance spectroscopy (EIS) in Fig. S9 (Supporting information) revealed a lower charge-transfer resistance for mesocrystalline nanorods [36]. The improved kinetics can be ascribed to: (1) Optimized electrolyte infiltration enabled by hierarchical porosity; (2) Enhanced structural integrity due to single-crystalline building units, effectively mitigating cyclic degradation. Notably, the composition of the electrolyte plays a decisive role in the stability of the electrode/electrolyte interface [37,38]. This provides an innovative strategy for the synergistic optimization of microcrystalline NiCo2O4 electrodes: their unique 3D interconnected grain boundary network structure can form a strong interfacial coupling effect with novel electrolytes, thereby significantly enhancing the electrochemical performance of the electrode material.
Figure 2
Figure 2.
The Li+-storage properties of the NiCo2O4 nanorods. (a) Rate performance from 0.1 A/g to 5 A/g. (b) Cycle stability at 2 A/g. (c) Separation of capacitive (gray part) and diffusion (red line part) controlled contribution at 0.8 mV/s and (d) pseudocapacitive contribution ratio at a series of scan rates for the mesocrystalline nanorods. Ex-situ TEM images of (e-h) mesocrystalline and (i-l) polycrystalline NiCo2O4 nanorods at various lithiation voltages.
To elucidate the mechanical response mechanisms of mesocrystalline and polycrystalline NiCo2O4 nanorods during lithiation processes, we systematically tracked individual nanorods via ex-situ TEM. We concentrate on the critical first lithiation stage, revealing the unique advantages of the mesocrystalline structure in suppressing initial irreversible volumetric expansion by comparing the evolution of mechanical strength between mesocrystalline and polycrystalline nanorods. This design is based on two considerations: (1) Structural damage caused by the first lithiation is the primary cause of electrode performance degradation; (2) Our previous study has already confirmed the stability in subsequent cycles [1], thus the current work prioritizes addressing the more pressing issue of the initial cycle. The results demonstrated that NiCo2O4 nanorods synthesized on two different substrates (NF and CC) exhibited typical transformational electrochemical behavior, manifested by progressive volumetric expansion with increasing lithiation depth [1,39,40]. Specifically, the radial expansion of mesocrystalline nanorods grown on NF ranged from 11% to 30% when the lithiation potential was decreased from 1.25 V to 0.01 V (vs. Li+/Li) (Figs. 2e-h and Fig. S10 in Supporting information), while those on CC exhibited a higher expansion gradient of 20% to 48% (Figs. 2i-l and Fig. S10). Notably, mesocrystalline NiCo2O4 nanorods demonstrated a 37.5% reduction in volumetric expansion ratio compared to their polycrystalline nanorods at 0.01 V. This discrepancy can be attributed to the unique structural advantages of mesocrystalline nanorods The hierarchical organization of nanocrystalline subunits enables superior stress buffering capacity, while synergistic deformation mechanisms at internal grain boundaries effectively redistribute anisotropic stresses generated during lithiation.
Figs. 3a-d illustrate the substrate-driven growth process of mesocrystalline NiCo2O4 nanorods on NF. At a low temperature (30 ℃), rapid formation of non-uniform lamellar structures was observed in Fig. 3a, confirming that the abundant active sites on the NF surface significantly facilitated heterogeneous nucleation. Upon increasing the temperature to 90 ℃, structural densification of nanosheets with intertwined sheet-rod features emerged (Figs. 3b and c), which can be attributed to surface energy anisotropy-driven preferential growth along specific crystallographic planes. Further elevating the temperature to 120 ℃ triggered structural reorganization via Ostwald ripening, resulting in short rod-like nanostructures (Fig. 3d). Notably, extending the reaction time to 12 h eventually yielded well-aligned mesocrystalline nanorods (Figs. 1a-d). This morphological evolution suggests that the NF substrate not only reduces the nucleation energy barrier but also enables synergistic regulation of rapid low-temperature nucleation and oriented growth. In contrast, the CC substrates driven a polycrystalline growth pathway (Figs. 3e-h). At 30 ℃, only sporadic nanoparticles were formed (Fig. 3e), while increasing the temperature to 60 ℃ promoted particle proliferation without structural ordering (Fig. 3f). At 90 ℃, surface energy minimization drove particle assembly into nanosheets with a thickness of 325 nm (Fig. 3g). Subsequent heating to 120 ℃ induced anisotropic etching, thinning the nanosheets to 75 nm (Fig. 3h). After 12 h of reaction, polycrystalline nanorods with morphology comparable to the NF system were obtained (Figs. 1e-h). This transformation highlights that crystal growth on CC is controlled by diffusion-controlled rather than interface-directed mechanisms.
Figure 3
Figure 3.
SEM images and corresponding schematic diagrams illustrating the structural evolution during the growth of (a-d) mesocrystalline and (e-h) polycrystalline NiCo2O4 nanorods. (a, e) 30 ℃-1 h; (b, f) 60 ℃-1 h; (c, g) 90 ℃-1 h; (d, h) 120 ℃-1 h.
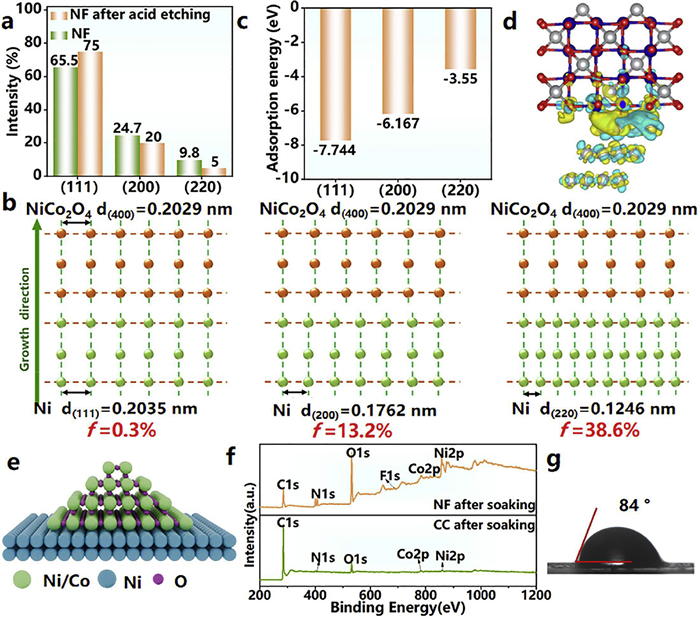
Substrate-crystal interface analysis and theoretical calculations further revealed the differential regulatory mechanisms of acid treatment on the surface properties and crystal growth behaviors of NF and CC substrates. XRD analysis demonstrated that acid treatment induced a preferential crystallographic orientation on the NF surface. Specifically, the diffraction peak intensity of the (111) plane increased about 10%, whereas those of the (200) and (220) planes decreased about 5% (Fig. 4a and Fig. S11 in Supporting information). This phenomenon is attributed to the preferential dissolution of Ni (200) and Ni (220) planes with higher surface energy during the acid etching process, thereby promoting the exposure of the low-index Ni (111) plane [41]. In contrast, no significant changes in diffraction peak intensities were observed for acid-treated CC (Fig. S12 in Supporting information), confirming the negligible influence of acid treatment on the crystalline structure of carbon fibers. When the lattice mismatch (f) is below 5%, minimization of interfacial energy facilitates effective heteronucleation and epitaxial growth [25,42,43]. Lattice mismatch calculations elucidated the key mechanism of heteroepitaxial growth: the mismatches between Ni (200)/Ni (220) planes and the NiCo2O4 (400) plane (the dominant exposed facet of mesocrystals in Fig. 1c) were as high as 13.2% and 38.6%, respectively. Contrastingly, the mismatch between the Ni (111) plane and NiCo2O4 (400) plane was only 0.3% (Fig. 4b), significantly below the critical threshold required for epitaxial growth.
Figure 4
Figure 4.
NF-driven epitaxial growth mechanism facilitates nucleation and growth. (a) Crystal facet ratio of NF and NF after acid etching. (b) The lattice mismatch of Ni (111), Ni (200) and Ni (220) to NiCo2O4 (400). (c) Adsorption energies of NiCo2O4 (400) on Ni (111), Ni (200) and Ni (220). (d) Charge density difference of NiCo2O4 (400) on Ni (111). (e) Schematic diagram of lattice matching-induced epitaxial growth. (f) XPS spectrum of NF after soaking and CC after soaking. (g) Static water contact-angle measurement of NF after acid etching.
DFT calculations revealed that the Ni (111) facet exhibited the most favorable thermodynamic stability with an adsorption energy of −7.744 eV, significantly lower than those of Ni (200) (−6.167 eV) and Ni (220) (−3.550 eV) systems (Fig. 4c) [44–47]. This energetic superiority suggests stronger thermodynamic driving forces for interfacial bonding in the Ni (111) structure. Depending on the stacking patterns, the structure c in Fig. S13 (Supporting information) is the most thermodynamicly stable while the structure b has the most potential to be formed by depositing NiCo2O4 on the (111) surface of Ni (Fig. S14 in Supporting information). Interfacial charge density difference analysis (Fig. 4d and Fig. S15 in Supporting information) demonstrated substantial charge redistribution at the Ni (111)/NiCo2O4 (400) heterointerface, with a charge transfer quantity of 0.21 e/Å3, representing approximately 10%−20% enhancement compared to other configurations [44,47]. The electrons surrounding the Co and O atoms of NiCo2O4 are highly delocalized on the Ni (111) plane, thereby resulting in a stronger chemical interaction between the NiCo2O4 molecule and Ni. This pronounced electronic reconstruction originates from the exceptional geometric coherency matching between Ni (111) and NiCo2O4 (400), as evidenced by the ultralow lattice mismatch (0.3%, Fig. 4b). In short, acid-treated NF optimizes lattice coherence between the (111) plane and NiCo2O4 (400) plane (Fig. 4e), thereby reducing the nucleation activation energy to a thermodynamically favorable range. In comparison, CC lacks effective lattice-matching sites (Fig. S16 in Supporting information), leading to the formation of disordered polycrystalline structures under high-temperature thermodynamic driving forces (Figs. 1f-h). X-ray photoelec-tron spectroscopy (XPS) analysis were conducted to investigate the chemical states of substrate surfaces after solution immersion to clarify the role of substrate-solution interfacial chemistry in crystal growth. As shown in Figs. S17 and S18 (Supporting information) characteristic peaks of urea and NH4F were detected in C 1s, N 1s, and F 1s spectra, confirming the physical adsorption of precursors on both substrates. Deconvolution of Ni 2p3/2 and Co 2p3/2 spectra revealed that both Ni and Co existed in the +2 oxidation state (Fig. S19 in Supporting information), ruling out redox reactions during solution immersion. Notably, the characteristic peak intensities on CC were significantly lower than those on NF (Fig. 4f and Figs. S17-S19 in Supporting information). Combined with contact angle measurements (CC: 124°; NF: 84°; Δθ = 40°, Fig. 4g and Fig. S20 in Supporting information), this indicates that the three-dimensional interconnected network of NF enhances solution wettability (48% reduction in contact angle) [45], thereby optimizing reactant adsorption and mass transport kinetics to promote efficient nucleation and oriented growth at low temperatures.
Based on the above experimental verification and DFT computational analysis, we have revealed the synergistic regulation mechanism between the surface characteristics of porous substrates and the growth kinetics of oxide crystals. When employing NF as a substrate for synthesizing mesocrystalline NiCo2O4 nanorods, three synergistic effects were identified: (1) Ni (111) crystal planes exposed by acid treatment formed a low lattice-mismatched interface with the NiCo2O4 (400) planes (mismatch factor f = 0.3%), significantly reducing the activation energy for heterogeneous nucleation. (2) The three-dimensional porous network structure remarkably enhanced electrolyte wettability (contact angle decreased by 48%), optimizing the spatial distribution of precursors and mass transfer kinetics. (3) The low-index crystallographic planes facilitated the preferential epitaxial growth of crystals along the [100] direction. This lattice-matching-driven heteroepitaxial mechanism enables rapid kinetics-controlled nucleation and oriented assembly under mild reaction conditions (30–60 ℃), ultimately yielding mesocrystalline nanorods with single-crystal-like characteristics (Figs. 3a-d and Figs. 1a-d). In contrast, CC substrates result in surface diffusion-dominated crystal growth due to their chemically inert surface and lack of lattice-matching sites. This necessitates a thermodynamic driving force provided by high temperatures (120 ℃) to overcome energy barriers, resulting in typical polycrystalline nanostructures (Figs. 3e-h and Figs. 1e-h).
In conclusion, the controlled synthesis of mesocrystalline/polycrystalline NiCo2O4 nanorods were achieved through substrate interface engineering, systematically elucidating the interaction mechanism between substrate-crystal interfacial directional growth. This substrate-driven growth mechanism challenges the long-held view of thermodynamically dominated in classical crystal growth theory, establishing a quantitative correlation between substrate functionalization and crystal growth pathways. More importantly, we propose a universal design paradigm: "Lattice matching → Nucleation kinetics → Mesostructure → Electrochemical performance" to reconcile the long-term trade-off between Li+ transport kinetics and structural stability in energy storage materials. These findings provide important insights for developing next-generation high-energy-density, fast-charging batteries.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This project was financially supported by the National Nature Science Foundation of China (No. 52401273), and Science and Technology Department of Henan (Nos. 242102241007, 252102320178 and 252102321067), and Training Program for Young Backbone Teachers in Higher Education Institutions in Henan Province (No. 2024GGJS101).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.111660.
[1]
J.J. Liu, Y.F. Yuan, X.W. Guo, et al., Adv. Energy Mater. 11 (2021) 2100503. doi: 10.1002/aenm.202100503
H. Yang, K.K. Fang, J.W. Duan, et al., Energy Storage Mater. 75 (2025) 103995. doi: 10.1016/j.ensm.2024.103995
[46]
H.L. Peng, Y. Han, L. Zhang, et al., Angew. Chem. Int. Ed. 64 (2025) e202502088. doi: 10.1002/anie.202502088
[47]
Y.X. Yu, P. Li, X.Y. Xie, et al., Energy Storage Mater. 74 (2025) 103904. doi: 10.1016/j.ensm.2024.103904
Scheme 1
The epitaxial growth of (a) oriented mesocrystalline NiCo2O4 with highly connected and matched interfacial lattices (present work) and (b) traditional polycrystalline NiCo2O4 without highly connected and matched interfacial lattices.
Figure 2
The Li+-storage properties of the NiCo2O4 nanorods. (a) Rate performance from 0.1 A/g to 5 A/g. (b) Cycle stability at 2 A/g. (c) Separation of capacitive (gray part) and diffusion (red line part) controlled contribution at 0.8 mV/s and (d) pseudocapacitive contribution ratio at a series of scan rates for the mesocrystalline nanorods. Ex-situ TEM images of (e-h) mesocrystalline and (i-l) polycrystalline NiCo2O4 nanorods at various lithiation voltages.
Figure 4
NF-driven epitaxial growth mechanism facilitates nucleation and growth. (a) Crystal facet ratio of NF and NF after acid etching. (b) The lattice mismatch of Ni (111), Ni (200) and Ni (220) to NiCo2O4 (400). (c) Adsorption energies of NiCo2O4 (400) on Ni (111), Ni (200) and Ni (220). (d) Charge density difference of NiCo2O4 (400) on Ni (111). (e) Schematic diagram of lattice matching-induced epitaxial growth. (f) XPS spectrum of NF after soaking and CC after soaking. (g) Static water contact-angle measurement of NF after acid etching.

 DownLoad:
DownLoad:


 下载:
下载:






 下载:
下载:
