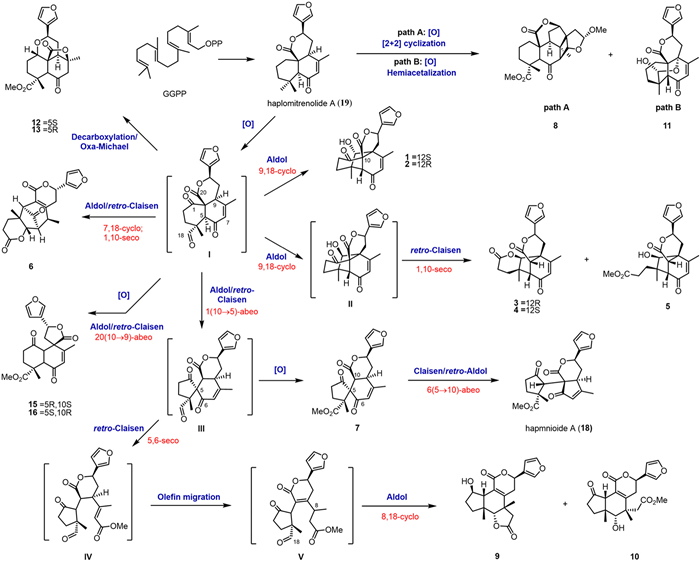
Scheme 1.
Plausible biogenetic pathways for compounds 1–13, 15, 16, 18, 19.
Haploides A–G: Diterpenoids featuring four new carbon skeletons via non-carbocation-driven skeletal diversification
Ming-Zhu Zhu , Ze-Jun Xu , Jiao-Zhen Zhang , Li-Zhi Shi , Zong-Xu Gao , Yi Li , Shuang-Zhi Yuan , Yan-Rong Ma , Ai-Xia Cheng , Hong-Xiang Lou

Terpenoid natural products, characterized by stereochemically complex architectures, have long served as inspiration sources for therapeutic agent development [1,2]. Among diterpenoid derivatives, labdanes represent a structurally unique diterpenoid subclass encompassing pharmaceutically significant metabolites, including andrographolide, forskolin, and chlorolissoclimide. Labdane-type diterpenoids demonstrate remarkable structural plasticity through diverse skeletal rearrangements (Fig. S1 in Supporting information) [3,4]. Although carbocation-driven skeletal reorganizations represent well-characterized biochemical transformations in terpenoid biosynthesis [5–8], diversity-oriented transformations mediated by aldolase-type cascade mechanisms constitute an emerging area of research in natural product biogenesis, with limited characterized examples reported to date [9,10].
As ancient representatives of the embryophytic lineage, liverworts occupy a phylogenetically pivotal position as the earliest-diverging land plants. This taxonomically rich division, consisting of over 23,000 taxonomically validated species globally, represents a prolific source of structurally diverse specialized metabolites, particularly bisbibenzyls and terpenoids [11–19]. Notably, labdane-derived Haplomitrium diterpenoids are isolated exclusively from the genus Haplomitrium and have been identified as chemical markers for this liverwort lineage [18]. Our phytochemical exploration of Haplomitrium mnioides (Lindb.) R.M.Schust., collected from Xiangling Mountain within Ya'an City, Sichuan Province, China, yielded 19 diterpenoid derivatives, including 16 novel compounds designated haploides A–P (1–16). Structural characterization revealed four unprecedented carbon skeletons: 9,18-cyclolabdanes (1 and 2), 1,10-seco-9,18-cyclolabdanes (3–5), 7,18-cyclolabdanes (6), and 1,10-seco-1,5-cyclolabdanes (7). Seven additional labdanes (8–14) featuring our previously identified scaffolds [17,18], two labdane-related diterpenoids (15 and 16), and three known analogs (17–19) (Scheme 1), were isolated (Fig. 1). Moreover, four pairs of novel diastereomers, haploides A and B (1 and 2), C and D (3 and 4), L and M (12 and 13), and O and P (15 and 16) were simultaneously discovered. A plausible aldol-type cascade biogenetic pathway was deduced for the formation of these novel skeleton labdanes. Anti-inflammatory assays revealed that haploide O (15) and haplomitrenolide C (17) significantly inhibited the secretion of proinflammatory cytokines interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α), and interleukin-6 (IL-6). Herein, we report the isolation, structural characteristics, proposed biosynthetic pathways, and anti-inflammatory activity of these diterpenoids.
Haploide A (1) was obtained in the form of colorless crystals. High-resolution electrospray ionization mass spectrometry (HRESIMS) at m/z 379.1155 (calcd. for C20H20O6Na+ 379.1152) indicated that its molecular formula is C20H20O6, indicating 11 indices of hydrogen deficiency. Nuclear magnetic resonance (NMR) spectra indicated the presence of two methyl groups [δH 2.27 (s), 1.00 (s)]; three methylenes; three methines, including two oxygenated methines [δH 5.67 (dd, J = 11.3, 6.2 Hz), δC 77.0; δH 4.29 (t, J = 2.2 Hz), δC 81.7]; three quaternary carbons; three carbon-carbon double bonds [δH 6.02 (s), 6.60 (brs), 7.50 (t, J = 1.6 Hz), 7.64 (brs); δC 128.7, 166.4, 126.2, 110.1, 144.9, 142.1], two keto carbonyls (δC 198.4, 207.5); and one ester carbonyl (δC 170.5). It was deduced that haploid A (1) contained five rings. The NMR data of 1 were similar to those of haplomitrenonolide A, except for the existence of a C−C bond between C-9 and C-18, one hydroxy group at C-18, and one keto carbonyl at C-1. These deductions were confirmed based on heteronuclear multiple bond correlations (HMBCs) from H3–19 (δH 1.00) to C-3 (δC 37.0), C-4 (δC 45.9), C-5 (δC 61.7), and C-18 (δC 81.7); from H3–17 (δH 2.27) to C-7 (δC 128.7), C-8 (δC 166.4), and C-9 (δC 57.4); from H-18 (δH 4.29) to C-3, C-5, C-8, and C-11 (δC 34.0); and from H2–3 (δH 1.98) to C-1 (δC 207.5) (Fig. 2A). Thus, the planar structure for 1 was unequivocally confirmed as having a novel 9,18-cyclolabdane skeleton.
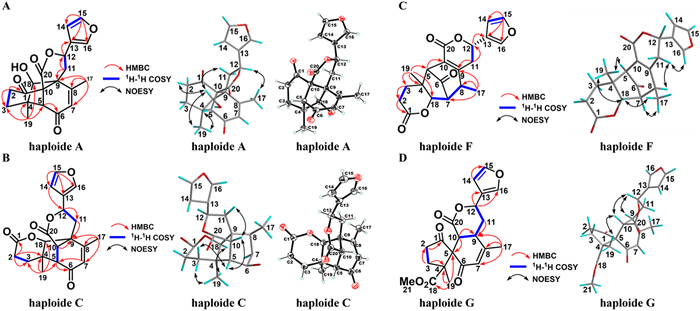
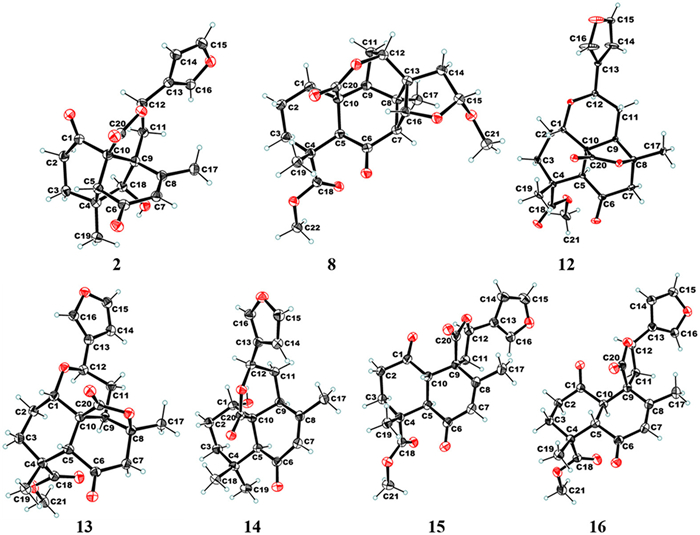
Additionally, the signals of H-18/H3–19, H3–19/H-5, H-18/H2–2, H-18/H2–11, and H3–17/H-12 in the nuclear Overhauser enhancement spectroscopy (NOESY) results indicated the cofacial nature of these protons (Fig. 2A). Ultimately, a single-crystal X-ray diffraction analysis, considering a Flack parameter of 0.00 (11) and using Cu Kα radiation (CCDC 2344630), finally established the absolute structure as 4R,5R,9S,10R,12S,18S. Haploide B (2) shared the same molecular formula and exhibited nearly identical NMR data as those of 1, except for the chiral inversion at C-12, as affirmed based on the NOESY signal of H3–19/H-5, H-18/H2–2, H-18/H2–11, electronic circular dichroism (ECD) calculations (Fig. S2 in Supporting information), and X-ray diffraction (Cu Kα radiation, CCDC 2344631) (Fig. 3). Thus, haploide B (2) was determined to be the C-12 epimer of 1, with an absolute configuration of 4R,5R,9S,10R,12R,18S.
Haploide C (3) was assigned the same molecular formula as that of haploide B (2). However, several structural differences were noted: The A-ring was cleaved between C-1 and C-10, and the lactone ring was formed between C-1 and C-18 in 3, supported by HMBCs from H2–2 (δH 2.39, 2.54) to C-1 (δC 171.1); from H2–3 (δH 1.82, 2.22) to C-1; and from H-10 (δH 3.00) to C-4 (δC 43.3), C-5 (δC 59.9), C-8 (δC 163.2), C-9 (δC 54.0), C-18 (δC 88.7), and C-20 (δC 169.3); as well as the key 1H–1H correlation spectroscopy (COSY) correlations of H-5 (δH 3.36)/H-10 and H-10/H-18 (δH 4.15) (Fig. 2B). The relative configuration was inferred based on the NOESY correlations of H3–19/H-18, H3–19/H-3α, H-3β/H-5, H-5/H-10, and H-10/H-11β. Ultimately, the absolute structure of 3 was determined based on single-crystal X-ray diffraction using Cu Kα radiation (CCDC 2344632), indicating the absolute stereochemistry to be 4R,5S,9S,10S,12R,18R. Haploide D (4) was identified as the C-12 epimer of 3 based on their similar NMR spectrum and NOESY signals of H3–19/H-18, H-10/H-12, H3–19/H-3α, H-3β/H-5, H-5/H-10, and H-10/H-11β in 4. The consistency of experimental and calculated CD curves (Fig. S2) determined the absolute configuration of 4 as 4R,5S,9S,10S,12S,18R.
Haploide E (5) had one fewer degree of unsaturation than 3, and it has a similar planar structure to 3, except for the lactone ring being cleaved between C-1 and C-18, and one carbomethoxy unit formed at C-1 in 5. These deductions were supported by HMBCs from H3–21 (δH 3.66) to C-1 (δC 174.4); and from H-18 (δH 4.26) to C-3 (δC 37.7), C-9 (δC 52.1), C-10 (δC 50.9), and C-11 (δC 30.9); along with the 1H−1H COSY signal of H-5 (δH 37.7)/H-10 (δH 37.7). The NOESY correlations of H3–19/H-18, H-5/H-10, and H-10/H-11α determined the relative configuration of 5. Comparing the experimental and calculated ECD data (Fig. S2), the absolute configuration of 5 was established as 4R,5S,9S,10S,12R,18S.
Haploide F (6) exhibited the same molecular formula as 4. The NMR spectrum of 6 was extremely similar to that of 4, except for the intramolecular cyclization between C-7 and C-18 in 6, unlike that between C-9 and C-18 in 4. Additionally, the double bond shifted from C7–C8 to C9–C10 in 6. These deductions were confirmed by HMBCs from H3–17 (δH 1.59, d, J = 7.4 Hz) to C-7 (δC 55.6), C-8 (δC 46.2), and C-9 (δC 155.7); from H2–11 (δH 2.58, 2.86) to C-9 and C-10 (δC 124.2); from H-5 (δH 3.30) to C-9 and C-10; from H-18 (δH 4.47) to C-7; and from H-7 (δH 2.95) to C-4 (δC 43.8) (Fig. 2C); as well as the 1H–1H COSY signals of H-8 (δH 3.28)/H-7 and H-7/H-19 (Fig. 2C). Thus, the planar structure for 6 was unequivocally clarified as a novel 7,18-cyclolabdane skeleton. The NOESY correlations of H3–17/H-7, H-5/H3–19, H3–19/H-18, H-18/H3–17, H-8/H-11β, and H-11β/H-16 suggested the relative configuration of 6. From the perspective of the biogenic pathway, the structure of 6 was determined considering the absolute configuration of 4 and their similar ECD curves. Ultimately, ECD calculations (Fig. S2) identified the absolute configuration of the 6 as 4R,5S,7S,8S,12S,18S.
Haploide G (7) was determined to have the molecular formula C21H22O7 via HRESIMS (m/z 409.1251 [M + Na]+, calcd. 409.1258), with 11 degrees of unsaturation. 1H and 13C NMR spectra suggested its high similarity to haplomitrenolide C [20], except for the rearrangement of the A-ring from a six-membered to a five-membered carbon ring, connected to the B-ring at C-5 (δC 66.6), and introduction of one carbonyl group at C-1 (δC 213.3). These results were confirmed based on HMBCs from H3–19 (δH 1.31) to C-3 (δC 32.0), C-4 (δC 56.1), C-5, and C-18 (δC 171.8); and from H-10 (δH 3.50) to C-1, C-5, and C-9; as well as the key 1H–1H COSY correlations of H-9/H-10. NOESY correlations of H3–19/H-10, H-10/H-12, and H-12/H-9 (Fig. 2D) suggested two possible relative configurations for compound 7, designated as 7 (4R,5R,9R,10S,12R) and 7 (4S,5S,9R,10S,12R). ECD calculations were performed for both diastereomers, and the results revealed that the experimental ECD spectrum of compound 7 showed a better agreement with the calculated spectrum of 7 (4R,5R,9R,10S,12R) than that of 7 (4S,5S,9R,10S,12R) (Fig. S2). To further validate their configurations, seven low-energy conformers of compound 7 within 3.0 kcal/mol (Fig. S3 and Table S4 in Supporting information) with a specific gravity of Boltzmann distribution >1.0% were refined and considered for density functional theory (DFT) and Gauge-Independent Atomic Orbital (GIAO) NMR shift calculation at the mPW1PW91/6–31G(d) level of theory [21,22]. The correlation coefficient was the key parameter applied for quantifying the agreement between the calculated and experimental 1H and 13C NMR data, and the results further supported the assignment of 7 (4R,5R,9R,10S,12R) (Fig. S4 in Supporting information). Ultimately, the absolute structure was determined as 4R,5R,9R,10S,12R.
The 1H and 13C NMR spectra of haploide H (8) were similar to those of haplomintrin A [17], except for the presence of one methoxy (δH 3.39; δC 55.1) and the absence of a double bond. HMBCs from H3–21 (δH 3.39) to C-15 (δC 109.8); and from H-15 (δH 5.29) to C-14 (δC 37.3), C-21 (δC 55.1), C-13, and C-16; along with the 1H–1H COSY signals of H2–14/H-15 suggested that the double bond at C-14 was reduced, and one methoxy was connected at C-15 in 8. NOESY correlations of H3–19/H-3β, H-3α/H-5, H-5/H-9, H-9/H3–17, H3–17/H-7, H-7/H3–21, and H-7/H-16 indicated the relative configuration of 8. Ultimately, the absolute structure of 8 was determined to be 4R,5S,7S,8R,9S,10S,12R,13S,15R,16R, based on the single-crystal X-ray diffraction analysis with Cu Kα radiation (CCDC 2344633) (Fig. 3).
Haploide Ⅰ (9) was identified as the C-5 epimer of hapmnioide C [18], based on their similar NMR spectra and NOESY signals of H3–17/H-18, H-18/H3–19, H3–19/H-5, H-18/H-3β, H-3α/H-1, H3–17/H-11β, and H-11β/H-14. The absolute configuration of 9 was confirmed to be 1R,4R,5S,8S,12R,18R based on ECD calculations (Fig. S2). The NMR data of haploide J (10) suggested a similar planar structure to that of 9. However, the five-membered lactone ring was hydrolyzed, and the carboxyl group at C-6 underwent esterification, thereby introducing a methyl ester group. Additionally, the hydroxyl group at C-1 was oxidized to the carbonyl group in 10. These predictions were determined based on the HMBC signals from H3–21 (δH 3.62) to C-6 (δC 171.2), from H2–7 (δH 2.43, 2.83) to C-6, from H2–2 (δH 2.31, 2.49) to C-1 (δC 215.7). The relative configuration of 10 was confirmed based on the NOESY signals of H3–17/H-18, H-18/H3–19, H3–19/H-5, H3–17/H-11β, and H-11β/H-14. Accordingly, the configuration of 10 was determined to be 4R,5S,8S,12R,18R based on the ECD calculations (Fig. S2) and biosynthetic pathways.
Haploide K (11) showed one fewer degree of unsaturation than 2, and its NMR data were similar to those of 2, except for the presence of one hemiacetal signal (δH 4.70; δC 97.1) and the absence of the ketone group. HMBCs from H3–19 to C-3, C-4, C-5, and C-18 (δC 97.1); from H3–17 to C-7, C-8, and C-9 (δC 75.5); along with the key 1H–1H COSY signal of H2–1 (δH 1.73)/H2–2, suggested that the ketone at C-1 was reduced, and epoxidation occurred between C-9 and C-19, unlike 2. The relative configuration of 11 was established based on the NOESY correlations of H-5/H3–19, H3–19/H-18, and H-1/H-12. Comparing the experimental and calculated ECD curves (Fig. S2), the absolute configuration of 11 was determined to be 4R,5R,9S,10R,12R,18R.
Haploides L and M (12 and 13) were identified as a pair of C-5 epimers, sharing the same molecular formula of C21H24O7. The NMR spectra of 12 closely resembled those of cacofuran B [23], except for the presence of one keto carbonyl at C-6 (δC 203.4), two ester carbonyls at C-18 (δC 177.7) and C-20 (δC 177.0), one oxygenated methine at C-1 (δC 65.2), one oxygenated methyl attached to C-18, and one ether bond between C-1 and C-12, as confirmed by HMBCs from H-12 (δH 4.99) to C-1; from H-1 (δH 3.86) to C-12; from H3–19 (δH 1.26) to C-3 (δC 31.7), C-4 (δC 42.4), C-5 (δC 59.3), and C-18 (δC 177.7); from H3–21 (δH 3.70) to C-18; and from H-5 (δH 3.68) to C-6 (δC 203.4) and C-20 (δC 177.0). Its relative configuration was determined based on the NOESY signals of H3–19/H-3β, H-3β/H-1, H-3α/H-5, H-5/H-9, H-9/H3–17, H-9/H-12. The absolute configuration (1S,4R,5S,8S,9R,10S,12R) of 12 was conclusively determined by combining X-ray diffraction results (Cu Kα radiation, CCDC 2344628) (Fig. 3). The NOESY signals (H3–19/H-5, H-5/H-3β, H-3β/H-1, H-9/H3–17, H-9/H-12) of 13 indicated that it represents the C-5 epimer of 12. Finally, the absolute configuration of 13 was determined as 1S,4R,5R,8S,9R,10S,12R by single-crystal X-ray diffraction using Cu Kα radiation (CCDC 2344629) (Fig. 3).
The NMR data of 14 closely resembled those of 19 [20], with the only difference being the chemical shift at C-1 (δC 34.2 in 19 vs. δC 66.5 in 14), revealing the presence of a hydroxyl group at this position. This was supported by the 1H–1H COSY signals of H-1 (δH 4.43)/H-2 (δH 1.74, 1.91). The relative configuration of 14 was determined based on the NOESY signals of H-5/H-9, H-11β/H-1, H-1/H-12, and H-14/H3–17. Ultimately, the absolute structure of 14 was determined to be 1S,5S,9S,10S,12R based on the single-crystal X-ray diffraction using Cu Kα radiation (CCDC 2344634) (Fig. 3).
Haploides O and P (15 and 16) were identified as a pair of C-5 and C-10 isomers based on their identical molecular formula of C21H22O7. The NMR data of 15 suggested that its planar structure was similar to that of haplomitrenolide C [20], except that the six-membered lactone ring was replaced by a five-membered lactone ring that was connected to the B-ring by spiroC-9, and one ketone was introduced at C-1 (δC 207.3). These deductions were confirmed by key HMBCs from H3–17 to C-9 (δC 51.0); from H2–2 to C-1; from H-10 to C-9; from H2–11 to C-9, C-12, C-13, and C-20; from H-12 to C-9, C-11, and C-13; along with the 1H–1H COSY signals of H2–11/H-12. The relative configuration of haploide O (15) was deduced from NOESY correlations of H-5/H2–11 and H-10/H3–19. The absolute structure of haploide O (15) was finally confirmed to be 4R,5R,9R,10S,12R through single-crystal X-ray diffraction using Cu Kα radiation (CCDC 2344624) (Fig. 3). Moreover, the absolute structure of haploide P (16) was established as 4R,5S,9R,10R,12R, according to the NOESY correlations (H3–19/H-5 and H2–11/H-10) and Cu Kα single-crystal X-ray diffraction analysis (CCDC 2344625) (Fig. 3).
A biogenetic network was postulated for 1–19, rationalizing their skeletal diversification through aldol-type cascade transformations (Scheme 1). The biosynthetic origin can be traced to 19, which is derived from geranylfarnesyl pyrophosphate via a series of enzymatic reactions and then undergoes oxidization/[2 + 2] cyclization or oxidization/hemiacetalization to afford 8 and 11, respectively. Central to this network is the C-1 oxidation event, which generates key intermediate Ⅰ. Regioselective aldolase-mediated aldol/retro-Claisen rearrangements at divergent positions of intermediate Ⅰ account for the structural divergence observed in compounds 1–7. Specifically, C9-specific aldol/retro-Claisen transformations of intermediate Ⅰ produce the labdane-derived derivatives 15 and 16. Structural elaboration of 7 through Claisen/retro-aldol reactivity at the C10-adjacent ester moiety culminates in the formation of 18. For 9 and 10, intermediate Ⅲ undergoes a tandem sequence cascade involving retro-Claisen cleavage, olefinic double-bond migration, and subsequent Claisen-type reorganization at C8 to establish their characteristic frameworks.
All isolated compounds were systematically screened for their anti-inflammatory potential, focusing on their ability to modulate the secretion of key proinflammatory cytokines. Given the pivotal role of proinflammatory cytokines, such as IFN-γ, TNF-α, and IL-6, in mediating inflammatory responses through immune cell activation and tissue damage amplification [24], the anti-inflammatory efficacy of the compounds was evaluated by measuring their ability to suppress these cytokines in ConA-induced murine spleen cells. Notably, haploide O (15) and haplomitrenolide C (17) demonstrated significant anti-inflammatory activity, effectively inhibiting the secretion of these proinflammatory cytokines at a concentration of 10 µmol/L (Fig. S15 in Supporting information). These findings highlight the therapeutic potential of these compounds in mitigating inflammatory responses.
In conclusion, four unprecedented labdane scaffolds were isolated and identified from H. mnioides: 9,18-cyclolabdanes (1 and 2), 1,10-seco-9,18-cyclolabdanes (3–5), 7,18-cyclolabdanes (6), and 1,10-seco-1,5-cyclolabdanes (7), along with seven novel labdanes (8–14), two novel halimane diterpenoids (15 and 16), and three known congeners (17–19). Interestingly, four pairs of novel diastereomers, haploides A and B (1 and 2), C and D (3 and 4), L and M (12 and 13), and O and P (15 and 16) were simultaneously isolated. In addition, the plausible biosynthetic pathways of compounds 1–18 were deduced to originate from 19 as the starting material through an aldol-type cascade process. Anti-inflammatory assays revealed that haploide O (15) and haplomitrenolide C (17) significantly inhibited the secretion of the proinflammatory cytokines IFN-γ, TNF-α, and IL-6 in ConA-induced murine spleen cells. These findings provide valuable insights into the chemical diversity and biological activity of labdane diterpenoids.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Ming-Zhu Zhu: Writing – original draft, Methodology, Investigation, Formal analysis, Data curation. Ze-Jun Xu: Writing – review & editing, Supervision, Investigation, Formal analysis. Jiao-Zhen Zhang: Investigation, Formal analysis. Li-Zhi Shi: Investigation. Zong-Xu Gao: Investigation. Yi Li: Investigation. Shuang-Zhi Yuan: Investigation. Yan-Rong Ma: Investigation. Ai-Xia Cheng: Investigation. Hong-Xiang Lou: Writing – review & editing, Supervision, Project administration, Methodology, Investigation, Funding acquisition, Formal analysis, Data curation.
Financial support was provided by the National Natural Science Foundation of China (Nos. 82293682, 82173703, and 82273807), and Shandong Provincial Natural Science Foundation (No. ZR2022YQ74).
Supplementary material associated with this article can be found, in the online version, at doi:
T. Rodrigues, D. Reker, P. Schneider, G. Schneider, Nat. Chem. 8 (2016) 531–541. doi: 10.1038/nchem.2479
Y.J. Wang, P. Tang, W.C. Tu, et al., Chin. Chem. Lett. 36 (2025) 109955.
L. Xiong, Q.M. Zhou, Y.K. Zou, et al., Org. Lett. 17 (2015) 6238–6241. doi: 10.1021/acs.orglett.5b03227
O. Antoniuk, A. Maranha, J.A.R. Salvador, et al., Nat. Prod. Rep. 41 (2024) 1858–1894. doi: 10.1039/d4np00021h
D.J. Tantillo, Nat. Prod. Rep. 28 (2011) 1035–1053. doi: 10.1039/c1np00006c
A. Minami, T. Ozaki, C. Liu, et al., Nat. Prod. Rep. 35 (2018) 1330–1346. doi: 10.1039/c8np00026c
P. Luo, J.M. Lv, H.T. Zhen, et al., Chin. Chem. Lett. 36 (2025) 111042.
G.B. Tabekoueng, H. Li, B. Goldfuss, et al., Angew. Chem. Int. Ed. 63 (2024) e202413860.
C. Zeymer, R. Zschoche, D. Hilvert, J. Am. Chem. Soc. 139 (2017) 12541–12549. doi: 10.1021/jacs.7b05796
Y.Q, Li, M.J. Cong, W.G. Wang, et al., Angew. Chem. Int. Ed. 63 (2024) e202403365.
H.D. Zinsmeister, H. Becker, T. Eicher, Angew. Chem. Int. Ed. 30 (1991) 130–147.
T. Shen, X.N. Wang, H.X. Lou, Nat. Prod. Rep. 26 (2009) 916–935. doi: 10.1039/b905960a
J.B. Qu, L.M. Sun, H.X. Lou, Chin. Chem. Lett. 24 (2013) 801–803.
M. Rempt, G. Pohnert, Angew. Chem. Int. Ed. 49 (2010) 4755–4758. doi: 10.1002/anie.201000825
L.N. Wang, J.Z. Zhang, X. Li, et al., Org. Lett. 14 (2012) 1102–1105.
J.Z. Zhang, R.X. Zhu, G. Li, et al., Org. Lett. 14 (2012) 5624–5627. doi: 10.1021/ol302295a
J.C. Zhou, J.Z. Zhang, A.X. Cheng, et al., Org. Lett. 17 (2015) 3560–3563. doi: 10.1021/acs.orglett.5b01664
J.C. Zhou, J.Z. Zhang, R.J. Li, et al., Org. Lett. 18 (2016) 4274–4276. doi: 10.1021/acs.orglett.6b01854
Y. Li, Z.J. Xu, R.X. Zhu, et al., Org. Lett. 22 (2020) 510–514. doi: 10.1021/acs.orglett.9b04270
Y. Asakawa, M. Toyota, T. Masuya, Phytochemistry. 29 (1990) 585–589.
L. Grauso, R. Teta, G. Esposito, et al., Nat. Prod. Rep. 36 (2019) 1005–1030. doi: 10.1039/c9np00018f
X.X. Li, J. Fu, Y.H. Li, et al., Org. Lett. 24 (2022) 1388–1393. doi: 10.1021/acs.orglett.2c00242
J. Tanaka, G. Marriott, T. Higa, et al., J. Nat. Prod. 64 (2001) 1468–1470.
M.F. Neurath, Nat. Rev. Immunol. 24 (2024) 559–576. doi: 10.1038/s41577-024-01008-6

Figure 2 Key 1H−1H COSY, HMBC, NOESY correlations of haploide A, C, F, G, and ORTEP drawings of haploides A, C.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:


 下载:
下载: