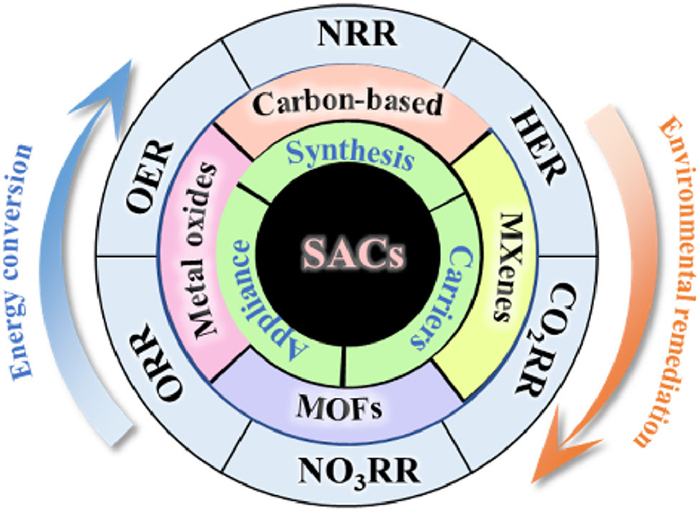
Figure 1.
The detailed outline diagram specifically designed for this comprehensive review.
Support engineering of single-atom electrocatalysis: Mechanism analysis and application expansion
Mingying Chen , Junjie Ma , Xiyong Chen , Qian Liu , Yanhong Feng , Xijun Liu

In the contemporary era, the global community is confronted with pressing challenges in clean energy conversion and environmental protection, driven by the urgent need to address climate change and resource depletion [1–3]. As the demand for sustainable development intensifies, electrocatalytic technology has emerged as a cornerstone of scientific research and technological innovation [2,4,5]. However, traditional electrocatalysts, which often suffer from inefficiencies in atomic utilization, limited active sites, and insufficient durability, are increasingly inadequate in meeting the rigorous demands for high activity, selectivity, and stability required by modern energy and environmental applications [6]. These shortcomings collectively impede the development of electrocatalytic systems. The advent of single-atom catalysts (SACs) has marked a revolutionary breakthrough in electrocatalysis. SACs, characterized by their unique atomic-level dispersion of active metal sites, ensure maximal utilization of each metal atom, significantly enhancing atomic efficiency and offering unprecedented opportunities to overcome the limitations of conventional electrocatalysts [1,7–9]. As an essential structural component of SACs, the support materials not only provide physical anchoring for single-atom active sites but also play a critical role in modulating their electronic structure, geometric configuration, and local reaction environment [10–12]. In recent years, with the continuous optimization of SACs' structures, researchers have discovered that the synergistic effect existing between metal active centers and support materials can significantly enhance catalytic performance. This synergy arises not only from traditional electronic interactions but, more importantly, from the cooperative participation of both metal sites and support atoms in the catalytic reaction mechanism, forming unique strong metal-support interactions (SMSI) [13]. Due to the direct bonding between the metal atoms and the supports, the supports can exert significant influence on the electronic properties and geometric configuration of individual metal atoms, thereby optimizing their catalytic activity and selectivity. Furthermore, catalytic performance can be further refined through co-catalysis strategies. This metal-support synergistic catalytic mechanism not only provides new insights for designing highly efficient catalysts but also establishes a theoretical foundation for a deeper understanding of the structure-activity relationships in SACs [10,14]. By rationally designing support materials (e.g., tuning defect structures, surface chemical properties) and optimizing metal-support interactions, researchers can further unlock the potential of SACs, driving their widespread application in energy conversion (e.g., water splitting, CO2 reduction) and environmental remediation (e.g., pollutant degradation). The choice of support material, ranging from carbon-based frameworks and metal oxides to MXenes and metal-organic frameworks (MOFs), imparts distinct catalytic properties to SACs, making it imperative to systematically investigate the behavior and performance of SACs supported on diverse supports in various electrocatalytic reactions [7,10,15–17]. Over the years, our research team has dedicated extensive efforts to the study of SACs, accumulating profound insights into the role of support engineering in regulating single-atom electrocatalytic activity. Through innovative experimental designs and advanced characterization techniques, we have successfully developed a series of support materials with tailored structures and optimized properties, which have been effectively applied to the synthesis and catalytic applications of SACs [4,18,19]. These efforts have not only enhanced our understanding of the structure-activity relationships in SACs but also expanded the scope of their practical applications. Concurrently, we have closely monitored and critically evaluated advancements made by our peers in this rapidly evolving field, engaging in active academic exchanges and collaborative research initiatives to broaden our perspectives and refine our methodologies.
This review systematically examines the critical function of support engineering in regulating SACs' performance. We present a detailed account of our team's achievements in the development of SACs, alongside a comprehensive review of the progress made by other researchers in related areas. This provides a comprehensive overview of common synthetic methods for SACs, evaluating the advantages and limitations of each approach. We also systematically review the impact of various support materials, such as carbon-based structures, metal oxides, MXenes, and MOFs, on the catalytic performance of SACs, with a particular focus on their structural characteristics, electronic properties, and interaction mechanisms with active sites. Through detailed case studies of typical electrocatalytic reactions, including the oxygen reduction reaction (ORR), hydrogen evolution reaction (HER), oxygen evolution reaction (OER), carbon dioxide reduction reaction (CO2RR), nitrogen reduction reaction (NRR) and nitrate reduction reaction (NO3RR), the enhanced performance and inherent advantages of SACs enabled by advanced support engineering are thoroughly elucidated. Moreover, this work delves into the promising applications of SACs in renewable energy conversion, energy storage systems, and environmental remediation, highlighting their transformative potential in addressing global energy and sustainability challenges (Fig. 1). We point out the current limitations and unresolved controversies of SACs. By addressing these challenges, our research aims to provide strategic guidance for the rational design and development of high-performance SACs, thereby accelerating advancements in related technological fields. Through the establishment of a robust theoretical framework and the provision of actionable insights, this study seeks to serve as a foundational reference for future research of SACs, inspiring innovative breakthroughs in this dynamic and rapidly progressing field.
The synthesis of SACs is intrinsically linked to the preparation of their supporting materials, with the two processes often occurring concurrently or being intricately interdependent. The choice of synthesis method and the properties of the support materials significantly influence the dispersion, stability, and catalytic performance of the single-atom active sites. Currently, researchers predominantly utilize well-established and widely applied techniques for SACs synthesis, including impregnation, co-precipitation, ball milling, chemical etching, and high-temperature pyrolysis methods [20–22]. Each of these methods presents distinct mechanisms for anchoring single metal atoms onto supports, furnishing invaluable insights and practical references for the development of highly efficient SACs supported on diverse materials. In this section, we will conduct a systematic and in-depth elaboration on these prevalent synthesis methods. We will not only expound upon their fundamental principles but also conduct a thorough examination of their respective merits and limitations. Through a comprehensive and meticulous analysis of these methods, our objective is to endow researchers in related fields with a solid theoretical underpinning and practical guidance. The synthesis methods are thoroughly described in Sections S1-S5 (Supporting information).
SACs are a class of advanced catalytic materials composed of isolated metal atoms anchored onto a supporting support. The catalytic activity of SACs primarily originates from the isolated metal atoms, which may also interact with adjacent atoms or functional groups on the support surface. Due to their high surface energy, single metal atoms are inherently unstable and prone to migration and aggregation, often forming clusters that diminish their catalytic efficiency. SMSI is employed to immobilize these single atoms on suitable supports, ensuring their stability and dispersion [23]. The catalytic performance of SACs is profoundly influenced by the nature of these interactions and the support's electronic and structural properties [24]. Therefore, a comprehensive understanding of the mechanisms by which support modulates the performance of SACs is crucial for the rational design and optimization of high-performance catalysts. This section offers a comprehensive and systematic review of the influence of diverse support materials, such as carbon-based materials, metal oxides, MXenes, and MOFs, on the catalytic performance of SACs. Particular attention is given to the structural characteristics, electronic properties, and interaction mechanisms between the supports and the active sites. By evaluating the performance of these supports in critical catalytic reactions, such as ORR, HER, OER, CO2RR, NRR and NO3RR to elucidate the distinct advantages and inherent limitations associated with each support type. The insights derived from this analysis are intended to inform and guide future research endeavors, facilitating the development of optimized SACs.
Carbon-based materials have emerged as one of the most prevalent supports for SACs due to their exceptional properties, including high specific surface area, superior electrical conductivity, remarkable chemical stability, and tunable surface chemistry. Commonly employed carbon-based supports encompass graphene, carbon nanotubes, ordered mesoporous carbon, and carbon fibers [25–27]. The synthesis of SACs on these supports can be achieved through either bottom-up or top-down approaches. The bottom-up strategy involves directly introducing single-atom catalytic sites onto pre-existing carbon supports via methods such as impregnation. Conversely, the top-down approach entails the initial preparation of polymeric precursors through self-assembly, followed by carbonization and acid etching to successfully anchor SACs onto the carbon supports [28,29]. These methodologies offer the flexibility to precisely modulate the surface architecture of the carbon supports, thereby inducing alterations in the electronic structure of the metal active sites through the creation of intrinsic defects. This, in turn, leads to significant variations in catalytic activity and selectivity. Furthermore, these supports facilitate the formation of stable coordination structures with metal single atoms]through defect sites (e.g., vacancies, edge sites) and heteroatom doping (e.g., N, S, P), thereby effectively regulating the catalytic performance [30]. Additionally, the π-conjugated system of carbon materials facilitates electron transfer, which can modulate the electronic structure of the single atoms and enhance their catalytic activity [31]. In recent years, carbon-based SACs have demonstrated remarkable performance in ORR, CO2RR and NRR [32–36]. It has been demonstrated that the active centers of SACs typically consist of isolated metal atoms forming stable coordination structures with coordinating atoms (e.g., N, O, S) on the support. This configuration not only effectively stabilizes the metal single atoms but also optimizes the adsorption behavior of reaction intermediates by modulating the electronic structure, thereby significantly enhancing catalytic performance. For instance, manganese (Mn)-based SACs with Mn-O sites exhibit exceptional catalytic activity in the NRR due to the appropriate energy and symmetry of their 3d orbitals, which facilitate the adsorption and activation of N2 molecules. Our team successfully constructed Mn-O3N1 sites anchored on porous carbon (Mn-O3N1/PC) through precise regulation of Mn-O bonding conditions. This catalyst achieved an NH3 yield of 66.41 µg h‒1 mgcat‒1 at −0.35 V (vs. RHE), approximately four times higher than that of the Mn-N4/PC control catalyst (Fig. 2a). This performance enhancement is attributed to the unique geometry and electronic structure of the Mn-O3N1 site, which not only promotes N2 adsorption and activation but also significantly reduces the free energy change in the potential-determining step [37]. Similarly, Tran et al. [38] discovered that positively charged Co single atoms supported on N-doped carbon nanofibers serve as highly efficient active sites for catalyzing the reduction of N2 to NH3, achieving an NH3 yield of 67.6 µg h‒1 mg‒1 at 0.1 V with a maximum Faradaic efficiency (FE) of 56.9%. Density functional theory (DFT) calculations revealed that charge transfer between the Co single atom and the support modulates the local electronic structure, inducing the formation of Lewis acid pairs and enabling the Co site to exhibit high intrinsic activity for NRR (Fig. 2b). Additionally, porous carbon materials are considered excellent supports for SACs due to their gas storage capacity and hydrophobicity, which enhance the N2 concentration near the single-atom catalytic sites [39]. Building on this, Liao et al. [40] immobilized Mo on the quadruple hollow sites of phosphotungstic acid embedded in carbon nanotubes (CNTs) to obtain the Mo-PTA@CNT electrocatalyst. Electrochemical testing revealed that this catalyst achieved an NH3 yield of 51 µg h‒1 mg‒1 at −0.1 V with an FE of 83%. Similarly, niobium (Nb)-based SACs have also demonstrated remarkable performance in NO reduction reactions (NORR). For example, Nb SACs supported on B and N co-doped carbon nanotubes (Nb-SA/BNC) achieved NH3 yield of 8.2 × 10‒8 mol cm‒2 s‒1 in room-temperature electrochemical denitrification. The introduction of B and N heteroatoms not only improved the electrical conductivity of the catalyst but also provided abundant anchor sites to stabilize the Nb single atoms and optimize their electronic structure. Field emission scanning electron microscopy (FESEM) and transmission electron microscopy (TEM) characterizations revealed that Nb-SA/BNC possesses a nanotube array structure with a specific surface area of 875.5 m2/g and hierarchical micro/mesoporous features, facilitating mass transfer and reactant diffusion, thereby accelerating the electrocatalytic process. Theoretical calculations further indicated that the Nb single-atom sites promote NO adsorption and lower the energy barrier of the potential-determining step [41]. Considering the ordered porous carbon matrix facilitates the exposure of active sites and the bulk transport of electrolytes during electrolysis. A N, S co-doped ordered porous carbon-supported Pd SACs (PdSA/N, S-OPC) was prepared using a template-assisted wet chemistry method. This catalyst exhibited exceptional hydrogen oxidation reaction (HOR) activity and stability under alkaline conditions, outperforming commercial Pt/C catalysts and ranking among the highest reported performances. Experimental and theoretical analyses suggested that this superior HOR performance stems from the synergistic effect of the Pd-N4 coordination structure and N/S doping, which optimizes the adsorption behavior of Had species, significantly enhancing catalytic performance (Fig. 2c) [30]. Fan et al. [42] synthesized mesoporous Fe-N-C materials through a self-assembly and pyrolytic carbonization strategy, which demonstrated high activity in the electrocatalytic reduction of nitrate to N2 (Fig. 2d). In the context of CO2RR and ORR bifunctional catalysts, Nb SACs supported on nitrogen-doped ordered mesoporous carbon (Nb-N-C) achieved a FECO of up to 90% and an ORR half-wave potential of 0.84 V. When applied in zinc-air batteries (ZABs), the peak power density reached 115.6 mW/cm2 (Fig. 2e). DFT calculations revealed that the formation of Nb-N coordination bonds effectively lowered the energy barriers for the generation of key intermediates (*COOH and *O) in CO2RR and ORR, thereby enhancing catalytic performance [34]. In addition, Fe-based SACs have shown excellent performance in NRR. For example, N, S co-doped graphene (NSDG)-loaded Fe SACs (Fe-SAS/NSDG) significantly enhanced the NRR activity through the Fe-N3S active center. The introduction of S enhanced the affinity of the material for N2 and optimized the adsorption and activation process of N2 molecules. As shown in Figs. 2f-i, the lowest adsorption energy is found when the N2 tip is adsorbed on Fe-N3S [43].
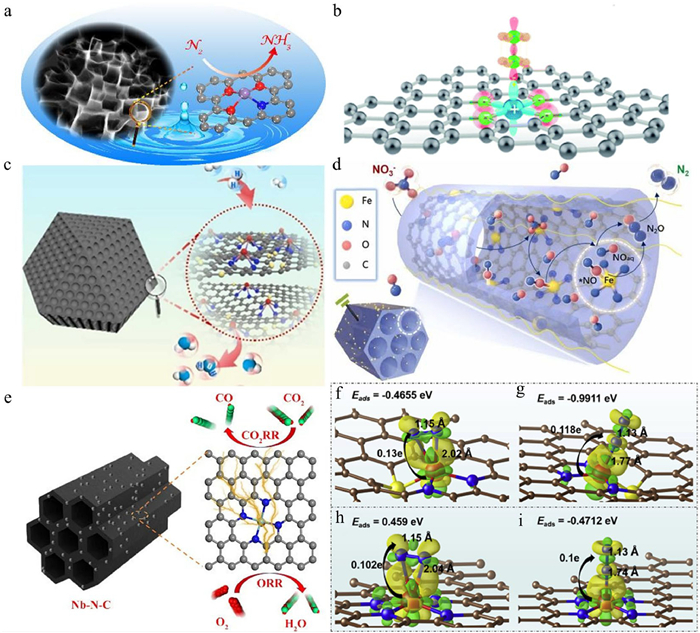
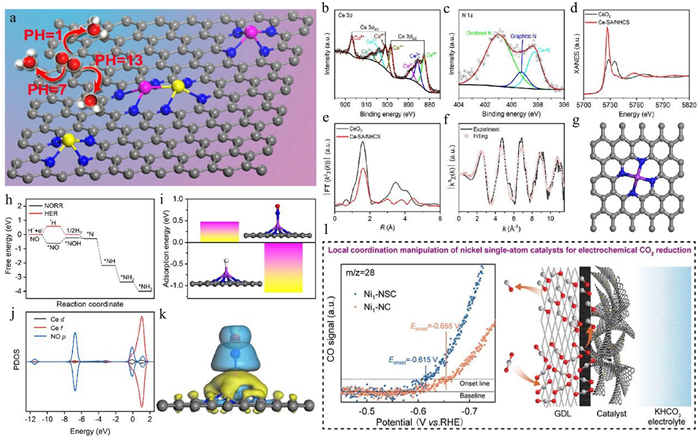
Similarly, the Fe-Zn diatomic catalyst (Fe-Zn-SA/NC) anchored on a porous N-doped carbon skeleton exhibited excellent ORR performance in the full pH range (Fig. 3a) [44]. B and N co-doped carbon supports loaded with single-atom Mn sites exhibit excellent HER and hydrazine oxidation reactions (HzOR) bifunctional catalytic performance in alkaline electrolytes [32]. As confirmed by XPS analyses, N-doped carbon black-supported Ag SACs (SA-Ag/NC) exhibited enriched pyridine N species on their surface. These sites facilitated the stable dispersion of single atoms through strong coordination with Ag atoms, resulting in exceptional NH3 yield and FE in electrocatalytic reactions [35]. In addition, N-doped hollow carbon sphere-supported Ce SACs demonstrated remarkable performance in the NORR in acidic solutions, achieving a FENH3 of 91% at −0.7 V (vs. RHE) with a yield of 1023 µg h‒1 mgcat‒1. Characterization analyses confirmed the existence of the Ce-N4 coordination structure (Figs. 3b-g). Theoretical simulations indicated (Figs. 3h-k) that the Ce-N4 active site not only activated NO molecules through strong electronic interactions but also lowered the free energy barrier for the transition of *NO to *NOH intermediates, enhancing NORR kinetics and suppressing competitive hydrogen evolution [12]. In NRR, Fe supported on N-doped frameworks exhibited exceptionally high catalytic activity (ISAS-Fe/NC). The introduction of N atoms altered the electron cloud distribution of the carbon material, creating partially positively charged neighboring carbon atoms that facilitated the adsorption and immobilization of Fe single atoms. The formation of the Fe-N4 active center optimized the adsorption and activation process of reactant molecules, significantly improving NRR catalytic activity. Additionally, the high conductivity of carbon-based materials promoted rapid electron transfer between the catalyst and reactants, reducing the reaction overpotential and further enhancing catalytic efficiency [45]. Xia et al. [46] achieved highly selective CO2-to-CO conversion by constructing an asymmetric ligand-structured Ni-N3-S atomic site catalyst (Ni1—NSC), which exhibited a FECO exceeding 99% at a high current density of −320 mA/cm2 (Fig. 3l). Spherical aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) revealed that Ni atoms were uniformly dispersed on the carbon support surface. In situ attenuated total reflection surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS) and differential electrochemical mass spectrometry (DEMS) analyses demonstrated that the Ni-N3-S sites exhibited weaker binding strength to *CO and lower kinetic overpotential for CO2-to-CO conversion compared to conventional Ni-N4 sites. DFT calculations further confirmed that the enhanced activity and selectivity of the Ni1—NSC catalyst were attributed to the lower Gibbs free energy of the *COOH intermediate on the Ni-N3-S sites.
Despite the significant advantages of carbon-based supports in SACs, their stability under high temperatures or strongly oxidizing environments remains a challenge. Carbon corrosion can lead to the agglomeration and loss of single atoms, as well as limited interaction strength with highly reactive metals. Therefore, the development of novel support materials and the optimization of single-atom anchoring strategies are critical directions for future research, aiming to achieve more efficient and stable SACs designs.
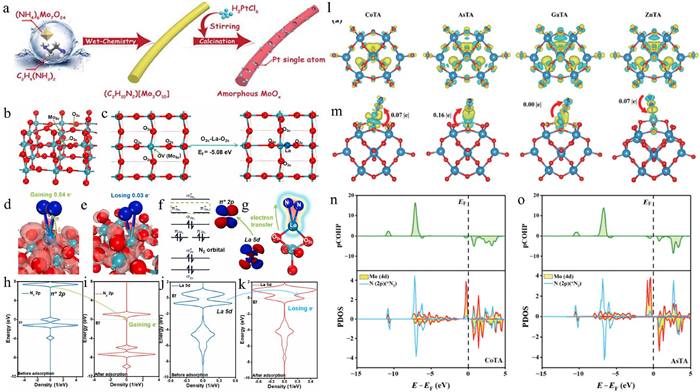
Metal oxides have garnered significant attention as ideal supports for SACs due to their structurally diverse crystalline frameworks, customizable surface characteristics, and adaptable electronic properties [47]. These materials demonstrate exceptional versatility in catalytic applications, particularly through their three primary categories of support materials: Titanium-based oxides (Ti-based oxides), tungsten-based oxides (W-based oxides), and molybdenum-based oxides (Mo-based oxides). A critical feature of these oxide supports lies in their inherent defect engineering capabilities, which manifest as oxygen vacancies (Ov), metal vacancies (Mv), crystallographic edge irregularities, and atomic-scale step discontinuities. These structural imperfections serve as favorable anchoring sites that stabilize isolated metal atoms through strong coordination bonds, effectively preventing atomic aggregation during catalytic processes. The functional superiority of metal oxide-supported SACs stems from dynamic interfacial interactions between the support matrix and immobilized metal species. Through synergetic electron transfer mechanisms, the oxide substrate can either donate or accept electron density from anchored single atoms, thereby inducing charge redistribution at the atomic interface. This electronic modulation significantly alters the d-band center position of metal active sites, a critical parameter governing catalytic behavior [23]. For instance, electron-deficient metal centers may exhibit optimized binding energies for specific reaction intermediates, while electron-rich configurations could facilitate the desorption of products. Amorphous α-MoOx supported on nitrogen-doped carbon (Pt-SA/α-MoOx) demonstrates exceptional catalytic performance in the HER (Fig. 4a). This catalyst achieves an ultra-high mass activity of 52.0 A/mgpt at an overpotential of 50 mV, primarily attributed to the synergistic effect between the amorphous α-MoOx support and Pt single atoms. Electron energy loss spectroscopy (EELS) analysis confirms the presence of abundant oxygen vacancies in the α-MoOx support, a finding corroborated by electron paramagnetic resonance (EPR) results. These oxygen vacancies form strong interactions with Pt single atoms, optimizing the adsorption and desorption processes of hydrogen atoms (H*), thereby significantly enhancing HER activity. Furthermore, the reduction of Mo6+ to lower valence states (Mo5+, Mo4+, Mo3+, and Mo2+) in α-MoOx induced lattice-electron coupling distortions, further boosting electrochemical activity. The introduction of nitrogen-doped carbon not only improved the conductivity of the support but also facilitated charge transfer during electrocatalysis, underpinning the high performance of Pt-SA/α-MoOx. The significance of amorphous α-MoOx lay in its high defect concentration, tunable electronic structure, and strong synergistic effects with single atoms. These unique properties not only stabilized single-atom active sites but also significantly enhanced catalytic performance by optimizing the local electronic environment [48]. In another study, Gao et al. [49] successfully reduced the bandgap and enhanced the conductivity of TiO2 by introducing Nb single atoms. Characterization results revealed that Nb doping significantly improved the adsorption capacity of N2 on the Ti surface, while the Nb-Ti-O structure suppressed the HER and accelerated electron transfer in the NRR. At a potential of −0.5 V (vs. RHE), the catalyst achieved an NH3 production rate of 213 µg h‒1 mg‒1 with a FE of 9.2%. Moreover, researchers synthesized MgO nanosheets and their supported single-atom Pt catalyst (Pt-SA/MgO) for toluene oxidation. Compared to pure MgO, Pt-SA/MgO demonstrated significantly enhanced catalytic activity. Studies showed that the surface of Pt-SA/MgO readily generates oxygen vacancies, which facilitate the activation of molecular oxygen and the formation of reactive oxygen species. Based on experimental data and theoretical calculations, a mechanism for reactive oxygen-promoted toluene oxidation was proposed: In the presence of H2O, molecular oxygen more readily dissociates on the oxygen vacancies of Pt-SA/MgO to form *OH radicals, which are the primary reactive oxygen species [50]. Currently, two main approaches are employed to enhance the activity of SACs supported on metal oxides. Constructing oxygen vacancies to increase the anchoring capacity of the support for single atoms, thereby improving single-atom loading, and introducing dopants to further enhance the electronic regulation of the support on single atoms. For example, Liu et al. [51] loaded La, which has unsaturated orbitals, onto oxygen-deficient MoO3−x. The strong interaction between La and two coordinated lattice oxygens in MoO3−x (O2-La-Ox coordination) effectively prevented the detachment or aggregation of single atoms, avoiding catalyst deactivation (Figs. 4b-k). Tang et al. [52] presented a protonated iridium oxide (H3.8Ir1−xRuxO4) encapsulating monotonically dispersed ruthenium atoms, which exhibits excellent activity and stability in acidic water oxidation. Single Ru doping facilitates the induction of localized oxygen vacancies in the Ir-O lattice, which synergistically enhances the adsorption of OOH* intermediates and improves the OER activity. In addition, the preferential oxidation of Ru and the electronegativity of the oxygen vacancies significantly stabilized the Ir-O active site and improved the OER stability. As a result, the H3.8Ir1−xRuxO4 catalyst has an overpotential of 255 mV at 10 mA/cm2 and exhibits excellent catalytic durability in acidic electrolytes for more than 1100 h, representing a significant increase in stability by an order of magnitude compared to pristine H3.8IrO4. The proton exchange membrane electrolyzer using H3.8Ir1−xRuxO4 catalyst as the anode exhibited stable performance for more than 1280 h at a high current density of 2 A/cm2. Introducing dopants to further enhance the electronic regulation of the support on single atoms. Based on DFT calculations, Lin et al. [53] proposed that introducing heteroatoms (e.g., Fe, Co, Ni, Cu, Zn, Ga and Ge) into Keggin-type polyoxometalate supports can adjust the coordination environment of Mo single atoms, promoting the electrocatalytic NRR. The charge distribution of O atoms at the 4-H site was closely related to the type of central heteroatom. When the central heteroatom was Co or As, more electrons were transferred from W atoms to O atoms. Charge density difference analysis indicated that Mo atoms on CoTA and ASTA exhibit stronger electronegativity, allowing more electrons to flow from the supported Mo atoms to the N≡N antibonding orbital, effectively activating the N≡N bond (Figs. 4l and m). Partial crystal orbital Hamiltonian population (COHP) and PDOS analysis of the N—N bond demonstrate that electron transfer from Mo to N2 antibonding orbitals activates the N2 molecule and extends the N=N bond length (Figs. 4n and o). Therefore, the Mo atoms supported by CoTA and AsTA have a more significant activation effect on N2 than those supported by GaTA and ZnTA.
In summary, metal oxide supports offer high design flexibility. By constructing oxygen vacancies or introducing heteroatoms, their electronic structures can be significantly improved, optimizing the strength of their interactions with single atoms. However, their loadings are generally low. In practical applications, the choice of oxide support, dopant elements, and single-atom species can be tailored to specific needs, enabling precise control and optimization of catalytic performance.
MXenes were first synthesized in 2011 by Prof. Yury Gogotsi's group through selective etching of Ti3AlC2 MAX phase precursors using hydrofluoric acid (HF), yielding the novel 2D layered material Ti3C2Tx, later named "MXenes" to denote two-dimensional transition metal carbides, nitrides, or carbonitrides [54]. Their general formula is Mn+1XnTx, where M represents transition metals (e.g., Sc, Ti, V), X denotes carbon/nitrogen, and Tx corresponds to surface terminations (e.g., -O, -OH, -F). Both the M and X layers in MXenes are chemically tunable, while the diverse surface terminations enable the creation of a vast MXenes family with distinct properties. As 2D materials, MXenes possess atomic-level thickness and structural diversity, while maintaining stability in harsh environments (acidic, alkaline, and organic solvents). At the same time, MXenes have excellent electrical properties, similar to their precursor MAX, MXenes without surface group saturation mostly exhibit metallic properties, and the d electronic state of the transition metal atoms has an energy band through the Fermi energy level, which can conduct electrons [55]. After being saturated by the surface group, the surface group forms a new chemical bond with the transition metal atoms, and the d-orbitals of the transition metal atoms are partially occupied by the electrons of the surface group. The d-orbitals of the transition metal atoms hybridize with the valence electron orbitals of the surface group, and the energy of the hybrid orbitals decreases. MXenes without surface group saturation will become semiconductors after surface saturation, while some MXenes retain their metallic properties [56]. These attributes confer MXenes with unique catalytic advantages. Specifically, their ultrahigh surface area, tunable bandgap, mechanical robustness, and abundant surface defects establish MXenes as ideal platforms for SACs. The formation of stable coordination bonds between surface terminations and metal single atoms enhances both the dispersion and stability of active sites. Simultaneously, charge redistribution at the metal-support interface modulates the electronic structure of catalytic centers, enabling optimized adsorption energies for reaction intermediates and superior electrocatalytic performance [57,58]. A highly efficient and stable electrochemical carbon monoxide reduction (COR) catalyst was developed by anchoring copper single atoms on Ti3C2Tx MXenes nanosheets, achieving an unprecedented selectivity of 98% for multicarbon products. In COR, Cu-SA/Ti3C2Tx exhibited superior C2 product selectivity (98%) compared to Cu-NP/Ti3C2Tx. Notably, at −0.7 V (vs. RHE), the FE for C2H4 reached 71%, one of the highest values reported for Cu-based COR catalysts. Furthermore, Cu-SA/Ti3C2Tx demonstrated remarkable electrochemical stability over 68 h. X-ray absorption fine structure (XAFS) analysis confirmed the stable anchoring of Cu single atoms via O coordination on the Ti3C2Tx substrate (Figs. 5a-c). Theoretical simulations revealed (Figs. 5d-i) that atomically dispersed Cu-O3 sites significantly promote C—C coupling of CO molecules, generating the critical *CO—CHO intermediate and reducing the free energy barrier of the potential-determining step, thereby explaining the catalyst's high activity and selectivity [59]. A novel electrocatalyst comprising atomically dispersed Pt anchored on MXenes through Pt-O and Pt-Ti coordination bonds (denoted as Pt-SA/MXenes) was developed for efficient HER. The asymmetric coordination environment induces local electric field polarization, endowing the Pt-SA/MXenes with exceptional alkaline HER performance. Specifically, the catalyst achieves a current density of 10 mA/cm2 at a mere overpotential of 33 mV and demonstrates remarkable stability over 27 h of continuous operation. Notably, the mass activity reaches 23.5 A/mgPt at 100 mV overpotential, surpassing that of commercial Pt/C by a factor of 29.4. Theoretical calculations based on DFT suggested that the polarized electric field effectively modulates the electronic structure of Pt-SA/MXenes, thereby lowering the energy barrier for H* intermediate adsorption/desorption and enhancing the overall HER catalytic efficiency [60]. Additionally, we proposed a novel self-driven dual hydrogen production system based on oxygen-functionalized Ti3C2Ox MXenes-supported single Rh atoms (Rh-SA/Ti3C2Ox). This bifunctional catalyst exhibited outstanding performance in both pH-universal HER and HzOR. DFT calculations indicated that the introduction of single Rh atoms optimized the electronic structure of Ti3C2Ox, facilitating hydrogen adsorption in HER and dehydrogenation in HzOR (Figs. 5j and k) [61]. Recent advancements have led to the development of MXene-supported metal catalysts, such as Ru/Mo2CTx [62], Ti3C2Ox-PtSA [63] and Pt-on-Pd/Ti3C2Ox [64], which have been successfully applied in electrochemical reactions. For instance, Du et al. [65] synthesized V4C3Tx-supported Ni nanoparticle (Ni@MX) composites via a two-step freeze-drying and hydrogen thermal reduction process. These catalysts demonstrated exceptional activity for electrocatalytic NRR in 0.1 mol/L KOH, achieving an NH3 yield of 21.29 µg h‒1 mg‒1 and an FE of 14.86%. DFT calculations revealed that the high activity stemmed from synergistic effects between Ni sites and oxygen vacancies on V4C3Tx, which enhanced nitrogen adsorption and reduced Gibbs free energy barriers. Moreover, the V4C3Tx MXenes support suppressed the competing HER, further boosting NRR activity. Single-atom Ru sites anchored on Ti3C2Tx MXenes nanosheets were discovered to function as trifunctional electrocatalysts for acidic HER, OER, and ORR. Theoretical calculations showed that isolated Ru-O2 sites optimize reactant/intermediate adsorption and lower the energy barrier of the potential-determining step, accelerating HER, OER, and ORR kinetics [66]. Although MXenes have been synthesized by liquid-phase and molten-salt etching methods, they still suffer from slow reaction kinetics for the removal of A species from the MAX phase as well as long production times (5–48 h). Yang's team [58] developed a minute-scale production method to produce MXenes by selectively etching the MAX phase (Ti2AlC) under metal chloride (ZnCl2) vapor (Ti2CClx) (Fig. 5l). By this method, the etching time is less than one-sixtieth of the liquid phase etching and molten salt etching methods. The resulting MXenes can be directly used as an efficient platform for immobilizing single-atom Pt, with good HER electrocatalytic performance, a Tafel slope as low as 31 mV/dec, and a cycling stability of up to 5000 cycles. Although MXenes are often used as substrates for SACs, the knowledge of the interactions between single atoms and MXenes is still relatively scarce. Based on the abundant intrinsic defects on the surface of MXenes and the tunable functional groups, Prof Rogach's team [67] innovatively constructed a variety of single-atom Co-based catalysts (Co@V2CTx, Co@Nb2CTx, and Co@Ti3C2Tx). The HER and OER overpotentials of Co@V2CTx are 35 mV and 242 mV at a current density of 10 mA/cm2, respectively. Through synchrotron radiation spectroscopy, DFT calculations, and comparative analysis of the electronic structures and oxidation states of Co active sites on different MXenes substrates, the study revealed strong electron-orbital coupling and enhanced charge transfer dynamics in Co@V2CTx, which synergistically improved both HER and OER activities (Fig. 5m). This work systematically elucidates the interaction mechanisms between single atoms and MXenes substrates, offering critical insights for the rational design of SACs.
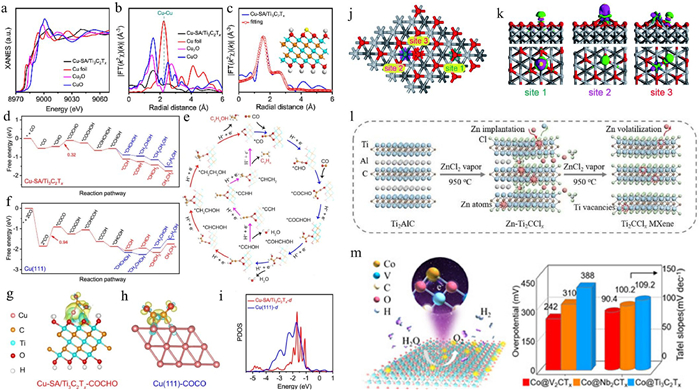
Despite their high conductivity, tunable surface chemistry, and mechanical stability, MXenes face challenges in synthesis and processing, and their long-term stability under electrochemical conditions requires further investigation.
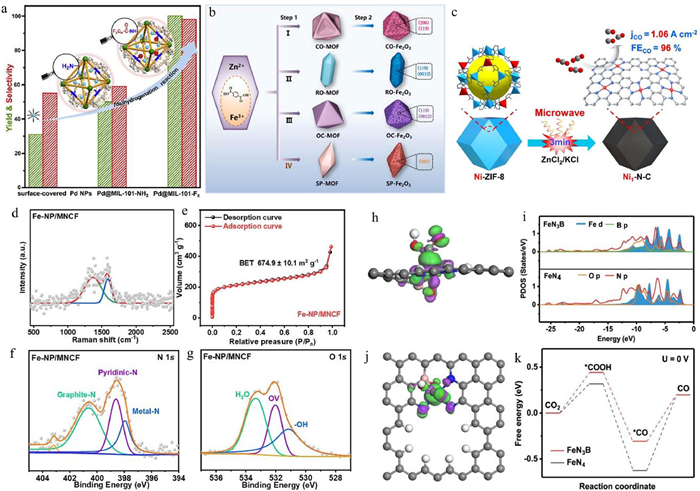
MOFs, characterized by their porous crystalline structure formed through the self-assembly of metal ions or clusters with organic ligands, offer an exceptional platform for SACs. MOFs were first reported by Yaghi's team in 1995 [68]. Since then, a vast number of MOFs have been successfully synthesized, with several classical MOFs becoming research hotspots in materials science. To date, researchers have developed over 20,000 distinct MOFs structures with varied morphologies through the combination of diverse metal ions and organic ligands. Their unique architectural features include ultra-high specific surface area, tunable pore size, and versatile coordination environments [17]. By immobilizing metal single atoms within the MOFs framework via coordination bonds, high dispersion and stability of active sites can be achieved, making MOFs particularly advantageous for catalytic reactions requiring precise control over the coordination environment. Furthermore, catalytic performance is intrinsically governed by the microenvironment surrounding active sites, making the rational design of advanced SACs pivotal for achieving high-efficiency catalytic systems. For instance, Jiang et al. [69] designed a Pd NPs@MOFs composite by modifying the ligand branches of MOFs, integrating metal active sites with tailored microenvironments to optimize interactions between Pd nanoparticles (NPs) and substrates (Fig. 6a). This composite demonstrated superior catalytic activity and selectivity in the dehydrocoupling of organosilanes and dehydrogenation of halogenated nitrobenzenes, outperforming conventional Pd NPs supported on surfactants or other supports. The morphology of MOFs significantly influences their catalytic properties, as different structures expose distinct lattice planes, thereby affecting reactivity [70,71]. The diversity in MOF morphology arises from the coordination dynamics between metal ions, ligands, and solvent molecules [71]. Solvent properties, such as polarity, intermolecular forces, and boiling point, further modulate crystal growth [72]. For example, Bai et al. [73] synthesized Fe-MOFs with controllable morphologies via a one-step solvothermal method by regulating competitive coordination in the metal-solvent system (Fig. 6b). Upon pyrolysis, these Fe-MOFs transformed into Fe2O3 with varied morphologies and exposed active crystalline planes, leading to differentiated electrocatalytic performances. Although MOFs exhibit significant potential in the field of catalysis, their limited chemical stability, mechanical stability, and electrical conductivity have restricted their widespread application. Through pyrolysis treatment, MOFs can be transformed into MOFs-derived carbon materials, which not only retain the porous structure and high specific surface area of the MOFs precursors but also significantly enhance chemical stability, mechanical stability, and electrical conductivity. Additionally, the porous structure of MOFs can be utilized to encapsulate metal nanoparticles, which, after pyrolysis, are uniformly dispersed on carbon or oxide supports. By precisely tuning the structure of the MOFs precursors, the morphology and electronic structure of the derived materials can be meticulously designed, thereby optimizing metal-support interactions and further improving catalytic performance. This strategy provides a novel direction for the development of highly efficient catalysts [74,75]. For instance, Jiang et al. developed a nitrogen-doped graphene-coated PdNiCo alloy catalyst derived from MOFs, where trace palladium atoms enhanced electron transfer efficiency, and the interface between the alloy and nitrogen-doped carbon synergistically modulated hydrogen adsorption energy, significantly boosting HER activity. This catalyst exhibited performance comparable to commercial Pt/C [76]. In addition, considering that MOFs can effectively transfer their advantages to carbon-based materials during pyrolysis, Wen et al. [77] developed a microwave-assisted rapid pyrolysis method to prepare nitrogen-doped carbon-supported single-atom nickel catalysts (Ni1–N-C) from Ni-ZIF-8 precursors within 3 min (Fig. 6c), achieving a FECO of 96% in CO2RR. Similarly, our team synthesized Fe-NP/MNCF, a bifunctional catalyst for ORR and CO2RR, by pyrolyzing Fe-doped ZIF-8 modified with phosphomolybdic acid hydrate (PMo). Fe-NP/MNCF demonstrated exceptional ORR activity, surpassing commercial Pt/C, and remarkable stability in CO2 electrolysis driven by ZABs. Raman spectroscopy revealed that PMo introduction increased defect density, enhancing electrocatalytic activity (Fig. 6d), while BET analysis confirmed a high specific surface area of 674.85 m2/g, facilitating reactant mass transfer (Fig. 6e). XPS analysis identified graphite-N, pyridine-N, and metal-N species, with the latter forming Fe-N-C structures that optimized ORR performance by promoting 4-electron pathways and inhibiting H2O2 formation (Fig. 6f). In addition, no metal-O peaks were detected in Fe-NP/MNCF (Fig. 6g), suggesting that it effectively avoids the loss of active sites due to Fe oxidation. It was shown that polyacid doping not only significantly increased the surface charge distribution and defect density of the catalyst, but also optimized the multilevel pore structure of ZIF-8, providing more channels for electron transfer. This innovative MOF modification strategy opens a new way to design efficient ORR and CO2RR catalysts [75]. Chen et al. utilized Zn/Mo-MOFs to anchor Mo single atoms and ultra-small MoC clusters on nitrogen-doped porous carbon, demonstrating strong N2 adsorption capabilities for NRR [78]. However, challenges such as high costs and metal aggregation during carbonization remain. Alternative approaches, such as coupling MIL-53(Fe) with TiO2, have achieved notable NH3 yields without carbonization [79], while Liu et al. [80] prepared Fe-anchored B- and N-co-doped carbon substrates from ferrocenyl boronic acid-coated ZIF-8, exhibiting exceptional activity in CO2RR and Zn-CO2 batteries (Figs. 6h-k).
A comparative analysis of the four support types reveals distinct advantages and limitations in different electrocatalytic reactions. Carbon-based materials excel in ORR and HER due to their high conductivity and tunable surface chemistry, while metal oxides and MOFs show superior performance in OER and CO2RR, respectively. MXenes, with their unique combination of conductivity and surface functionality, demonstrate potential across multiple reactions but require further development. The catalytic activity of SACs is influenced by several key factors, including the dispersion and stability of single atoms, the electronic interaction between supports and active sites, and the mass transfer properties of the support. Optimizing these factors through innovative support design and synthesis strategies is essential for enhancing SACs performance.
Through precise regulation of active sites via support engineering, SACs exhibit significant potential in renewable energy conversion and storage. Their unique structures and exceptionally high atomic utilization rates make them pivotal materials in various energy technologies, offering innovative solutions for achieving efficient and cost-effective energy conversion and storage. In fuel cells, high-performance single-atom ORR catalysts can substantially enhance the efficiency and durability of fuel cells while reducing costs, thereby accelerating the commercialization of fuel cell technology [81–83]. In water electrolysis for hydrogen production, SACs with superior HER and OER catalytic properties can significantly improve electrolysis efficiency, reduce energy consumption, and facilitate large-scale green hydrogen production, laying a solid foundation for the development of the hydrogen energy industry [84–86]. Furthermore, in energy storage systems such as metal-air batteries and lithium-ion batteries, SACs are expected to play a critical role by enhancing battery performance and cycle life, providing new approaches to addressing energy storage challenges [34,87–89].
In summary, SACs with precisely regulated active sites through support engineering demonstrate vast application potential in fields such as fuel cells, water electrolysis, metal-air batteries, and lithium-ion batteries. Their remarkable characteristics: high efficiency, low cost, and controllability, provide essential technical support for the efficient conversion and storage of renewable energy. These advancements are likely to drive the rapid development and widespread adoption of clean energy technologies. Looking ahead, as research into the regulation mechanisms of SACs active sites deepens and breakthroughs in large-scale preparation technologies emerge, SACs are poised to play an even more critical role in the energy sector, offering robust technical solutions for achieving carbon neutrality goals.
In the field of environmental remediation, SACs, with their exceptional activity, selectivity, and atomic utilization efficiency, demonstrate immense potential, particularly in wastewater treatment and exhaust gas purification. By precisely regulating active sites through support engineering, SACs can efficiently catalyze the conversion of environmental pollutants, providing innovative solutions for achieving green and sustainable environmental remediation. For instance, in electrocatalytic carbon dioxide reduction, SACs engineered for CO2RR can convert carbon dioxide into valuable chemicals or fuels. This not only enables the resource utilization of carbon dioxide but also effectively mitigates greenhouse gas emissions, contributing significantly to global climate change mitigation [90–93]. In wastewater treatment, SACs can efficiently degrade persistent organic pollutants such as dyes, pesticides, and pharmaceutical residues into harmless small molecules like CO2 and H2O through processes such as the electrochemical oxidation reaction, thereby achieving water purification and offering a powerful tool for water resource protection and recycling [94–96]. Additionally, due to their excellent atomic efficiency and tunable electronic structure, SACs exhibit unusual molecular adsorption and electron transfer capabilities, which are also valuable in the detection and degradation of polluted gases [97–100].
In summary, SACs with precisely regulated active sites through support engineering present broad application prospects in environmental remediation. Their outstanding performance in carbon dioxide resource utilization, wastewater treatment, and exhaust gas purification provides critical technical support for achieving sustainable environmental protection [101–104]. In the future, as research into the regulation mechanisms of SACs active sites advances and large-scale preparation technologies mature, SACs are expected to play an even more pivotal role in environmental remediation, offering robust technical guarantees for building a clean and healthy ecological environment.
This review provides a comprehensive and systematic exploration of the pivotal role of support engineering in modulating the activity of SACs. By meticulously examining common synthesis methods for SACs, this work critically evaluates the strengths and limitations of each approach, thereby offering essential theoretical insights and practical guidance for future research endeavors. Furthermore, the article elucidates the transformative impact of advanced support engineering on enhancing the performance and intrinsic advantages of SACs through detailed case studies of representative electrocatalytic reactions. In terms of practical applications, this work underscores the immense potential of SACs in renewable energy conversion, energy storage systems, and environmental remediation. However, despite significant progress in tuning the electrocatalytic activity of SACs through support engineering, several critical challenges remain:
• The relatively low loading capacity of single atoms severely limits the scalability of SACs for industrial applications. Future research must focus on developing innovative synthesis strategies capable of achieving high single-atom loading while maintaining optimal dispersion and stability.
• Achieving precise selectivity control in complex reaction environments remains a pressing issue, necessitating a deeper atomic-level understanding of the interaction mechanisms between supports and single atoms, as well as the effects of reaction conditions on catalytic pathways.
• The long-term stability of SACs under harsh operational conditions requires further improvement, demanding the design of more robust support materials and the optimization of synthesis protocols.
• Looking ahead, future research should prioritize the following directions:
• Optimizing the anchoring mechanisms and electronic structure of single atoms through innovative support design frameworks, such as hierarchical porous architectures, heteroatom doping, and surface functionalization.
• Advancing synthesis technologies, including atomic layer deposition and microwave-assisted synthesis, to achieve precise control over single-atom loading and distribution.
• Exploring novel support materials, such as covalent organic frameworks (COFs) and two-dimensional material heterojunctions, to expand the versatility and application scope of SACs.
• Promoting interdisciplinary collaboration to drive transformative breakthroughs in energy conversion, environmental remediation, and green chemical engineering, ultimately contributing to global sustainable development efforts.
In conclusion, this article thoroughly examines the critical role of support engineering in enhancing the activity of SACs. It reviews significant advancements in SACs development, evaluates common synthesis methods, and analyzes the impact of various support materials on catalytic performance. The article also highlights the potential of SACs in renewable energy, energy storage, and environmental applications. However, challenges such as low single-atom loading, selectivity control, and long-term stability persist. Future research should focus on innovative support designs, advanced synthesis technologies, and the exploration of novel materials to overcome these limitations and further advance SACs applications in sustainable development.
The authors declare no conflict of interest exists and state that the article is original, unpublished, and not being considered for publication elsewhere.
Mingying Chen: Writing – original draft. Junjie Ma: Writing – original draft. Xiyong Chen: Conceptualization. Qian Liu: Resources. Yanhong Feng: Data curation. Xijun Liu: Supervision.
This work was financially supported by the Guangxi Natural Science Fund for Distinguished Young Scholars (No. 2024GXNSFFA010008), the Special Fund for Science and Technology Development of Guangxi (No. AD25069078), and the National Natural Science Foundation of China (No. 22469002).
Supplementary material associated with this article can be found, in the online version, at doi:
H. Wang, S. Wang, Y. Song, et al., Angew. Chem. Int. Ed. 63 (2024) e202402950. doi: 10.1002/anie.202402950
X. Wang, Z. Kang, D. Wang, et al., Nano Energy 121 (2024) 109268. doi: 10.1016/j.nanoen.2024.109268
M. Chen, J. Ma, C. Chen, et al., Chem. Eng. J. 498 (2024) 155302. doi: 10.1016/j.cej.2024.155302
K. Chen, J. Xiang, Y. Guo, et al., Nano Lett. 24 (2024) 541–548. doi: 10.1021/acs.nanolett.3c02259
L. Ma, X. Gao, X. Liu, et al., Chin. Chem. Lett. 34 (2023) 107735. doi: 10.1016/j.cclet.2022.08.015
J. Ma, F. Huang, A. Xu, et al., Adv. Sci. 11 (2024) 2306858. doi: 10.1002/advs.202306858
Y. Li, Z. Wei, Z. Sun, et al., Small 20 (2024) 2401900.
K. Wang, K. Wei, X. Wang, J. Ge, Electrochim. Acta 513 (2025) 145624. doi: 10.1016/j.electacta.2024.145624
T. Wei, J. Zhou, X. An, Mater. Rep. : Energy 4 (2024) 100285.
L. Gloag, S.V. Somerville, J.J. Gooding, R.D. Tilley, Nat. Rev. Mater. 9 (2024) 173–189. doi: 10.1038/s41578-023-00633-2
P. Liu, Y. Liu, K. Wang, et al., Nano Res. 17 (2024) 7957–7966. doi: 10.1007/s12274-024-6799-7
W. Zhang, X. Qin, T. Wei, et al., J. Colloid Interface Sci. 638 (2023) 650–657. doi: 10.1016/j.jcis.2023.02.026
K. Qi, M. Chhowalla, D. Voiry, Mater. Today. 40 (2020) 173–192. doi: 10.1016/j.mattod.2020.07.002
H. Zhao, W. Qi, X. Tan, et al., Chin. Chem. Lett. 36 (2025) 110898. doi: 10.1016/j.cclet.2025.110898
S. Marimuthu, N.R.K. Yabesh, G. Maduraiveeran, Mater. Today Chem. 37 (2024) 102035. doi: 10.1016/j.mtchem.2024.102035
X. Zhao, K. Gao, S. Xue, et al., Chin. Chem. Lett. 36 (2025) 110309. doi: 10.1016/j.cclet.2024.110309
X. Liu, C. Jia, G. Jiang, et al., Chin. Chem. Lett. 35 (2024) 109455. doi: 10.1016/j.cclet.2023.109455
C. Guo, N. Li, S. Gao, et al., Fuel. 367 (2024) 131416. doi: 10.1016/j.fuel.2024.131416
Y. Zhang, Z. Li, K. Chen, et al., Adv. Energy Mater. 14 (2024) 2402309. doi: 10.1002/aenm.202402309
Y. Huang, J. Xiong, Z. Zou, Z. Chen, Adv. Mater. 37 (2025) 2312182. doi: 10.1002/adma.202312182
R. Su, Z. Wang, Z. Liu, et al., J. Water Process. Eng. 68 (2024) 106319. doi: 10.1016/j.jwpe.2024.106319
K. Chida, T. Yoshii, R. Kawaguchi, et al., Green Chem. 26 (2024) 8758–8767. doi: 10.1039/d4gc02055c
R. Li, L. Luo, X. Ma, W. Wu, et al., J. Mater. Chem. A 10 (2022) 5717–5742. doi: 10.1039/d1ta08016d
S. Zhao, Y. Wen, X. Peng, et al., J. Mater. Chem. A 8 (2020) 8913–8919. doi: 10.1039/d0ta00190b
C. Zhang, W. Yuan, Q. Wang, et al., ChemNanoMat 6 (2020) 1474–1478. doi: 10.1002/cnma.202000337
Z. Wang, Y. Yan, Y. Zhang, et al., Carbon Energy 5 (2023) e306. doi: 10.1002/cey2.306
S. Gao, H. Li, Z. Lu, et al., Carbon Energy 7 (2025) e637. doi: 10.1002/cey2.637
X. Zheng, J. Hao, Z. Zhuang, et al., Nanoscale 16 (2024) 4047–4055. doi: 10.1039/d3nr05331h
X. Liu, M. Chen, J. Ma, et al., China Powder Sci. Technol. 30 (2024) 35–45.
H. Liu, J. Fu, H. Li, et al., Appl. Catal. B: Environ. 306 (2022) 121029. doi: 10.1016/j.apcatb.2021.121029
L. Han, S. Song, M. Liu, et al., J. Am. Chem. Soc. 142 (2020) 12563–12567. doi: 10.1021/jacs.9b12111
X. Peng, J. Hou, Y. Mi, et al., Nanoscale 13 (2021) 4767–4773. doi: 10.1039/d0nr09104a
L. Han, P. Ou, W. Liu, et al., Sci. Adv. 8 (2022) eabm3779. doi: 10.1126/sciadv.abm3779
S. Gao, T. Wang, M. Jin, et al., Sci. China Mater. 66 (2023) 1013–1023. doi: 10.1007/s40843-022-2236-8
Y. Chen, R. Guo, X. Peng, et al., ACS Nano 14 (2020) 6938–6946. doi: 10.1021/acsnano.0c01340
M. Li, Y. Xie, J. Song, et al., Chin. J. Catal. 60 (2024) 42–67. doi: 10.1117/12.3044233
L. Han, M. Hou, P. Ou, et al., ACS Catal. 11 (2021) 509–516. doi: 10.1021/acscatal.0c04102
N. Tran, X. Liu, Y. Cho, et al., J. Mater. Chem. A 10 (2022) 8432–8439. doi: 10.1039/d2ta00308b
Y. Wang, X. Cui, J. Zhao, et al., ACS Catal. 9 (2019) 336–344. doi: 10.1021/acscatal.8b03802
W. Liao, L. Qi, Y. Wang, et al., Adv. Funct. Mater. 31 (2021) 2009151. doi: 10.1002/adfm.202009151
X. Peng, Y. Mi, H. Bao, et al., Nano Energy 78 (2020) 105321. doi: 10.1016/j.nanoen.2020.105321
J. Fan, Y. Chen, X. Chen, et al., Appl. Catal. B: Environ. 320 (2023) 121983. doi: 10.1016/j.apcatb.2022.121983
S. Chen, M. Bu, Z. Zhou, et al., Mater. Today Energy 25 (2022) 100954. doi: 10.1016/j.mtener.2022.100954
J. Xu, S. Lai, D. Qi, et al., Nano Res. 14 (2021) 1374–1381. doi: 10.1007/s12274-020-3186-x
F. Lü, S. Zhao, R. Guo, et al., Nano Energy 61 (2019) 420–427. doi: 10.1016/j.nanoen.2019.04.092
Z. Chen, C. Wang, X. Zhong, et al., Nano Lett. 23 (2023) 7046–7053. doi: 10.1021/acs.nanolett.3c01808
A. Jamadar, R. Sutar, S. Patil, et al., Mater. Rep. : Energy 4 (2024) 100283.
J. Xu, C. Zhang, H. Liu, et al., Nano Energy 70 (2020) 104529. doi: 10.1016/j.nanoen.2020.104529
Y. Gao, Y. Yang, L. Hao, et al., Chem. Catal. 2 (2022) 2275–2288.
S. Zhao, Y. Wen, X. Liu, et al., Nano Res. 13 (2020) 1544–1551. doi: 10.1007/s12274-020-2765-1
X. Liu, Y. Luo, C. Ling, et al., Appl. Catal. B: Environ. 301 (2022) 120766. doi: 10.1016/j.apcatb.2021.120766
J. Tang, X. Liu, X. Xiong, et al., Adv. Mater. 36 (2024) 2407394. doi: 10.1002/adma.202407394
L. Lin, F. Wei, R. Jiang, et al., Nano Res. 16 (2023) 309–317. doi: 10.1007/s12274-022-4800-x
A. Feng, Y. Yu, Y. Wang, et al., Mater. Design 114 (2017) 161–166. doi: 10.1016/j.matdes.2016.10.053
V. Kamysbayev, A. Filatov, H. Hu, et al., Science 369 (2020) 979–983. doi: 10.1126/science.aba8311
M. Khazaei, A. Ranjbar, M. Arai, T. Sasaki, et al., J. Mater. Chem. C 5 (2017) 2488–2503. doi: 10.1039/C7TC00140A
S. Razzaq, S. Faridi, S. Kenmoe, et al., J. Am. Chem. Soc. 147 (2025) 161–168. doi: 10.1021/jacs.4c08518
Y. Guo, Q. Zhu, Z. Wang, et al., Adv. Energy Mater. 14 (2024) 2304149. doi: 10.1002/aenm.202304149
H. Bao, Y. Qiu, X. Peng, et al., Nat. Commun. 12 (2021) 238. doi: 10.1038/s41467-020-20336-4
X. Peng, H. Bao, J. Sun, et al., Nanoscale 13 (2021) 7134–7139. doi: 10.1039/d1nr00795e
X. Peng, Y. Mi, X. Liu, et al., J. Mater. Chem. A 10 (2022) 6134–6145. doi: 10.1039/d1ta07375c
Y. Wu, L. Wang, T. Bo, et al., Adv. Funct. Mater. 33 (2023) 2214375. doi: 10.1002/adfm.202214375
J. Zhang, E. Wang, S. Cui, et al., Nano Lett. 22 (2022) 1398–1405. doi: 10.1021/acs.nanolett.1c04809
C. Yang, Q. Jiang, H. Liu, et al., J. Mater. Chem. A 9 (2021) 15432–15440. doi: 10.1039/d1ta01784e
C. Du, L. Yang, K. Tang, et al., Mater. Chem. Front. 5 (2021) 2338–2346. doi: 10.1039/d0qm00898b
X. Peng, S. Zhao, Y. Mi, et al., Small 16 (2020) 2002888. doi: 10.1002/smll.202002888
X. Zhao, X. Zheng, Q. Lu, et al., EcoMat 5 (2023) e12293. doi: 10.1002/eom2.12293
O. Yaghi, H. Li, J. Am. Chem. Soc. 117 (1995) 10401–10402. doi: 10.1021/ja00146a033
L. Li, Z. Li, W. Yang, et al., Chem. 7 (2021) 686–698. doi: 10.1016/j.chempr.2020.11.023
J. Ji, Z. Li, C. Hu, et al., ACS Appl. Energy Mater. 12 (2020) 40204–40212. doi: 10.1021/acsami.0c06931
X. Cheng, X. Dao, S. Wang, et al., ACS Catal. 11 (2021) 650–658. doi: 10.1021/acscatal.0c04426
M. Ren, H. Zang, S. Cao, et al., J. Mater. Chem. A 11 (2023) 10435–10444. doi: 10.1039/d3ta01432k
Y. Niu, Y. Yuan, Q. Zhang, et al., Nano Energy 82 (2021) 105699. doi: 10.1016/j.nanoen.2020.105699
L. Jiao, W. Yang, G. Wan, et al., Angew. Chem. Int. Ed. 59 (2020) 20589–20595. doi: 10.1002/anie.202008787
T. Wang, Q. Zhang, K. Lian, et al., J. Colloid Interface Sci. 655 (2024) 176–186. doi: 10.1016/j.jcis.2023.10.157
S. Xu, F. Yang, S. Han, et al., CrystEngComm 22 (2020) 6063–6070. doi: 10.1039/d0ce01030h
M. Wen, N. Sun, L. Jiao, et al., Angew. Chem. Int. Ed. 63 (2024) e202318338. doi: 10.1002/anie.202318338
L. Chen, C. He, R. Wang, et al., Chin. Chem. Lett. 32 (2021) 53–56. doi: 10.1016/j.cclet.2020.11.013
Z. Sun, J. Lin, S. Lu, et al., Langmuir 40 (2024) 5469–5478. doi: 10.1021/acs.langmuir.3c04025
S. Liu, M. Jin, J. Sun, et al., Chem. Eng. J. 437 (2022) 135294. doi: 10.1016/j.cej.2022.135294
M. Pang, M. Yang, H. Zhang, Y. Shen, et al., Nano Res. 17 (2024) 9371–9396. doi: 10.1007/s12274-024-6923-8
D. Zhang, Z. Wang, F. Liu, et al., J. Am. Chem. Soc. 146 (2024) 3210–3219. doi: 10.1021/jacs.3c11246
G. Chen, R. Lu, C. Li, et al., Adv. Mater. 35 (2023) 2300907. doi: 10.1002/adma.202300907
R. Yao, K. Sun, K. Zhang, et al., Nat. Commun. 15 (2024) 2218. doi: 10.1038/s41467-024-46553-9
W. Shi, T. Shen, C. Xing, et al., Sci. 387 (2025) 791–796. doi: 10.1126/science.adr3149
J. Yang, F. Zhang, J. Chen, China Powder Sci. Technol. 30 (2024) 161–170.
J. Wang, L. Jia, H. Lin, Y. Zhang, ChemSusChem 13 (2020) 3404–3411. doi: 10.1002/cssc.202000702
S. Shah, T. Najam, M. Bashir, et al., Energy Storage Mater. 45 (2022) 301–322. doi: 10.1016/j.ensm.2021.11.049
Y. Liu, L. Tian, M. Huang, et al., Environ. Sci. Technol. 59 (2025) 880–891. doi: 10.1021/acs.est.4c06608
S. Wei, J. Zhu, X. Chen, et al., Nat. Commun. 16 (2025) 1652. doi: 10.1038/s41467-025-56271-5
Z. Yang, R. Chen, L. Zhang, et al., Ind. Chem. Mater. 2 (2024) 533–555. doi: 10.1039/d3im00109a
Z. Peng, Q. Zhang, G. Qi, et al., Chin. J. Struct. Chem. 43 (2024) 100191.
X. Li, W. Hu, China Powder Sci. Technol. 31 (2025) 81–90.
K. Zuo, S. Garcia-Segura, G. Cerrón-Calle, et al., Nat. Rev. Mater. 8 (2023) 472–490. doi: 10.1038/s41578-023-00564-y
C. Zhai, Y. Chen, X. Huang, et al., Environ. Funct. Mater. 1 (2022) 219–229. doi: 10.1177/0308518x211055181
X. Zhao, X. Niu, X. Liu, et al., Mater. Rep. : Energy 4 (2024) 100271.
L. Liu, K. Yung, H. Yang, B. Liu, Chem. Sci. 15 (2024) 6285–6313. doi: 10.1039/d4sc01030b
Y. Zhang, J. Zhao, S. Lin, Chin. J. Struct. Chem. 43 (2024) 100415.
Y. Liu, Y. Guo, Y. Jiang, et al., Mater. Rep. : Energy 4 (2024) 100252.
S. Lv, X. Han, J. Wang, et al., Angew. Chem. Int. Ed. 59 (2020) 11583–11590. doi: 10.1002/anie.202001510
Y. Tian, B. Li, J. Wang, et al., Chem. Eng. J. 490 (2024) 151704. doi: 10.1016/j.cej.2024.151704
W. Ma, T. Jian, J. Ma, et al., China Powder Sci. Technol. 30 (2024) 1–14. doi: 10.1109/tgrs.2024.3442470
M. Yang, J. Ding, Z. Wang, et al., Chin. Chem. Lett. 36 (2025) 110861. doi: 10.1016/j.cclet.2025.110861
G. Zhang, A. Wang, L. Niu, et al., Adv. Energy Mater. 12 (2022) 2103511. doi: 10.1002/aenm.202103511

Figure 1 The detailed outline diagram specifically designed for this comprehensive review.

Figure 2 (a) Illustration of the electrochemical NH3 synthesis mechanism utilizing Mn-O3N1/PC catalysts. Reproduced with permission [37]. Copyright 2021, American Chemical Society. (b) Conceptual representation of N2 capture and activation facilitated by Co single atoms, driven by the lone-pair electron pulling effect. Reproduced with permission [38]. Copyright 2022, Royal Society of Chemistry. (c) PdSA/N, S-OPC catalysts for efficient HOR. Reproduced with permission [30]. Copyright 2022, Elsevier. (d) Proposed reaction pathways for electrocatalytic denitrification on Meso-Fe-N-C catalysts. Reproduced with permission [42]. Copyright 2023, Elsevier. (e) Schematic diagram of bifunctional Nb-N-C catalysts. Reproduced with permission [42]. Copyright 2022, Springer Nature. Comparative analysis of N2 activation on Fe-N3S and Fe-N4 coordination structures: (f, g) Optimized side-on and end-on adsorption geometries of N2 on atomically dispersed Fe-N3S sites; (h, i) Corresponding side-on and end-on adsorption configurations of N2 on Fe-N4 coordination centers. Reproduced with permission [43]. Copyright 2022, Elsevier.

Figure 3 (a) Atomic Fe-Zn dual-metal sites designed for high-efficiency pH-universal ORR. Reproduced with permission [44]. Copyright 2020, Springer Nature. Chemical characterization of Ce-SA/NHCS: (b, c) High-resolution Ce 3d and N 1s XPS spectra; (d) XANES spectra of Ce-SA/NHCS and CeO2 at the Ce L3-edge; (e) Fourier-transformed EXAFS (FT-EXAFS) spectra of Ce-SA/NHCS and CeO2 at the Ce L3-edge; (f, g) FT-EXAFS fitting curves in k-space and the corresponding atomic structure of Ce-SA/NHCS. Theoretical simulations: (h) Free energy diagrams for the HER and NORR on the Ce-SA/NHCS surface; (i) Calculated adsorption energies for H and NO, along with their corresponding geometries; (j) Projected density of states (PDOS) of the protonated NO molecule and its bonded Ce-N4 site; (k) Charge density differences of the adsorbed NO molecule on the Ce-N4 moiety. Reproduced with permission [12]. Copyright 2023, Elsevier. (l) Schematic illustration of efficient CO2 electrolysis to CO through local coordination manipulation. Reproduced with permission [46]. Copyright 2023, American Chemical Society.

Figure 4 (a) Schematic illustration of the synthesis process for Pt-SA/α-MoOx. Reproduced with permission [48]. Copyright 2020, Elsevier. (b) The optimized structural configuration of MoO3−x. (c) Top view of the MoO3−x structure and the optimized placement of a La atom at a terminal oxygen vacancy site. (d) The electron density difference between N and La/MoO3−x, and (e) MoO3−x, with the red isosurface indicating charge accumulation. (f) Schematic representation of the nitrogen π *2p orbital. (g) Illustration of electron transfer from La to the adsorbed nitrogen molecule. Density of states (DOS) of the N2 2p orbital (h) before and (i) after adsorption on La/MoO3−x. DOS of the La 5d orbital (j) before and (k) following N2 adsorption. Reproduced with permission [51]. Copyright 2022, Elsevier. (l) Charge differential density maps for CoTA, AsTA, GaTA, and ZnTA. (m) Charge differential density maps upon N2 adsorption on MO—CoTA, MO-AsTA, MO-GaTA, and MO-ZnTA. PDOS and COHP of N2 on (n) Mo-CoTA and (o) Mo-AsTA. Reproduced with permission [53]. Copyright 2022, Springer Nature.

Figure 5 (a) XANES spectra at the Cu K-edge, with CuO, Cu2O, and Cu foil as reference materials. (b) k2-weighted Fourier transform (FT) EXAFS curves, where χ (k) represents the EXAFS oscillation function. (c) EXAFS fitting curve of Cu-SA/Ti3C2Tx, with an inset illustrating the structural configuration of Cu-SA/Ti3C2Tx. Free energy diagrams of CO reduction on (d) Cu-SA/Ti3C2Tx and (e) Cu (111). (f) Schematic representation of COR pathways. Charge density difference of the *COCHO-adsorbed and *COCO-adsorbed configurations on (g) Cu-SA/Ti3C2Tx and (h) Cu (111), respectively. (i) PDOS of d-orbitals in Cu-SA/Ti3C2Tx and Cu (111), with aligned Fermi levels. Reproduced with permission [59]. Copyright 2021, Springer Nature. First-principles calculations of HER and HzOR: (j) Three distinct active sites on the Rh-SA/Ti3C2Ox catalyst surface: 1 (O), 2 (Rh), and 3 (Rh-O). (k) Side and top views of the three-dimensional charge density difference for H-adsorbed configurations at different sites on the Rh-SA/Ti3C2Ox catalyst surface. Reproduced with permission [61]. Copyright 2021, Royal Society of Chemistry. (l) Schematic representation of the etching process of Ti2AlC to produce vacancy-enriched Ti2CClx MXenes. Reproduced with permission [58]. Copyright 2024, John Wiley and Sons. (m) Mechanism of electrocatalytic enhancement for cobalt single atoms anchored on various MXenes substrates in HER and OER. Reproduced with permission [67]. Copyright 2022, John Wiley & Sons.

Figure 6 (a) Influence of varying microenvironments on the catalytic efficiency within MOFs. Reproduced with permission [69]. Copyright 2021, Elsevier. (b) Schematic representation of the synthesis procedure for Fe2O3 with diverse morphologies and structural configurations. Reproduced with permission [73]. Copyright 2021, Elsevier. (c) Schematic depiction of the microwave-assisted rapid fabrication of MOF-derived Ni1—N-C catalysts for CO2RR. Reproduced with permission [77]. Copyright 2024, John Wiley and Sons. (d) Raman spectroscopic analysis of Fe-NP/MNCF. (e) N2 adsorption-desorption isotherms of Fe-NP/MNCF. XPS spectra of (f) N 1s, and (g) O 1s for Fe-NP/MNCF. Reproduced with permission [75]. Copyright 2024, Elsevier. Differential charge density distribution of FeN3B with adsorbed *COOH from (h) side and (i) top perspectives. (j) Density of states (DOS) for FeN4 and FeN3B. (k) Free energy profiles for CO2RR on FeN4 and FeN3B. Reproduced with permission [80]. Copyright 2022, Elsevier.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
 下载:
下载: