Department of Pharmaceutics, Wuya College of Innovation, Shenyang Pharmaceutical University, Shenyang 110016, China
b.
Joint International Research Laboratory of Intelligent Drug Delivery Systems of Ministry of Education, Shenyang Pharmaceutical University, Shenyang 110016, China
c.
Department of Pharmacy, The First Affiliated Hospital of Jinzhou Medical University, Jinzhou 121000, China
Received Date:
26 March 2025 Accepted Date:
18 July 2025 Revised Date:
16 July 2025 Available Online:
15 January 2026
Abstract:
Despite demonstrating significant anti-tumor potential as an artemisinin derivative, artesunate faces delivery efficiency challenges due to low water solubility and insufficient targeting specificity. To improve the delivery efficiency, we engineered three artesunate (ART) derivatives, AC15-L (linear), AC15-B (branched), and AC15-C (cyclic) with distinct aliphatic chain architectures. Unexpectedly, we observed that AC15-C exhibited superior cytotoxicity against 4T1 breast cancer cells, and had the highest binding affinity for Lon protease 1 (LONP1) (−72.6 kcal/mol). Subsequently, disulfide bond-containing lipid-PEG (DSPE-SS-PEG2K) modified chain architecture-engineered ART derivatives nanoassemblies (NAs) were developed to mitigate solubility-related limitations while enhancing targeting precision. Molecular docking and experimental validation demonstrated that ART derivatives inhibited LONP1 through hydrophobic interactions while preserved Fe2+-mediated Fenton-like reaction activity. In vitro and in vivo evaluations demonstrated that AC15-C NAs outperformed free ART and other NAs, suppressing 4T1 tumor growth via dual action: LONP1-directed mitochondrial proteostasis collapse and reactive oxygen species (ROS) amplification through Fe2+-ART interactions. This study elucidated a novel anti-tumor mechanism of ART through the rational design of derivatives with spatially configured aliphatic chains, and developed reduction-responsive NAs to provide an advanced delivery strategy.
Cancer ranks as the second leading cause of death globally, with treatment efforts are persistently hindered by chemoresistance, tumor heterogeneity, and systemic toxicity [1,2]. Conventional chemotherapeutics, such as platinum-based agents and taxanes, indiscriminately target rapidly dividing cells, leading to adverse effects like myelosuppression and neurotoxicity, while tumor microenvironments often develop oxidative stress tolerance to evade therapeutic insults [3-8]. Recent advances highlight reactive oxygen species (ROS)-based pro-oxidative therapy as a promising strategy for selectively eliminating cancer cells with intrinsically elevated ROS levels [9-12]. Artemisinin, a sesquiterpene lactone derived from Artemisia annua, and its semi-synthetic derivative artesunate (ART), harbors a unique endoperoxide bridge that reacts with tumor-associated Fe2+via Fenton-like reactions, generating cytotoxic ROS to induce oxidative damage [13-15]. Despite the broad-spectrum anti-tumor activity of ART against breast, ovarian, and colorectal cancers, its clinical utility remains constrained by short half-life and ill-defined molecular targets [16]. The historical studies have overemphasized ROS-mediated cytotoxicity while neglecting precise protein engagement mechanisms. Addressing these limitations is critical to unlocking the full anticancer potential of ART.
Mitochondrial proteostasis, a hallmark of cancer metabolic reprogramming, is governed by the ATP-dependent Lon protease 1 (LONP1) [17]. This mitochondrial matrix protease selectively degrades oxidatively damaged respiratory chain complex proteins to prevent cytotoxic aggregation while maintaining genomic stability by clearing misfolded proteins [18]. Inhibition of LONP1 disrupts this critical quality control mechanism, leading to the accumulation of misfolded and oxidatively damaged proteins within mitochondria, ultimately causing mitochondrial dysfunction and an increase in ROS levels [19]. LONP1 overexpression is strongly associated with poor prognosis and chemoresistance in breast, ovarian, and pancreatic cancers [17]. Previous studies have revealed that ART directly interacts with proteolytic domain of LONP1, accelerating degradation of its substrate CYP11A1 to suppress ovarian androgen synthesis, thereby ameliorating polycystic ovarian syndrome [20]. However, whether LONP1 mediates cytotoxicity of ART in non-hormone-dependent cancers (e.g., triple-negative breast cancer) and how the spatial configuration of ART dictates its binding affinity to LONP1 remains unresolved. This knowledge gap impedes rational drug design and demands systematic exploration of structure-target engagement relationship of ART.
To elucidate how configuration modulates LONP1 targeting, we pioneered the chain architecture-driven design strategy, departing from conventional electronic effect-dominated paradigms. Three ART derivatives were synthesized by conjugating a C15 aliphatic side chain in distinct spatial configurations: linear (AC15-L), branched (AC15-B), and cyclic (AC15-C). In 4T1 breast cancer cells, AC15-C demonstrated reduction in half maximal inhibitory concentration (IC50) compared to ART, which highlight the critical role of chain rigidity. Intriguingly, Fe2+ chelation assays revealed equivalent iron consumption across derivatives, refuting the traditional hypothesis of Fenton-like reaction efficiency as the primary determinant of efficacy. Molecular docking unveiled mechanistic insights that the AC15-C configuration complemented LONP1's hydrophobic active pocket forming stable interactions (affinity = −72.6 kcal/mol), whereas the AC15-L and AC15-B had weak binding affinity. Subsequent in vitro validation via immunofluorescence (IF) and Western blotting confirmed potent LONP1 inhibition, which directly correlated with its ROS amplification capability. These findings redefine ART derivatives' structure-activity relationships, establishing spatial topology as a pivotal driver of target engagement and therapeutic efficacy.
Despite the superior LONP1 affinity of AC15-C, its poor solubility and systemic exposure-related risks necessitate advanced delivery solutions. Conventional nanocarriers (e.g., liposomes, polymeric micelles) improve solubility but introduce synthetic excipient-associated biocompatibility concerns [21-25]. To overcome these limitations, three chain architecture-engineered ART derivatives nanoassemblies (NAs) without the assistance of any excipients was fabricated via one-step nanoprecipitation method [26-31]. Surface modification with reduction-reactive disulfide bond-containing lipid-PEG (DSPE-SS-PEG2K) endowed excellent stability, prolonged circulation and tumor-selective drug release triggered by the elevated glutathione (GSH) levels in the tumor microenvironment [32,33]. DSPE-SS-PEG2K shielding minimized drug release in normal tissues, while elevated intratumoral GSH triggered cleavage of disulfide bonds, enabling tumor-specific drug release [34-37]. In 4T1 tumor-bearing mice, AC15-C NAs achieved tumor suppression, which significantly surpassing free ART and other derivative NAs. Mechanistically, in vitro and in vivo demonstrated that NAs preserved the bioactivity of AC15-C, downregulating LONP1 expression and amplifying Fe2+-ROS cascades. This study elucidated a novel anti-tumor mechanism of ART through the rational design of derivatives with spatially configured aliphatic chains, and developed reduction-responsive NAs to provide an advanced delivery strategy (Fig. 1).
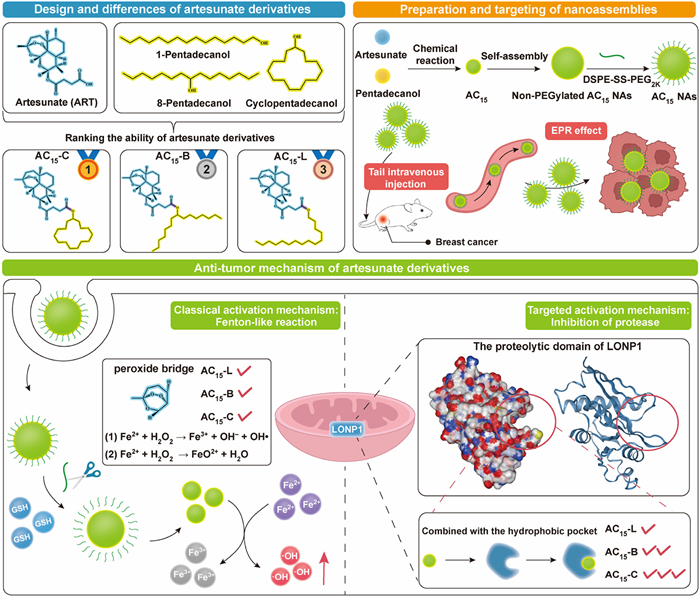
Figure 1
Figure 1.
Schematic diagram of ART derivatives for the treatment of breast cancer. First, ART derivatives were synthesized from ART with pentadecanols of three different configurations. All three derivatives could self-assemble into stable nanostructures. Then, AC15 NAs were constructed using DSPE-SS-PEG2K as PEG modifier. After endocytosis of AC15 NAs into tumor cells, AC15 can be rapidly decomposed and released to exert anti-tumor mechanisms: Fenton-like reaction and protease inhibition.
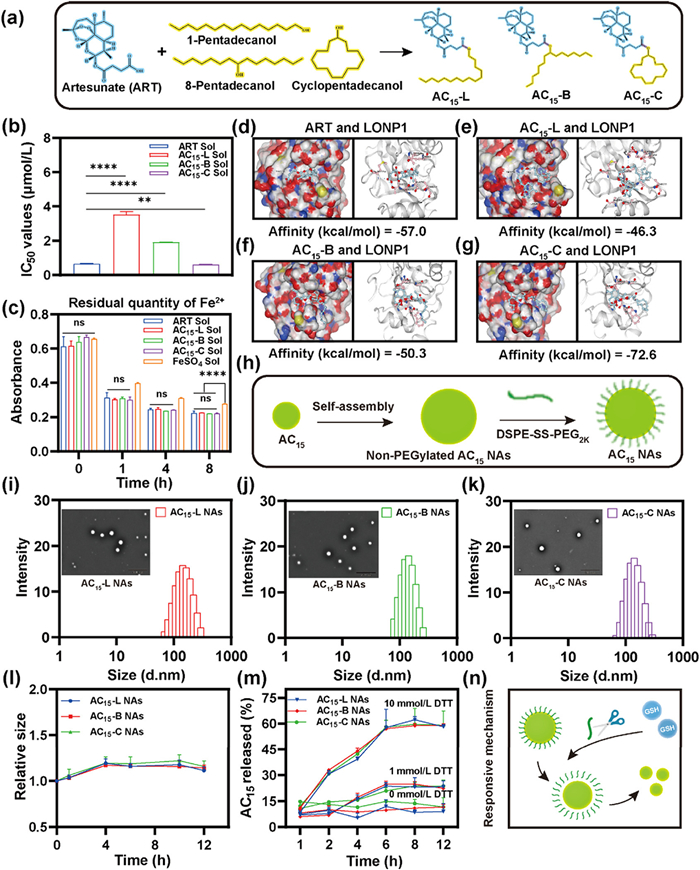
Three novel ART-fatty alcohol derivatives (AC15) were prepared via a straightforward one-step esterification between ART and C15 aliphatic side chains possessing identical carbon chain length (C15) but distinct configurations, yielding three structural variants: linear (AC15-L), branched (AC15-B), and cyclic (AC15-C) derivatives (Fig. 2a). The successful synthesis of AC15 were confirmed by mass spectrometry (MS) and proton nuclear magnetic resonance (1H NMR) (Figs. S1−S6 in Supporting information). Subsequently, the cytotoxicity of AC15 were systematically evaluated in 4T1 cells. Surprisingly, a clear hierarchy emerged in the results: AC15-C Sol > ART Sol > AC15-B Sol > AC15-L Sol (Fig. 2b and Fig. S7 in Supporting information). The canonical anti-tumor mechanism of ART involves peroxide bridge-mediated Fenton-like reactions with intracellular Fe2+, resulting in the generation of cytotoxic ROS [38]. To assess whether the structural modifications impacted ROS generation, we conducted an Fe2+ chelation experiment. After 8 h incubation, the Fe2+ consumption of AC15 was comparable to that of ART (Fig. 2c and Fig. S8 in Supporting information), suggesting that the modifications to the aliphatic acid chains did not interfere with the peroxide-bridge reactivity of AC15. This finding underscores the existence of a potentially novel anti-tumor mechanism of ART.
Figure 2
Figure 2.
Rational design, mechanism, and synthesis of architecture-engineered ART derivatives and NAs. (a) The structure of three aliphatic side chain modified ART derivatives with the same chain length and distinct configurations after synthesis. (b) IC50 values of ART and its derivatives in 4T1 cells. (c) Residual Fe2+ of ART and its derivatives during the reaction. (d–g) Simulated binding diagram and binding scores of ART and its derivatives with LONP1. (h) AC15 NAs self-assembly processes. (i–k) Particle sizes distribution profiles and transmission electron microscope (TEM) images of AC15-L NAs, AC15-B NAs and AC15-C NAs. Scale bar: 500 nm. (l) The colloidal stability of AC15 NAs cultured in PBS containing 10% FBS (pH 7.4) (n = 3). (m) In vitro drug release patterns of NAs incubated in release media with 0, 1 and 10 mmol/L DTT, respectively (n = 3). (n) The reduction-responsive mechanism of the disulfide bond. Data: mean ± SD (n = 3). ns: no significance. P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Given the emerging evidence regarding the role of artemisinin-targeted mitochondrial proteinases, we hypothesized that LONP1 proteinase could be a key factor responsible for the observed variations in cytotoxicity. Molecular docking simulations of the compounds with the LONP1 proteolytic domain revealed distinct structure-activity relationships: ART exhibited moderate binding affinity (docking score: −57.0 kcal/mol), followed by AC15-B (−50.3 kcal/mol) and AC15-L (−46.3 kcal/mol) (Figs. 2d–f). Notably, AC15-C demonstrated the highest binding affinity with a significant docking score of −72.6 kcal/mol (Fig. 2g). This finding further confirmed our conjecture that structural modification of ART by cyclic fat modification (AC15-C) enhanced its cytotoxicity by enhancing its binding affinity to LONP1.
Non-PEGylated AC15 NAs were fabricated via one step nanoprecipitation method (Tables S1 and S2 in Supporting information). In order to improve the stability and facilitate tumor-specific drug release, DSPE-SS-PEG2K (20%, w/w) were used as a surface modifier to prepare reduction-responsive NAs (AC15 NAs) (Fig. 2h and Table S3 in Supporting information). As shown in Figs. 2i−k and Fig. S9 (Supporting information), AC15 NAs (AC15-L NAs, AC15-B NAs, and AC15-C NAs) showed very similar morphology and particle size (~130 nm), with negative zeta potential of −30 ~ −40 mV. AC15 NAs showed good stability with negligibly changed in particle size at 4 ℃ for 7 days (Fig. S10 in Supporting information). Moreover, there were almost no significant particle size changes observed in AC15 NAs after incubation with phosphate buffered saline (PBS) (pH 7.4) and PBS (pH 7.4) containing 10% fetal bovine serum (FBS) for 12 h (Fig. 2l and Fig. S11 in Supporting information). These results suggested the important role of PEGylation decoration on the colloidal stability of NAs.
The molecular self-assembly mechanism of non-PEGylated AC15 NAs was investigated through computational molecular docking and nanoassembly disruption approaches (Fig. S12 in Supporting information). Two intermolecular interaction breakers-sodium dodecyl sulfate (SDS), and urea-were employed to disrupt NAs through selective interference with hydrophobic interactions, and hydrogen bonding, respectively. Notably, SDS-induced dramatic particle enlargement demonstrated the predominant contribution of hydrophobic forces to assembly stability (Fig. S13 in Supporting information). Therefore, we calculated the oil-water partition coefficient (logP) of ART derivatives by ALOGPS to judge their hydrophilicity and hydrophobicity. As shown in Fig. S14 (Supporting information), the logP values of AC15-L, AC15-B, and AC15-C, were 7.72, 7.55 and 7.15. In contrast, there was moderate size increases after co-incubation with urea, indicating that hydrogen bonding served as secondary factor contributing to molecular nanoassembly (Fig. S13).
On-demand disintegration of NAs at the tumor sites is crucial for achieving desirable anticancer outcomes. As previously mentioned, DSPE-SS-PEG2K was utilized to construct AC15 NAs. To evaluate the reduction-responsive disassembly property of NAs triggered by reductive stimuli, the changes in particle size of AC15 NAs in the presence of dithiothreitol (DTT, a common GSH simulant) was explored. The particle size of NAs underwent a significant alteration in the presence of 10 mmol/L DTT when compared that of 0 mmol/L DTT (Fig. S15 in Supporting information). As shown in Fig. 2m, AC15 NAs exhibited reduction-responsive release behavior strongly correlated with DTT concentration. Under the high reductive environment with 10 mmol/L DTT, approximately 60% of the AC15 payload was released within 8 h. In contrast, less release (~20%) occurred at a lower DTT concentration of 1 mmol/L within 12 h, while minimal release (~10%) was detected in blank culture medium containing 0 mmol/L DTT within 12 h. This pronounced concentration-dependent release profile highlights the precise control over payload release offered by the reduction-responsive AC15 NAs (Fig. 2n). These results suggested that DSPE-SS-PEG2K allowed reduction-responsive disassembly, which would contribute to low toxicity to normal tissues.
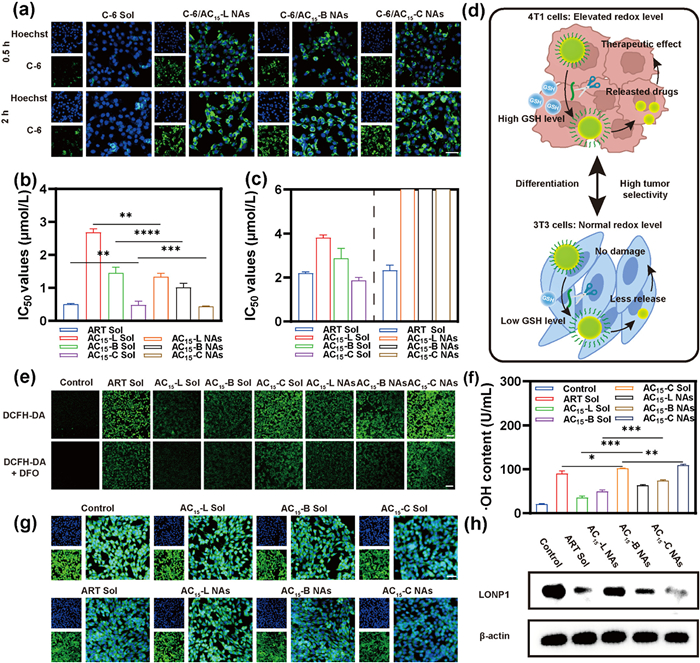
Given that effective cellular uptake is crucial for NAs to exert anti-tumor effects, we further explored the cellular uptake efficiency of NAs in 4T1 cells. Firstly, coumarin-6 (C-6)-labeled NAs (C-6/AC15 NAs) were prepared to study the cellular internalization characteristics. As shown in Fig. 3a, the fluorescence intensity of C-6 within 4T1 tumor cells treated with C-6/AC15 NAs was significantly higher compared to C-6 Sol at both 0.5 h and 2 h. Furthermore, the cellular uptake of NAs exhibited a time-dependent manner.
Figure 3
Figure 3.In vitro anti-tumor activity and mechanism. (a) The CLSM images of cellular uptake in 4T1 cells at 0.5 h and 2 h, respectively. Scale bar: 50 µm. (b) IC50 values of cytotoxicity against 4T1 cells with ART Sol, AC15 Sol, and AC15 NAs. (c) IC50 values of cytotoxicity against 3T3 cells with ART Sol, AC15 Sol, and AC15 NAs. (d) Different reduction states lead to specific activation of AC15 NAs in tumor cells with high selectivity. (e) Intracellular levels of ROS in 4T1 cells after treatments. Scale bar: 100 µm. (f) Intracellular levels of ·OH in 4T1 cells after treatments. (g) The expression and localization of LONP1 in 4T1 cells. Scale bar: 100 µm. (h) Western blot analysis results of intracellular LONP1 in 4T1 cells after treatments. Data: mean ± SD (n = 3). P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
In addition, we further assessed the in vitro anti-tumor activity of NAs using MTT assay. AC15-C NAs exhibited the most potent anti-tumor activity against 4T1 cells compared to AC15-C Sol (Fig. 3b and Fig. S16 in Supporting information). This superior efficacy should be attributed to its favorable nanoassembly properties, efficient cellular uptake and tumor-specific disassembly characteristics. In the previous exploration experiments, the cytotoxicity sequence of the solution was AC15-C Sol > ART Sol > AC15-B Sol > AC15-L Sol (Fig. 2b and Fig. S7 in Supporting information). As depicted in Fig. 3b, the cytotoxicity trend of NAs was similar to that of Sol, in order of AC15-C NAs > ART Sol > AC15-B NAs > AC15-L NAs. The observed differences in cytotoxicity among NAs were attributable to the distinct cytotoxic activities of AC15 in tumor cells, given that all NAs exhibited comparable cellular uptake and on-demand GSH-responsive drug disintegration profile. In addition, NAs demonstrated negligible cytotoxicity on 3T3 cells compared to ART Sol and AC15 Sol, indicating excellent biocompatibility (Fig. 3c and Fig. S17 in Supporting information). This once again demonstrated that AC15 NAs could be specifically activated in the tumor microenvironment (Fig. 3d), promising safer anti-tumor therapies.
Subsequently, we investigated multiple anti-tumor mechanisms of ART and its derivatives. As established, structural modifications to the aliphatic side chains preserved the reactivity of the peroxide bridge, enabling sustained intracellular Fe2+-dependent Fenton-like reactions. Since these reactions generated ROS including ·OH, we detected ·OH and total ROS levels using hydroxyl radicals (·OH) assay kit and DCFH-DA probe, respectively. As shown in Figs. 3e and f, ART Sol, AC15 Sol, and AC15 NAs all induced ·OH generation and ROS accumulation. Notably, AC15 NAs exhibited substantially higher ·OH and ROS levels than their solution counterparts (AC15 Sol) post-treatment. This enhancement was attributable to the superior cellular internalization efficiency of NAs, facilitating increased intracellular accumulation of AC15 and consequently amplifying ·OH and ROS generation. Critically, co-treatment with the iron chelator deferoxamine (DFO) attenuated ROS fluorescence intensity, confirming Fe2+-mediated Fenton-like reaction as an important anti-tumor mechanism (Fig. 3e). Notably, the differential ROS and ·OH accumulation levels across AC15-L, AC15-B, and AC15-C (Figs. 3e and f) aligned precisely with their respective LONP1 binding affinities observed earlier (Figs. 2e–g). Collectively, these findings imply the existence of an additional ROS-generating anti-tumor mechanism beyond Fenton-like reaction. As previously hypothesized, LONP1 likely serves as a key mediator of the observed cytotoxicity variations.
To investigate this, we assessed the expression of LONP1 in 4T1 cells using IF staining. As shown in Fig. 3g and Fig. S18 (Supporting information), compared with the control group, the expression level of LONP1 decreased accordingly with the increase of cytotoxic potency. This finding suggested that structural modifications of ART's aliphatic side chains alter its binding affinity to LONP1, thereby inducing varying degrees of LONP1 inhibition. Specifically, the cyclic structure of AC15-C exhibited optimal binding within the proteolytic domain of LONP1 via stable hydrophobic interactions, resulting in the most potent inhibition of the protein. Subsequently, the expression level of LONP1 in 4T1 cells was detected by Western blot and the results were consistent with those obtained by IF staining (Fig. 3h).
The in vivo drug delivery efficiency of NAs, particularly pharmacokinetics and biodistribution, critically determines their therapeutic efficacy. As hypothesized, AC15 NAs exhibited enhanced colloidal stability due to favorable colloidal stability through PEGylation, which significantly improved their systemic delivery performance. To validate this, pharmacokinetic profiling was conducted in Sprague-Dawley rats following intravenous administration of DiR-labeled AC15 NAs or free DiR Sol. As shown in Fig. 4a and Table S4 (Supporting information), DiR Sol demonstrated rapid systemic clearance. In contrast, AC15 NAs showed longer circulation time in the blood and higher area under the concentration–time curve (AUC) than that of DiR Sol. These findings confirmed that PEGylation optimizes NAs design, extending plasma residence to enhance EPR effect-mediated tumor accumulation. This study was approved by the Animal Experiment Ethics Committee of Shenyang Pharmaceutical University.
Figure 4
Figure 4.In vivo delivery and antitumor effects. (a) Pharmacokinetic behavior of AC15 NAs in vivo (n = 5). (b) Living fluorescence images of 4T1 tumor-bearing BALB/c mice after intravenous injection of DiR Sol and DiR/AC15 NAs (DiR equivalent dose 2 mg/kg) at different time intervals. (c) Ex vivo fluorescence images of major organs and tumors at the brightest point time. (d) Quantification of the average fluorescence intensity of (c) (n = 3). (e) Schematic illustration of treatment schedule. (f) The tumor growth profiles of 4T1 tumor-bearing BALB/c mice after different treatments (n = 5). (g) Tumor burden after treatment (n = 5). (h) Image of tumors after the last treatment. (i) IHC analysis and DHE staining sections of tumor after different treatments. Scale bar: 20 µm. Data: mean ± SD. P < 0.05, ***P < 0.001, ****P < 0.0001.
In order to achieve real-time evaluation on the biodistribution of NAs by fluorescence imaging, DiR was utilized to label the NAs (DiR/AC15 NAs). As shown in Fig. 4b, DiR Sol displayed weak fluorescence intensity in tumors within 60 h, attributed to its suboptimal pharmacokinetic behavior. In contrast, NAs exhibited significantly higher fluorescence signals in tumors compared to DiR Sol under similar conditions. Furthermore, ex vivo fluorescence imaging of DiR Sol and NAs in tumors and major organs was conducted at the time points of peak fluorescence signal. As showed in Figs. 4c and d, NAs demonstrated enhanced tumor accumulation compared to DiR Sol at 24 h post-injection. These results underscored that the precise fabrication of PEGylated NAs significantly optimized pharmacokinetics and biodistribution, thereby undoubtedly correlated with enhanced anti-tumor efficacy.
The excellent in vitro and in vivo drug delivery properties of AC15 NAs make them a promising nanomedicine for cancer therapy. We then evaluated the anti-tumor activity of the NAs in 4T1 breast cancer tumor-bearing mice (Fig. 4e). As showed in Figs. 4f–h, AC15 NAs showed obvious therapeutic advantages compared with ART Sol and had a strong inhibitory effect on tumor growth. This is because the NAs had better pharmacokinetic behavior and more tumor accumulation compared to the Sol, which had been successfully demonstrated in previous experiments. Furthermore, the results of hematoxylin and eosin staining (H & E), terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) and Ki67 staining were consistent with the above-mentioned tumor inhibition results, further verifying the good tumor growth inhibition ability of AC15-C NAs (Figs. S19–S21 in Supporting information). Building upon the potent in vivo anti-tumor efficacy of AC15 NAs, we investigated relevant biomarkers in tumor tissues to elucidate the underlying mechanisms. Immunohistochemical (IHC) analysis confirmed reduced LONP1 expression in vivo (Fig. 4i), consistent with our in vitro IF findings and Western blot analysis results (Figs. 3g and h). This reaffirmed that AC15 inhibited LONP1 activity through binding to its proteolytic domain. Dysregulation of LONP1 may perturb downstream effectors. Given the reported negative correlation between LONP1 and TDP-43, where LONP1 upregulation enhanced TDP-43 degradation, we concurrently assessed TDP-43 aggregation via IHC [39]. The results demonstrated that ART derivatives facilitated the formation of cytoplasmic TDP-43 aggregates through suppression of LONP1 (Fig. S22 in Supporting information).
As validated extensively in vitro, both Fenton-like reactions and LONP1 inhibition contributed to oxidative stress. Accordingly, we evaluated in vivo ROS levels in tumor sections using dihydroethidium (DHE) staining. Compared to the control group, all treatment groups exhibited elevated ROS (Fig. 4i). Furthermore, a preliminary assessment of therapeutic safety was conducted. As shown in Figs. S23–S25 (Supporting information), multiple treatments did not lead to significant abnormalities in hepatorenal function parameters, organ sections observed by H & E staining, or changes in body weight. Taken together, these results suggested that AC15-C NAs could be used as the highly efficient and safe NAs for tumor therapy.
This study established an innovative chain architecture engineering strategy that elucidated a novel anti-tumor mechanism of ART through the rational design of derivatives with spatially configured aliphatic chains, and provided a new delivery approach via excipient-free NAs. Through spatial modulation of the C15 aliphatic chain, the cyclic derivative AC15-C demonstrated superior binding affinity to LONP1 (−72.6 kcal/mol), while linear and branched counterparts exhibited suboptimal affinity. Moreover, the in vitro and in vivo efficacy of AC15-C correlated with configurational accessibility to LONP1 rather than iron consumption, redefining ART's structure-activity paradigm toward spatial topology-driven target specificity. Furthermore, these excipient-free NAs with reduction-reactive PEGylation achieved tumor-selective release, prolonged circulation, and enhanced tumor accumulation, driving potent 4T1 tumor suppression through dual mechanisms: LONP1 inhibition-mediated mitochondrial dysfunction and Fenton-like reaction. By bridging molecular optimization with nano delivery, this work deciphers a new antitumor mechanism of ART while offering a delivery strategy for enhanced anti-tumor efficacy.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was financially supported by the Liaoning Revitalization Talents Program (No. XLYC2403107), the Excellent Youth Science Foundation of Liaoning Province (No. 2024JH3/10200046) and the Basic Scientific Research Project of Liaoning Provincial Department of Education (No. LJ212410163015).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.111609.
[1]
Nat. Cancer 1 (2020) 1–2.
[2]
F. Bray, M. Laversanne, H. Sung, et al., CA Cancer J. Clin. 74 (2024) 229–263.
S. Shan, Z. Liu, S. Wang, et al., Ecotoxicol. Environ. Saf. 264 (2023) 115409. doi: 10.1016/j.ecoenv.2023.115409
Figure 1
Schematic diagram of ART derivatives for the treatment of breast cancer. First, ART derivatives were synthesized from ART with pentadecanols of three different configurations. All three derivatives could self-assemble into stable nanostructures. Then, AC15 NAs were constructed using DSPE-SS-PEG2K as PEG modifier. After endocytosis of AC15 NAs into tumor cells, AC15 can be rapidly decomposed and released to exert anti-tumor mechanisms: Fenton-like reaction and protease inhibition.
Figure 2
Rational design, mechanism, and synthesis of architecture-engineered ART derivatives and NAs. (a) The structure of three aliphatic side chain modified ART derivatives with the same chain length and distinct configurations after synthesis. (b) IC50 values of ART and its derivatives in 4T1 cells. (c) Residual Fe2+ of ART and its derivatives during the reaction. (d–g) Simulated binding diagram and binding scores of ART and its derivatives with LONP1. (h) AC15 NAs self-assembly processes. (i–k) Particle sizes distribution profiles and transmission electron microscope (TEM) images of AC15-L NAs, AC15-B NAs and AC15-C NAs. Scale bar: 500 nm. (l) The colloidal stability of AC15 NAs cultured in PBS containing 10% FBS (pH 7.4) (n = 3). (m) In vitro drug release patterns of NAs incubated in release media with 0, 1 and 10 mmol/L DTT, respectively (n = 3). (n) The reduction-responsive mechanism of the disulfide bond. Data: mean ± SD (n = 3). ns: no significance. P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Figure 3In vitro anti-tumor activity and mechanism. (a) The CLSM images of cellular uptake in 4T1 cells at 0.5 h and 2 h, respectively. Scale bar: 50 µm. (b) IC50 values of cytotoxicity against 4T1 cells with ART Sol, AC15 Sol, and AC15 NAs. (c) IC50 values of cytotoxicity against 3T3 cells with ART Sol, AC15 Sol, and AC15 NAs. (d) Different reduction states lead to specific activation of AC15 NAs in tumor cells with high selectivity. (e) Intracellular levels of ROS in 4T1 cells after treatments. Scale bar: 100 µm. (f) Intracellular levels of ·OH in 4T1 cells after treatments. (g) The expression and localization of LONP1 in 4T1 cells. Scale bar: 100 µm. (h) Western blot analysis results of intracellular LONP1 in 4T1 cells after treatments. Data: mean ± SD (n = 3). P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Figure 4In vivo delivery and antitumor effects. (a) Pharmacokinetic behavior of AC15 NAs in vivo (n = 5). (b) Living fluorescence images of 4T1 tumor-bearing BALB/c mice after intravenous injection of DiR Sol and DiR/AC15 NAs (DiR equivalent dose 2 mg/kg) at different time intervals. (c) Ex vivo fluorescence images of major organs and tumors at the brightest point time. (d) Quantification of the average fluorescence intensity of (c) (n = 3). (e) Schematic illustration of treatment schedule. (f) The tumor growth profiles of 4T1 tumor-bearing BALB/c mice after different treatments (n = 5). (g) Tumor burden after treatment (n = 5). (h) Image of tumors after the last treatment. (i) IHC analysis and DHE staining sections of tumor after different treatments. Scale bar: 20 µm. Data: mean ± SD. P < 0.05, ***P < 0.001, ****P < 0.0001.

 DownLoad:
DownLoad:


 下载:
下载:





 下载:
下载:
