Jiangsu Collaborative Innovation Center of Chinese Medicinal Resources Industrialization, National and Local Collaborative Engineering Center of Chinese Medicinal Resources Industrialization and Formulae Innovative Medicine, Jiangsu Provincial Key Laboratory of Functional Substances in Traditional Chinese Medicine Formulae and Innovative Drug Discovery, Nanjing University of Chinese Medicine, Nanjing 210023, China
b.
School of Medicine, Nanjing University of Chinese Medicine, Nanjing 210023, China
chunjie@njucm.edu.cn (C.-J. Bao). 1 These authors contributed equally to this work.
Received Date:
11 May 2025 Accepted Date:
18 July 2025 Revised Date:
17 July 2025 Available Online:
15 July 2026
Abstract:
Macrophage membrane-camouflaged nanoparticles (MmNPs) are emerging as efficient nanoplatforms for targeted delivery to inflammatory sites, tumors, and infected tissues due to the innate characteristics of macrophages. This biomimetic strategy effectively addresses several limitations of traditional targeted drug delivery systems, offering improved biocompatibility, extended blood circulation, immune evasion, and site-specific homing. Compared to conventional functionalization methods, MmNPs provide a simplified method for creating multifunctional nanoparticles. In this review, we explore the origins and functions of macrophages, highlighting how MmNP platforms are leveraged for precise drug delivery. The latest applications of MmNPs in targeted delivery are summarized, focusing on their intrinsic targeting properties, membrane surface modifications and designs for environmental stimulus response. Finally, we discuss the prospects and challenges in translating MmNP technology from experimental settings to clinical applications, aiming to inspire continued innovation in the design of MmNP for precise and effective drug delivery strategies against complex diseases.
Traditional targeted drug delivery systems, such as ferritin, liposomes, and extracellular vesicles, have been extensively studied, yielding significant advancements in therapeutic delivery strategies [1,2]. These systems are characterized by several key advantages, including efficient drug encapsulation, high biocompatibility, low systemic toxicity, and enhanced permeability and retention, making them suitable for treating diverse clinical conditions [3]. Despite these achievements, conventional nanoparticle-based delivery platforms still encounter significant limitations, such as limited delivery routes, suboptimal targeting precision, rapid blood clearance, and potential for off-target accumulation or unintended biodistribution [4-6].
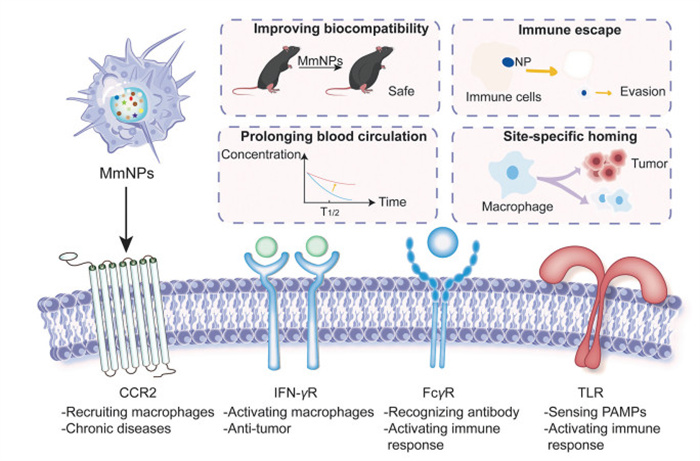
Recently, the strategy of cell membrane camouflaging nanoparticles to enhance drug delivery efficiency has become popular. Macrophages, as integral components of the innate immune system, express immune checkpoints and activate innate immune response through the receptors on surface [7,8]. The intrinsic biocompatibility of macrophage membrane enabled these biomimetic preparations less immune rejection and reduced toxicity, allowing them to cross biological barriers and avoid elimination of immune systems. Moreover, with the character of remarkable plasticity and native tumor-targeting ability, macrophage membrane are modifiable and promising [9,10], that is, macrophage membrane-camouflaged nanoparticles (MmNPs) are confirmed to have better tumor-targeting ability and the biocompatibility and safety of the formulation can be increased with modified macrophage membrane (Fig. 1).
Figure 1
Figure 1.
Schematic illustration of advantages and surface markers of MmNPs. MmNPs demonstrate improved biocompatibility, immune escape, prolonged blood circulation and site-specific homing. Main surface receptors on the macrophage surface include CCR2, interferon-γ receptor (IFN-γR), FcγR and TLR. These surface markers contribute to the versatility and functionality of MmNPs in therapeutic applications.
In this review, MmNPs has been divided into three principal systems according to their drug delivery mechanisms. We first provide a comprehensive overview of macrophage biology, focusing on their origins and regulatory mechanisms. We then discuss the functional properties of macrophages from the perspective of their membrane composition, exploring the potential of macrophage membranes as specialized targeting carriers. Next, we summarize recent advances in MmNPs, emphasizing their three primary applications for targeted drug delivery. Finally, we outline the emerging trends in the development of MmNPs, offering insights into potential future directions for research and innovation in this evolving field.
2.
Macrophages in development and regulation
2.1
Developmental origin of macrophages
The evolution of species diversity has driven the development of a sophisticated host defense system rooted in innate immunity [11]. Macrophages, as key innate immune cells, are mononuclear phagocytes essential for maintaining tissue integrity and homeostasis in vertebrates. Strategically distributed, they perform diverse functions, including clearing dead cells, debris, and foreign material, as well as orchestrating inflammatory responses [12]. While their immune and reparative roles are well-studied, their contribution to tissue development is equally vital. Understanding macrophage origins is crucial for elucidating their heterogeneity, tissue specialization, and broader developmental roles in MmNPs.
Macrophages originate from embryonic progenitors and bone marrow-derived monocytes. During embryogenesis, progenitor-derived macrophages migrate to developing tissues, regulating hematopoiesis, metabolism, tissue repair, and maturation [13,14]. Yolk sac-derived progenitors differentiate into tissue-resident macrophages (TRMs), which colonize organs such as the brain (microglia), liver (Kupffer cells), lungs (alveolar macrophages), and skin (Langerhans cells) from embryonic day 8.5 (E8.5), independent of the transcription factor MYB [15-17]. Fetal liver-derived monocytes, originating from hematopoietic stem cells (HSCs), also contribute to TRMs in a MYB-dependent manner from E9.5 [12]. Fate-mapping studies reveal that most tissue macrophage populations, including primitive macrophages and monocyte progenitors, are established prenatally and persist into adulthood [18-22].
It is now well-established that macrophages are typically regarded as long-lived, terminally differentiated cells that persist throughout adulthood [23]. Postnatally, tissue macrophages are replenished by bone marrow-derived monocytes, which differentiate and proliferate in response to developmental or pathological signals [19,24,25]. TRMs can self-maintain through local proliferation, independent of HSCs, potentially via intrinsic self-renewal mechanisms [26,27]. Low levels of MafB and c-Maf, in the presence of macrophage colony-stimulating factor (M-CSF), can activate macrophage self-renewal without altering their functional phenotype or inducing transformation [28].
2.2
Macrophage polarization
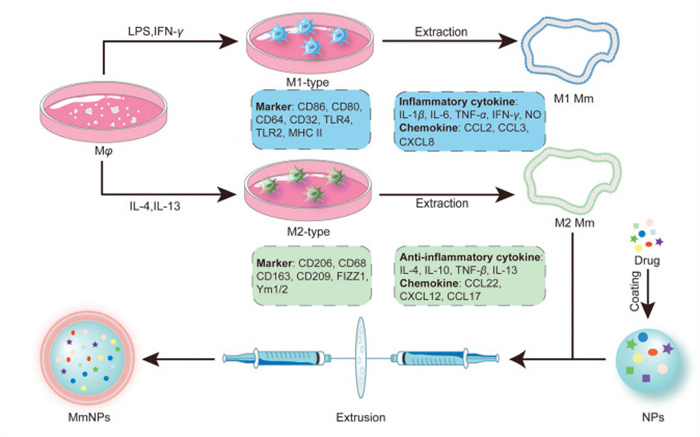
Under normal physiological conditions, naïve macrophages (Mφ) are widely distributed across tissues, maintaining homeostasis by clearing apoptotic debris. Their high plasticity enables polarization into distinct phenotypes in response to environmental or pathological stimuli (Fig. 2) [29].
Figure 2
Figure 2.
Schematic overview of macrophage polarization and the preparation of MmNPs. Macrophages are polarized into two major phenotypes, each characterized by distinct surface markers, cytokine profiles, and chemokine expression. Following polarization, macrophage membranes are isolated and used to camouflage pre-formed nanoparticles loaded with functional drug payloads. This process yields biomimetic nanoparticles (MmNPs), typically assembled via extrusion methods.
Classically activated M1 macrophages exhibit pro-inflammatory properties, driven by signals such as Toll-like receptor (TLR) ligands (e.g., lipopolysaccharide (LPS)), interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α), and granulocyte-macrophage CSF (GM-CSF). These cells secrete cytokines (e.g., interleukin-1β (IL-1β), IL-6, IL-12, TNF-α) and chemokines (e.g., C—C motif chemokine ligand 2 (CCL5), C-X-C motif chemokine ligand 2 (CXCL2)), and produce nitric oxide (NO) and reactive oxygen species (ROS) via inducible nitric oxide synthase (iNOS) and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase pathways, enhancing pathogen clearance and tumor suppression [29-31].
Alternatively, Mφ can polarize into M2 macrophages, which exhibit anti-inflammatory, pro-angiogenic, and tissue-repair functions. M2 polarization is induced by T helper 2 (Th2) cytokines (e.g., IL-4, IL-13), glucocorticoids, IL-10, and M-CSF, activating pathways such as Janus kinase–signal transducer and activator of transcription (JAK-STAT) and STAT3. M2 macrophages secrete anti-inflammatory mediators (e.g., IL-10, transforming growth factor-beta (TGF-β), CCL20) and promote tissue repair by recruiting Th2 cells, regulatory T cells (Tregs), and eosinophils [32-37].
Macrophage plasticity allows interconversion between M1 and M2 phenotypes in response to changing stimuli, highlighting their functional diversity. This dynamic behavior underscores the limitations of the binary M1/M2 classification, as macrophages often exhibit mixed roles in maintaining tissue homeostasis and resolving inflammation [38-40].
2.3
Macrophage infiltration
2.3.1
Chemotaxis
In innate immunity, circulating monocytes infiltrate inflamed or injured tissues, differentiating into macrophages or dendritic cells [41]. These monocyte-derived macrophages (MDMs) often overlap with tissue-resident phagocytes in immune responses. Macrophage infiltration is triggered by pathogen-associated molecular patterns (PAMPs), such as bacterial products, and damage-associated molecular patterns (DAMPs) released from damaged or dead cells due to trauma, ischemia, or injury [42]. Resident memory T cells can also recruit macrophages by secreting inflammatory cytokines and chemokines upon antigen activation [43].
TRMs express receptors like TLRs, NOD-like receptors (NLRs), and scavenger receptors to detect PAMPs and DAMPs [44-46]. Unlike MDMs, TRMs respond to microbial challenges by recruiting monocytes, which dominate inflammatory lesions [47].
2.3.2
Migration kinetics
Macrophages migrate in two-dimensional (2D) or three-dimensional (3D) modes. In 2D, they undergo processes like rolling, adhesion, and transcellular migration [48]. In 3D environments, such as the extracellular matrix (ECM), they use ameboid or mesenchymal migration. Ameboid movement, characterized by a round shape and actomyosin-driven motility, occurs in loose ECM, while mesenchymal migration, involving elongated cell shapes and protease-dependent ECM degradation, occurs in dense ECM [49,50]. Macrophages uniquely form podosome rosettes during mesenchymal migration, a feature absent in ameboid migration [51].
2.3.3
Cascade infiltration process
Macrophage infiltration is a cascade process, with outcomes varying based on tissue environment and pathological conditions.
(1) In the inflammatory tissue: Macrophage infiltration begins with weak, transient interactions between macrophages and venular endothelial cells near inflamed tissues, mediated by selectins (e.g., P-selectin, E-selectin) and integrins (e.g., vascular cell adhesion molecule-1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-1)) [52,53]. Chemokines on endothelial cells stimulate adherent macrophages, which then transmigrate or crawl along vessels using integrins to locate exit sites [54].
(2) In the tumor microenvironment: In the tumor microenvironment (TME), tumor-associated macrophages (TAMs) are highly heterogeneous and plastic, comprising up to 50 % of cells in some solid tumors [55,56]. Derived primarily from infiltrating monocytes, TAM recruitment is driven by tumor-derived factors like CCL2, CCL5, CSF-1, and hypoxia [57-61]. M2-like TAMs promote tumor initiation, progression, and metastasis by secreting inflammatory cytokines, remodeling the ECM via proteases, and inducing angiogenesis [62-65]. High TAM levels correlate with poor prognosis and treatment resistance.
(3) In the other pathological region: Macrophages play pivotal roles in diseases such as atherosclerosis, fibrosis, and cancer. In atherosclerosis, monocytes adhere to arterial endothelium, differentiate into lesional macrophages, and contribute to plaque progression or resolution, highlighting their dual roles in inflammation and repair [66,67]. Targeting macrophage plasticity offers therapeutic potential in these conditions.
2.4
Function of macrophages
Macrophages play essential roles in host defense and tissue homeostasis through chemotaxis, phagocytosis, antigen presentation, and tissue repair, protecting against pathogens and aiding tissue recovery.
2.4.1
Phagocytosis
Phagocytosis, a key macrophage function, involves the engulfment of pathogens, apoptotic cells, and debris via surface receptors like scavenger receptors, Fcγ receptors (FcγRs), and complement receptors (CRs) [68]. Following injury, macrophages clear cellular debris and secrete growth factors, reducing inflammation and promoting repair [69]. Specialized macrophages, such as bone marrow-derived macrophages (BMDMs) and TRMs, can phagocytose live, non-apoptotic cells, including infected or tumor cells, underscoring their role in maintaining tissue homeostasis [70].
2.4.2
Antigen presentation
Macrophages act as antigen-presenting cells (APCs) by presenting antigens via major histocompatibility complex class Ⅱ (MHCⅡ) molecules, activating Th cells. While less efficient than dendritic cells, proinflammatory macrophages exhibit cross-presentation capabilities, primarily through the vacuolar pathway [71]. However, identifying specific subsets and markers for cross-presenting macrophages remains challenging [72].
2.4.3
Tissue repair
Macrophage subsets exhibit specialized roles in tissue repair. MDMs often exacerbate inflammation, while embryonic-derived resident macrophages promote anti-inflammatory responses and tissue regeneration, highlighting their functional diversity in immune modulation and repair processes [73].
3.
Macrophage membrane and its advantages in the preparation of MmNPs
3.1
Characteristic of macrophage membrane
Macrophage membranes are highly dynamic, composed of lipids, proteins, and carbohydrates, with the lipid bilayer serving as the structural scaffold that facilitates their flexibility. Macrophage membranes retain the innate biological functionalities of their cellular origin, such as immune modulation, the ability to evade phagocytosis, extended circulation time in vivo, and enhanced targeting efficacy.
The proteins on the surface of macrophage membrane expressed by distinct macrophage populations exhibit functional heterogeneity. For instance, proteins on alveolar macrophages regulate lipid metabolism, while those on kupffer cells (KCs) play a role in blood coagulation [74]. In the context of the innate immune response, pattern recognition receptors (PRRs) are extensively expressed on macrophages, enabling their response to various PAMPs. Among these, the TLR family is particularly well-known, with studies indicating that TLRs are broadly expressed across macrophage populations, including intestinal macrophages, which express a wide array of TLRs, allowing for recognition of diverse pathogens [74].
Recruited macrophages typically express fewer PRRs but exhibit enhanced recognition capabilities for different PAMPs compared to TRMs. However, TRMs are characterized by a higher density of inflammation-associated receptors, including TLRs and IL-6 receptor family members. These inflammation-related receptors contribute to the superior inflammation-targeting capabilities of macrophage membranes, making them highly effective for developing inflammation-targeted nanocoatings [75].
Interestingly, the lipid composition of macrophage membranes can also serve as secondary signaling molecules during cellular signaling processes. A study by Schoeniger et al. demonstrated that an increase in unsaturated fatty acids within membrane lipids disrupts the membrane's microstructure, which in turn influences the activation of TLR4 receptors. This disruption can impact the overall efficacy of the PRR signaling pathway in pathogen recognition by TRMs [76,77].
3.2
Microenvironment-specific modifications
The therapeutic performance of conventional nanoparticles is frequently hindered by rapid clearance mediated by the host's phagocytic and immune systems. In contrast, MmNPs exhibit improved biocompatibility, immune evasion, and disease-specific targeting capabilities. These properties of MmNPs are influenced by the local microenvironment and may adapt accordingly under different physiological or pathological conditions. Within the TME, MmNPs leverage the innate properties of macrophage membranes to escape immune surveillance and respond to chemokine gradients, facilitating their accumulation within tumor tissues. Tumor-derived signals, such as elevated cytokine concentrations (e.g., CCL2, CSF-1) [78] and the acidic, hypoxic conditions, further enhance the preferential retention of MmNPs in tumor niches. Conversely, in inflammatory microenvironments marked by heightened oxidative stress, cytokine fluctuations, and acute or chronic immune activation, macrophage-associated receptors including folate receptors (FRs), CD44, and scavenger receptors (SRs) become upregulated during disease progression, thereby promoting targeted delivery to inflamed tissues [79]. These microenvironment-dependent variations underscore the necessity for rational design strategies, such as ligand conjugation and chemical surface engineering, to enhance targeting precision. Taken together, the unique properties of macrophage-derived membranes and their integration into nanoparticle-based delivery platforms offer promising opportunities for the development of next-generation therapeutics aimed at tumors, inflammatory disorders, and other pathologies characterized by complex microenvironments.
3.3
Advantages for preparing MmNPs
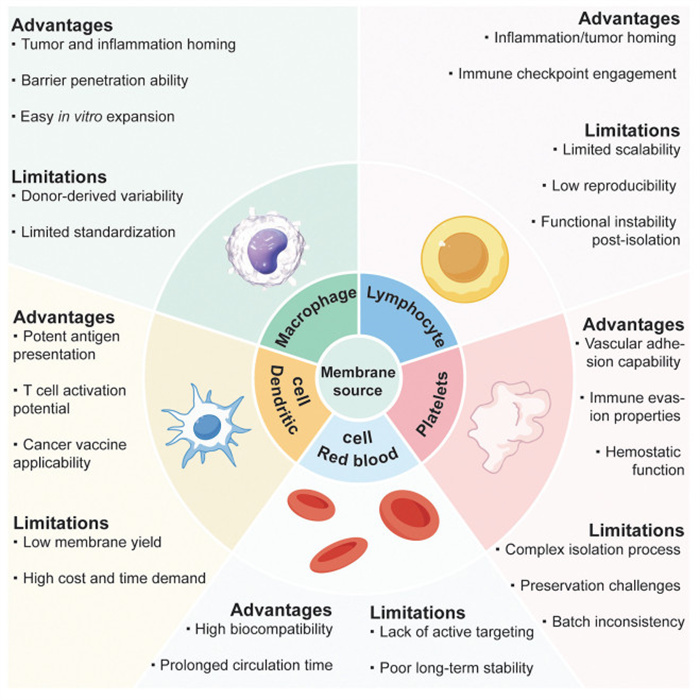
Recent advancements in drug delivery systems have employed bioinspired strategies to overcome challenges associated with clearance by the reticuloendothelial system (RES) and mononuclear phagocytic system (MPS), achieving enhanced biocompatibility and prolonged circulation. Among these, membrane- camouflaged nanoparticles have emerged as a promising approach for therapeutic delivery [80,81]. While various cell membranes, including those from red blood cells, platelets, dendritic cells and lymphocytes, have been explored, macrophage membranes have demonstrated increasing advantages due to their superior targeting capabilities and ease of preparation.
Membrane-camouflaged nanoparticles are typically fabricated by fusing a nanoparticle core with cell-derived membranes using methods such as extrusion, stirring, or sonication. However, the procedures for membrane isolation and application vary depending on the intrinsic characteristics of the source cells. The respective advantages and limitations of different membrane types are summarized in Fig. 3. For example, nanoparticles camouflaged with erythrocyte membranes have shown the capacity to prolong systemic circulation by leveraging the CD47-signal regulatory protein α (SIRP α) interaction, while also passively accumulating at lesion sites via the enhanced permeability and retention (EPR) effect. Nonetheless, their therapeutic precision remains limited due to the absence of inherent active targeting functionalities [82-85]. Similarly, platelet membrane-coated nanoparticles exhibit a natural affinity for injured vascular endothelium, offering potential in targeted vascular therapies. However, their clinical translation is challenged by the technical difficulties associated with membrane isolation, preservation, and functional stability [86-89]. Dendritic cell membranes possess intrinsic antigen-presenting capabilities and the potential to stimulate adaptive immune responses, making them attractive for immunotherapeutic applications. Nevertheless, the high cost, low yield, and labor-intensive expansion of dendritic cells significantly hinder scalability [90-92]. Lymphocyte membrane-coated nanoparticles display chemotactic behavior toward inflamed tissues, although concerns regarding unintended immune activation and autoimmune responses remain significant safety barriers [93-96]. In contrast, macrophage membranes are enriched with diverse surface receptors that recognize a broad range of pathological cues, including inflammatory chemokines, PAMPs, and tumor-associated adhesion molecules. Their relative ease of isolation and cost-effectiveness further enhances their practicality for membrane-coating strategies. Consequently, macrophage membrane-camouflaged nanoparticles offer dual-targeting capabilities toward both inflammatory microenvironments and tumor tissues, establishing them as a versatile and promising platform for therapeutic delivery.
Figure 3
Figure 3.
Comparative advantages and limitations of cell membranes for targeted drug delivery applications. Membranes from macrophages, dendritic cells, red blood cells, platelets and lymphocytes were discussed.
The fabrication of MmNPs involves several common steps are shown in Fig. 2. Firstly, primary macrophages are polarized into distinct phenotypes through cytokine stimulation, M1 macrophages via LPS and IFN-γ, and M2 macrophages via IL-4 and IL-13. Secondly, cellular membranes are isolated to obtain phenotype-specific membranes that retain the immunological signatures of their parent cells. M1-derived membranes are enriched with pro-inflammatory cytokines (e.g., IL-1β, IL-6, TNF-α, IFN-γ) and chemokines (e.g., CCL2, CXCL8), whereas M2-derived membranes exhibit elevated levels of anti-inflammatory mediators (e.g., IL-4, IL-10, TGF-β) and their corresponding chemokines. These functional membranes are then coated onto drug-loaded nanoparticles via extrusion, yielding biomimetic MmNPs with enhanced immune modulation and targeted delivery capabilities.
4.
MmNPs for targeted drug delivery
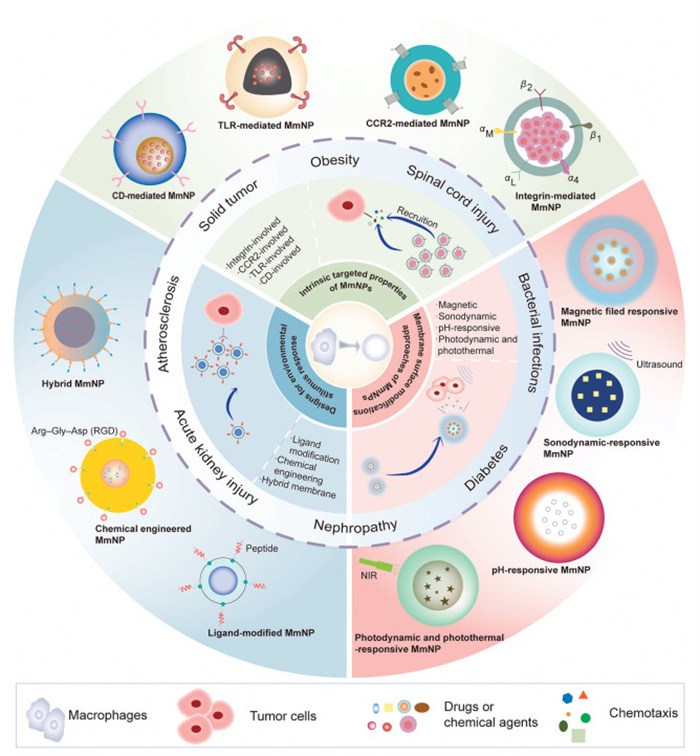
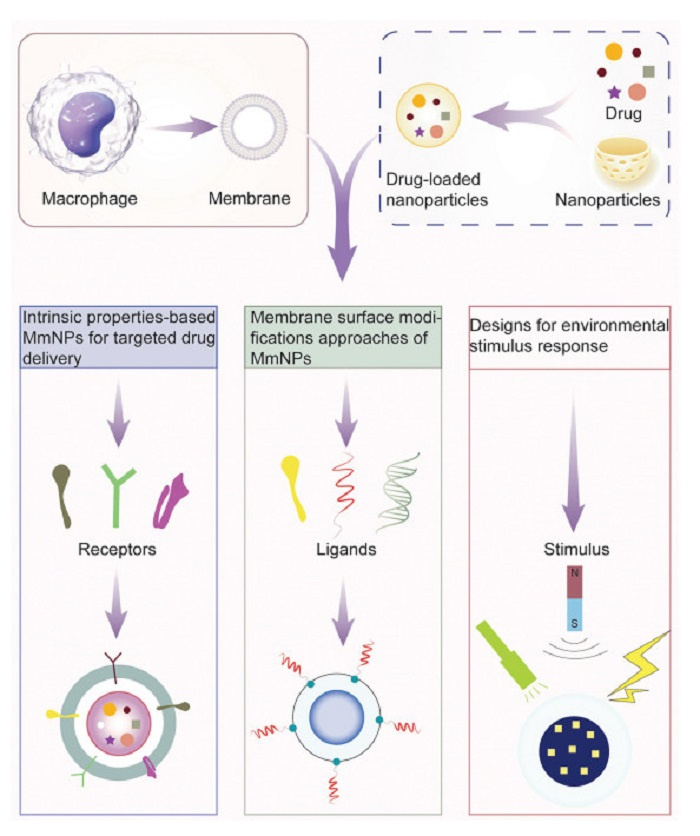
Given their intrinsic homing capabilities and excellent biocompatibility, MmNPs have emerged as a cutting-edge platform for precision drug delivery. As illustrated in Figs. 4 and 5, this review categorizes MmNPs into three primary systems: (1) Intrinsic property-based targeted delivery system utilizing the natural functionality of macrophage membranes, (2) membrane surface modifications-based targeted delivery system, and (3) environmental stimulus response-based targeted delivery system. These innovation approaches highlight the transformative potential of MmNPs in advancing precision medicine and targeted drug delivery.
Figure 4
Figure 4.
Overview of three categories of MmNPs for targeted drug delivery. This figure summarizes the three major categories of MmNPs, highlighting their design principles, mechanisms of action, and representative applications.
Figure 5.
Functional components enabling targeted delivery in MmNPs. This figure illustrates the design principles of three representative MmNP-based targeted delivery strategies, each driven by distinct targeting modalities. (ⅰ) Intrinsic properties-based MmNPs are achieved through native macrophage membrane receptors, such as integrins and chemokine receptors, which enable biomimetic recognition of pathological sites. (ⅱ) Active targeting involves surface modification with exogenous ligands (e.g., antibodies, peptides) to enhance selective binding to disease-specific markers. (ⅲ) Stimuli-responsive targeting incorporates environment-sensitive components that respond to internal or external triggers, such as magnetic fields, US, pH, or light, to enhance site-specific drug release.
4.1
Intrinsic properties-based MmNPs for targeted drug delivery
Macrophages, owing to their unique membrane composition, have emerged as effective targeting delivery systems for therapeutic nanoparticles owing to their intrinsic properties. Macrophage membrane markers not only enable recognition of tumor cells and foreign invaders but also possess innate homing characteristics that guide them toward tumor, inflamed, or infected regions via cell-cell communication mechanisms. The rapid growth of research in macrophage membrane-camouflaged nanoparticles has yielded significant advancements in tumor-targeted therapies [97], inflammation-targeted therapy [98], and applications against infection [99].
Nanoparticles cloaked with macrophage membranes demonstrate the ability to evade clearance by MDMs within the RES, thereby achieving prolonged systemic circulation. These nanoparticles are then recruited to specific sites with high precision due to their homologous targeting properties (Table S1 in Supporting information) [100]. Moreover, loading different kinds of drugs such as doxorubicin (DOX), polysaccharide [101,102] and other clinical anti-inflammatory and anti-tumor drugs [103] lead MmNPs to different types of diseases.
4.1.1
Integrins-involved MmNPs for targeted drug delivery
Integrins expressed on macrophages play critical roles in mediating adhesion, migration, and phagocytosis within the tumor microenvironment and sites of inflammation. The α4β1 integrin binds VCAM-1 on endothelial and tumor cells, facilitating macrophage recruitment and enhancing nanoparticle uptake via phagocytosis. Similarly, αLβ2 (LFA-1) interacts with ICAMs to support transendothelial migration, enabling macrophages to traverse the blood–brain barrier and home to inflamed tissues. αMβ2 (Mac-1), another β2 integrin, not only promotes adhesion and Fc receptor-mediated phagocytosis but also recognizes dsDNA, modulating TLR signaling. These integrin-ligand interactions collectively coordinate macrophage trafficking and functional responses in pathological contexts.
4.1.2
TLRs-related passive targeted delivery system
TLRs are broadly expressed on the membranes of activated macrophages, where they play a critical role in detecting and responding to diverse endogenous stimuli arising from injury, infection, or disease [104]. Beyond their selective ligand recognition capabilities, TLRs rely on auxiliary surface molecules such as CD14, myeloid differentiation factor 2 (MD2), and C-type lectin-like receptors to mediate proximal ligand binding and recognition [44,46,105]. TLRs recognize a broad spectrum of PAMPs, such as lipoteichoic acid (via TLR-2), double-stranded RNA (via TLR-3), bacterial lipopolysaccharide (via TLR-4), and single-stranded RNA (via TLR-7) [106]. As the first receptors activated during host-pathogen interactions, TLRs play an essential role in microbial detection and the induction of innate and adaptive immunity [107].
In innate immunity, TLRs initiate immune responses by dimerizing upon PAMP recognition, forming a supramolecular organizing center (SMOC) that coordinates multiple innate immune pathways. This structure acts as a hub for the activation of innate immune responses, ensuring a robust reaction to microbial challenges [108,109]. TLRs also have a critical function in adaptive immunity. They enhance antigen presentation by upregulating MHC molecules and co-stimulatory markers on macrophages, thereby facilitating effective T-cell activation [106,110]. Collectively, TLRs expressed on macrophages inherit the ability to engage with PAMPs or DAMPs on the target site.
4.1.3
Cluster of differentiation (CD) molecules-related passive targeted delivery system
Macrophages inherently target inflammatory sites through specific surface markers such as CD44 and CD11b, which mediate adhesion and signal transduction during inflammation [111]. Mechanistically, as the natural ligand for CD44 overexpressed on M1-like macrophages, CD44 mediates the homing of macrophages to inflammatory site [112].
4.1.4
Chemokine receptor-related passive targeted delivery system
In tumor microenvironments, TAMs are preferentially recruited by tumor cells, representing the largest subpopulation of immunosuppressive cells. The recruitment of TAMs is primarily facilitated by the CCL2/C—C chemokine receptor 2 (CCR2) chemokine axis, where CCL2 secreted by cancer cells attracts macrophages expressing the CCR2 receptor. Beyond tumor targeting, CCR2 receptors on macrophage membranes also enable macrophage-coated nanoparticles to localize rapidly to inflammatory sites, such as those observed in acute lung injury (ALI) [113], spinal cord injury [114], atherosclerotic [115,116] and acute respiratory distress syndrome (ARDS) [117].
4.1.5
Cytokines-related passive targeted delivery system
Main cytokines on macrophage membrane, including IL-receptor, TNF receptor and CSF1 receptor, are pivotal mediators of immune responses and represent valuable targets for therapeutic modulation. These receptors facilitate passive targeting in macrophage-based delivery systems. Their functional specificity and context-dependent roles necessitate precise receptor selection for effective treatment of cancers and inflammatory diseases.
4.2
Membrane surface modifications approaches of MmNPs
The rapid advancements in nanotechnology have enabled engineered nanomaterials to serve as an ideal platform for constructing actively targeted delivery systems through modifications of the macrophage membrane (Table S2 in Supporting information).
4.2.1
Ligand modification
The intricate network of signaling cascades orchestrates critical cellular processes such as growth, division, and programmed cell death, primarily mediated by stable receptor-ligand interactions. Transmembrane proteins play a pivotal role in these interactions, modulating complex signaling networks to maintain cellular equilibrium [118]. Leveraging these interactions, macrophage membranes can be extracted and modified with specific ligands to coat nanoparticles, thereby enhancing their recognition capabilities, regulating immune responses, facilitating intercellular communication, and diversifying functional applications.
4.2.2
Chemical engineering
Macrophage membrane metabolic modification using choline (Cho) analogs has emerged as a robust strategy for introducing functional chemical tags to facilitate drug delivery. Analogues such as azidoethylcholine (AECho) and azidopropylcholine (APCho) are metabolically incorporated into phosphatidylcholine (PC), the primary component of eukaryotic cell membranes. These modifications enable the surface expression of bioorthogonal groups, including azides and alkynes, which can be leveraged for targeted delivery through efficient chemistries such as azide-dibenzocyclooctyne (DBCO) copper-free click reactions [119-121].
4.2.3
Hybrid membrane
Hybrid membrane-coated nanoparticles represent a promising avenue for enhancing targeted delivery to glioblastoma tissues and other malignancies, leveraging the unique properties of tumor and macrophage cell membranes. These biomimetic nanoparticles combine the tumor-homing capabilities of cancer cell membranes with the immune evasion properties of macrophage membranes, offering a dual advantage in selective targeting and immune modulation [122].
4.3
Designs for environmental stimulus response
4.3.1
Magnetic field responsive MmNPs
Inorganic iron oxide nanoparticles (Fe3O4 NPs) have gained significant attention in photothermal cancer therapy due to their broad optical absorption within the near-infrared (NIR) range and their capacity to generate hyperthermia under NIR laser irradiation [123]. Notably, Fe3O4 NPs have received approval from the U.S. Food and Drug Administration (FDA) owing to their favorable biodegradability and biocompatibility. However, a major limitation of these nanoparticles is their rapid recognition and clearance by the immune system, particularly via the RES, which hampers their therapeutic efficacy in systemic circulation.
4.3.2
Sonodynamic responsive MmNPs
Ultrasound (US), as a mechanical wave, has garnered significant attention due to its advantages, including deep tissue penetration, high precision in targeting tumor tissues, absence of ionizing radiation, excellent biosafety profile, and minimal side effects [124-126]. Recently, sonodynamic therapy (SDT), leveraging the US-induced activation of sonosensitizers, has emerged as a promising therapeutic approach for addressing refractory diseases. By generating singlet oxygen (1O2), inducing cavitation, or producing hyperthermia, SDT holds potential for treating various conditions [126,127]. However, its broader application in combating cancer, cardiovascular diseases, and multidrug-resistant (MDR) bacterial infections is hindered by challenges such as low chemical and biological stability of sonosensitizers and insufficient accumulation in tumor tissues [128].
4.3.3
pH-responsive MmNPs
Tumor microenvironment-responsive therapies have garnered significant attention over recent years. The advancement of pH-responsive theranostic agents, which can be activated in the mildly acidic tumor microenvironment (pH 5.0–7.4), has demonstrated remarkable efficacy in antitumor applications [129].
Cationic polymers, including poly(methacrylamidoamine) (PMAMA) dendrimer, poly(β-amino ester), and poly(amino acid), are positively charged and exhibit a proton sponge effect due to their tertiary amine groups. In the acidic endosomal environment, these polymers capture protons, which induces an influx of water and chloride ions, leading to endosomal swelling and membrane rupture, allowing the material to escape the endosome.
Analogously, in the weakly acidic tumor microenvironment, the contents of membrane-camouflaged nanoparticles may rupture their coating membrane, enhancing therapeutic delivery.
4.3.4
Photodynamic and photothermal responsive MmNPs
PDT and PTT are light-activated therapeutic strategies that have been extensively explored for the treatment of cancer and inflammatory diseases, primarily through the generation of ROS or localized hyperthermia [130]. In PDT, photosensitizers are activated upon exposure to light, leading to the generation of ROS that mediate cytotoxic effects and induce immunogenic cell death [131]. In contrast, in PTT, NIR light is converted into heat by photothermal agents, resulting in the thermal ablation of pathological tissues. Both modalities offer precise spatial and temporal control, minimal invasiveness, and synergistic potential when combined with immunotherapies or chemotherapeutic regimens.
In conclusion, physical and chemical targeting delivery systems demand high technological precision, integrating multiple technologies to create multifunctional nanoparticles with added functionalities, such as real-time tracking. However, this level of technological advancement and instrumentation complexity currently limits large-scale production (Table S3 in Supporting information).
5.
Perspective
This review provides an in-depth exploration of macrophage origins, fundamental characteristics, and functional roles, with a particular emphasis on surface composition to clarify the mechanisms underlying the potential targeting properties of macrophage membranes. We further classify and discuss current methods for engineering macrophage membrane-camouflaged drug delivery systems, along with future directions in this advancing field.
Recent years have seen traditional oral drug formulations, such as tablets, capsules, and injections, play a central role in treating both acute and chronic diseases. However, advancements in "smart" drug delivery systems, including liposomes, polymers, and extracellular vesicles, have demonstrated improved efficiency and targeting precision [132]. To enhance tissue-specific drug targeting, controlled release, and dosage reduction, nanoparticles cloaked in macrophage membranes have shown significant promise.
Macrophage membrane-based delivery systems present both significant advantages and certain challenges in the field of targeted drug delivery. Macrophage membrane cloaking represents an innovative biomimetic approach to drug delivery [133], endowing MmNPs with unique immune functions and targeting abilities. Firstly, this membrane coating can markedly prolong the circulation time of nanoparticles by evading clearance from the mononuclear phagocyte system. Secondly, macrophage membranes exhibit tumor and inflammation homing ability, which enhance the targeting specificity of the nanocarriers toward diseased tissues. This targeting effect arises from the natural surface markers and chemokine receptors retained on the membrane. Thirdly, barrier penetration ability endows MmNPs more targets than common drug or nanoparticles. Moreover, MmNPs exhibit enhanced structural integrity, maintaining a more uniform and stable spherical morphology compared to their uncoated counterparts. Characterization studies have demonstrated that MmNPs preserve key membrane surface markers, including macrophage-specific antigens, thereby conferring improved targeting efficiency [134]. The biomimetic membrane serves as a protective barrier, attenuating the systemic toxicity of encapsulated therapeutic agents, particularly those with high intrinsic cytotoxicity, while enhancing localized therapeutic efficacy at disease sites.
Despite recent progress, macrophage membrane-camouflaged nanoparticles face notable challenges, including variability due to non-standardized extraction protocols that impede clinical translation. (ⅰ) Safety issues caused by the introduction of foreign objects and solutions: The processes of membrane isolation, hybridization [135], and surface modification carry the risk of introducing toxic impurities or compromising membrane integrity. The physicochemical instability of payloads such as photosensitizers [136], sonosensitizers, or metallic agents may further disrupt the native properties of the membrane during formulation [137,138]. Developing standardized extraction methods is essential to address this limitation. (ⅱ) Potential rejection reactions and solutions: Potential rejection of allogenic macrophages is a critical challenge, although autologous macrophage extraction, while still in its early stages, could reduce this risk. These challenges underscore the necessity for rigorous preclinical toxicological evaluation and rational formulation strategies to ensure both functional fidelity and biosafety.
To further address the clinical translational challenges mentioned above, we elaborate on more detailed solutions in four sections. (ⅰ) Control of cell origin: Poor batch-to-batch consistency and membrane preparation reproducibility, instability of membrane components, and limitations in manufacturing scalability remain major barriers to the clinical translation of MmNPs. The incorporation of automated membrane isolation technologies holds promise for minimizing batch-to-batch variability, while the application of advanced imaging modalities may enable real-time monitoring of drug biodistribution and therapeutic kinetics. Although commonly adopted techniques such as mechanical extrusion and ultrasonication have enabled membrane coating, they often result in variable encapsulation efficiency and compromise the functional integrity of the membrane. To address these limitations, automated membrane extraction and coating systems are under active development, while novel continues production method such as instruments for microfluidic formulation has been designed [139], offering the potential to improve industrial scalability, standardize production, and minimize operator-dependent variability. (ⅱ) Control of the membrane-coating process: Emerging research has shifted toward the development of "low-interference" functionalization strategies, such as lipid bilayer insertion, biomimetic peptide conjugation, and lipid-anchor integration, that aim to enhance targeting efficiency without compromising the structural or functional integrity of the macrophage membrane [140]. (ⅲ) Resolution of immunogenicity issues: Although macrophage membranes possess certain immunomodulatory properties, xenogeneic membranes may trigger host immune responses. Therefore, human-derived cells should be considered. Furthermore, conducting systematic toxicological and immunogenicity evaluations in both rodent and non-rodent models, focusing on antibody production, lymphocyte activation, and hypersensitivity reactions is quietly necessary [141]. (ⅳ) GMP-based quality control: The establishment of standardized biological sources, harmonized cell culture protocols, and stringent quality control parameters is increasingly regarded as essential for ensuring the reproducibility, scalability, and clinical viability of macrophage membrane-camouflaged nanotherapeutics. The quality control parameters should include vesicle/particle size, size distribution and other physicochemical properties.
Beyond these technical challenges, the integration of MmNPs with emerging technologies holds considerable promise for advancing clinical application. Interestingly, the membrane-level mimicry observed in MmNPs is conceptually aligned with the emerging paradigm of engineered macrophage-based cell therapies. Notably, chimeric antigen receptor macrophages (CAR-Ms) represent a cellular counterpart to MM-NPs, wherein macrophages are genetically reprogrammed to express tumor-specific CARs, enabling precise recognition, M1-polarization, and phagocytosis of malignant cells. Due to the high complexity of the tumor microenvironment in solid tumors, CAR-M therapy has been considered for years inspired by CAR-T therapy [142,143]. Modifying human macrophages with specific CARs to endow them the ability of phagocyting and presenting antigen against tumors, which had been in clinical trials (RR-M01, NCT04660929 CT-0508) for years and achieved good prognoses in personalized treatments. This therapy offers nanoparticles with a lower immune rejection response, significantly addressing the issue of the immunogenicity of allogeneic macrophages. Although the cost of personalized CAR-M therapy remains a barrier, the development of a universal CAR-M platform, similar to universal CAR-T, may provide a more feasible alternative. MmNPs represent a promising immuno-biomimetic delivery system with translational potential. Their mechanistic resemblance to CAR-engineered macrophages offers a compelling rationale for integrating membrane mimicry with cellular engineering in next-generation immunotherapy strategies.
Notably, artificial intelligence (AI) is expected to play a transformative role in this field. Machine learning algorithms can facilitate the rational selection of membrane sources and functionalization strategies, expediting the transition from empirical formulation to data-driven design. Furthermore, the combination of AI with multimodal high-throughput imaging enables real-time tracking of MmNPs' biodistribution and metabolic kinetics in vivo, providing critical insights for optimizing dosing regimens and delivery schedules. In addition, AI-driven modeling of tumor immune microenvironment heterogeneity may support the rational selection of membrane surface proteins and guide tailored surface modifications. These advances collectively pave the way for more precise, efficient, and personalized development of biomimetic nanomedicine platforms.
In conclusion, this interdisciplinary technology holds significant promise for drug delivery and therapeutic applications. In oncology, they enable targeted chemotherapy and immunomodulation within the tumor microenvironment. In infectious diseases, they can act as pathogen decoys or targeted antibiotic carriers, enhancing efficacy while minimizing off-target effects. Furthermore, their ability to home to inflamed tissues opens opportunities in autoimmune and cardiovascular diseases. The comprehensive application of MmNPs provides hope for the treatment of many diseases through target drug delivery that were previously incurable.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The authors acknowledge financial support from National Natural Science Foundation of China (Nos. 82405025, U21A20408, 82373710), Natural Science Foundation of Jiangsu Province (Nos. BK20230462, BK20240749), Natural Science Foundation of the Jiangsu Higher Education Institutions (Nos. 23KJB360010, 24KJB360017), Jiangsu Provincial Double-Innovation Doctor Program (Nos. JSSCBS20230146, JSSCBS0213), and the High-Level Key Discipline Construction Project of National Administration of Traditional Chinese Medicine (No. ZYYZDXK-2023083).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.111610.
Figure 1
Schematic illustration of advantages and surface markers of MmNPs. MmNPs demonstrate improved biocompatibility, immune escape, prolonged blood circulation and site-specific homing. Main surface receptors on the macrophage surface include CCR2, interferon-γ receptor (IFN-γR), FcγR and TLR. These surface markers contribute to the versatility and functionality of MmNPs in therapeutic applications.
Figure 2
Schematic overview of macrophage polarization and the preparation of MmNPs. Macrophages are polarized into two major phenotypes, each characterized by distinct surface markers, cytokine profiles, and chemokine expression. Following polarization, macrophage membranes are isolated and used to camouflage pre-formed nanoparticles loaded with functional drug payloads. This process yields biomimetic nanoparticles (MmNPs), typically assembled via extrusion methods.
Figure 3
Comparative advantages and limitations of cell membranes for targeted drug delivery applications. Membranes from macrophages, dendritic cells, red blood cells, platelets and lymphocytes were discussed.
Figure 4
Overview of three categories of MmNPs for targeted drug delivery. This figure summarizes the three major categories of MmNPs, highlighting their design principles, mechanisms of action, and representative applications.
Figure 5
Functional components enabling targeted delivery in MmNPs. This figure illustrates the design principles of three representative MmNP-based targeted delivery strategies, each driven by distinct targeting modalities. (ⅰ) Intrinsic properties-based MmNPs are achieved through native macrophage membrane receptors, such as integrins and chemokine receptors, which enable biomimetic recognition of pathological sites. (ⅱ) Active targeting involves surface modification with exogenous ligands (e.g., antibodies, peptides) to enhance selective binding to disease-specific markers. (ⅲ) Stimuli-responsive targeting incorporates environment-sensitive components that respond to internal or external triggers, such as magnetic fields, US, pH, or light, to enhance site-specific drug release.

 DownLoad:
DownLoad:

 下载:
下载:






 下载:
下载:
