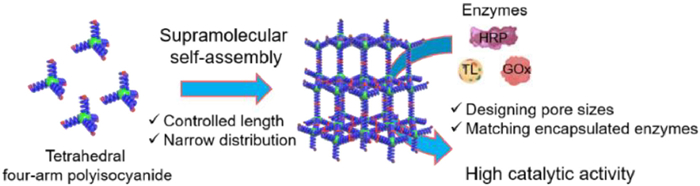
Scheme 1.
Schematic diagram of enzyme immobilization in SPFms.
Three-dimensional supramolecular polymer frameworks with precisely tunable and large apertures for enzyme encapsulation
Runtan Gao , Yang Zong , Tingting Li , Na Liu , Zongquan Wu

Fabricating porous frameworks with precisely controlled pore sizes is a long-standing research hotspot but still remains a formidable challenge, especially for the fabrication of frameworks with large pore sizes [1-4]. One possible approach to this challenge is utilizing long rigid macromolecules or polymers with controllable lengths as links. However, the interpenetration of these long links may result in amorphous materials with smaller and uncontrollable pore structures. Moreover, most polymer links have inefficient stiffness to support entire frameworks, potentially leading to collapsed or poorly ordered structures [5-8]. Therefore, rigid polymer links that can effectively support an entire framework, reduce structural defects, and enhance uniformity are urgently needed. Among the artificial polymers, polyisocyanide carrying substituent on every backbone atom and adopt a rodlike helical structure and may have great potential for the fabrication of frameworks [9-13]. Previously, we developed a family of alkyne-Pd(Ⅱ) catalysts enable the precise synthesie of helical polyisocyanides with precisely controlled molecular weight and extremely narrow distribution. Using the precisely synthesized polyisocyanides as links can fabricate frameworks with predesignable pore structures. The long rodlike polyisocyanides may lead to ordered frameworks with large pore size, which have great promise in enzyme capsulation.
Enzymes are excellent biocatalysts that show significant potential in the pharmaceutical and chemical industries due to their high activity and excellent selectivity [14-17]. However, owing to their inherent fragility, it is difficult for natural enzymes to maintain their favorable conformation and high catalytic activity in complex operable conditions and harsh environments [18-20]. Consequently, the direct utilization of enzymes in practical applications is often hampered by low activity, poor stability, hard recyclability, and limited operating conditions. One effective solution to overcome these drawbacks is the immobilization of natural enzymes onto solid supports [21-23]. In this context, porous frameworks with uniform channels are promising materials for enzyme immobilization. In addition to facilitating substrate transport and enrichment, porous frameworks also provide immobilized enzymes with selective permeability and structural stability [24,25]. The encapsulation of enzymes into porous materials involves two strategies: one is biomineralizing enzymes by self-assembly with porous material precursors, which overcomes the intrinsic pore size limitations of porous materials. However, the harsh medium required to crystallize the porous material is not very compatible with fragile enzymes [26-28]. A second strategy involves the infiltration of enzymes into pre-synthesized nanoscale porous materials, which enhances the mass transfer and stability of the enzymes. However, this strategy is generally limited to enzymes with sizes smaller than the internal pore aperture of the porous material, and larger enzymes are not easily encapsulated [29-32]. Furthermore, the pore size of a framework directly determines how it interacts with molecules, which influences its functionality and applications [33,34]. Compared to two-dimensional (2D) frameworks, three-dimensional (3D) frameworks provide more space and channels for enzyme encapsulation, leading to enhanced loading capacity and inhibited deactivation [32,35]. Therefore, the precise synthesis of 3D porous frameworks with controllable and large pore sizes to immobilize enzymes is essential.
Among the reported frameworks, supramolecular organic frameworks (SOFs) are composed of organic molecules linked via intermolecular supramolecular interactions. Due to the dynamic character of the supramolecular interactions, self-correction of the assemblies occurs and results in SOFs with highly ordered structures [36-38]. Hydrogen bonding is a popular supramolecular interaction featuring large bonding constants, high directionality, and good reversibility [39-42]. Therefore, constructing supramolecular organic polymer frameworks (SPFs) using rigid polyisocyanide with tunable length as links via hydrogen bonding connections may generate porous frameworks with bespoke shapes, sizes, and functionalities [25,43-45]. These porous frameworks could be used as "exoskeletons" to encapsulate fragile enzymes, generating a new type of metal-free crystalline biocomposite featuring superior catalytic activity and operational stability compared to free enzymes. Additionally, porous frameworks could be used to encapsulate multiple enzymes. This could enable enzymatic cascade reactions, which would eliminate the tedious isolation and purification of reaction intermediates.
Among the various supramolecular interactions, the hydrogen bonding interactiong between the 2-ureido-4[1H]-pyrimidinone (Upy) features high association constant. These attributes enable Upy to be widely used in supramolecular self-assembly and framework construction [46,47]. In this work, we designed and synthesized a series of 3D SPFs with tunable pore sizes. The frameworks with the pore size matchable to enzymes was applied as enzyme exoskeletons. First, tetrahedral four-arm star polyisocyanides with precisely controlled Mn and low Mw/Mn were prepared via the four-functional Pd(Ⅱ)-catalyzed living polymerization of isocyanide (Scheme 1). Then, a Upy unit was installed onto each chain end of the star polyisocyanide through post-polymerization functionalization via the terminal Pd(Ⅱ)-catalyzed Songashira cross coupling reaction. Leverage the intermolecular hydrogen bonding between the terminal Upy units, these tetrahedral star polyisocyanides self-assembly into ordered SPFms with uniformly porous networks. Remarkably, the pore size of these SPFs is proportional to the degree of polymerization (DP) of the polyisocyanide, enabling precise control of the pore aperture. Therefore, a family of SPFs with pore sizes of ca. 5.06, 7.27, 8.12, and 9.72 nm was precisely fabricated using four-arm polyisocyanides with DP of 10 (SPF10), 20 (SPF20), 30 (SPF30), and 40 (SPF40), respectively. The ordered SPFs possess uniform and tunable porosity, facilitating their usage as enzyme exoskeletons. The lipase (TL), horseradish peroxidase (HRP), and glucose oxidase (GOx) were respectively encapsulated into SPF10, SPF20, and SPF30 with high loading capacities of 26, 21, and 24 wt%. All the encapsulated enzymes display superior catalytic activity and operational stability compared to the corresponding free enzymes. Furthermore, these 3D SPFs faciliate the encapsulation of more than one enzymes, and realized a dual enzyme-catalyzed cascade reaction, which minimize the diffusion limitation of intermediates and improve the catalytic activity of the cascade reaction.
As shown in Fig. 1, tetrahedral four-arm alkyne–Pd(Ⅱ) catalysts (FA-Pd(Ⅱ)) were prepared from tetrakis(4-ethynylphenyl)methane (Fig. S1 in Supporting information). The living polymerization of isocyanide (1) initiated by FA-Pd(Ⅱ) with 1-to-Pd(Ⅱ) ratios fixed of 10, 20, 30 and 40 resulted in a series of four-arm polyisocyanides denoted FA-poly-1m-Pd(Ⅱ) (where m is theoretical DP, m = 10, 20, 30, and 40). These polyisocyanides had desired Mns and low Mw/Mns values, and they featured Pd(Ⅱ) terminals (Fig. S2 in Supporting information). Fig. 2a illustrates that these polymers exhibited single modal elution traces in size exclusion chromatography (SEC). A linear correlation between the Mn of the synthesized polymers and their 1-to-Pd(Ⅱ) catalyst ratio was observed. Additionally, the isolated FA-poly-1m-Pd(Ⅱ) displayed low dispersity, with Mw/Mn values of < 1.20 (Fig. 2b). To install Upy unit onto the chain-ends of FA-poly-1m-Pd(Ⅱ), the terminal Pd(Ⅱ) complex was activated by coordinating with a bidentate phosphine ligand (Wei-phos) and then catalyzed Sonogashira cross coupling reaction with Upy, followed the procedure reported by us previously [25]. The SEC analyses of the resulting FA-poly-1m-Upy bearing Upy terminals also showed single modal elution traces and maintained low dispersity (Mw/Mn < 1.20) (Fig. S3 and Table S1 in Supporting information). The structures of the isolated FA-poly-1m-Pd(Ⅱ) and FA-poly-1m-Upy were characterized by proton nuclear magnetic resonance (1H NMR). As displayed in Fig. S4 (Supporting information), three sharp resonances at downfield 10.25 (-NHCO-), 11.90 (-CONH-), and 13.12 (-NCNHC-) ppm were clearly observed, which can be ascribed to the Upy units in CDCl3. The 31P NMR spectrum of FA-poly-120-Pd(Ⅱ) showed a clear resonance at 13.1 ppm, which was attributed to the Pd-coordinated PEt3. whereas no 31P signal could be detected of FA-poly-120-Upy, suggesting that a Upy unit was installed on each polyisocyanide terminal (Fig. S5 in Supporting information). The Fourier transform infrared (FT-IR) spectrum of FA-poly-120-Upy showed vibrations at 2210 and 1630 cm‒1 attributed to the C≡C bond of FA-poly-120-Upy and C=N bond of the polyisocyanide backbone, respectively (Fig. S6 in Supporting information). Thermogravimetric analysis (TGA) revealed that both FA-poly-120-Upy and FA-poly-120-Pd(Ⅱ) had good thermal stability, with thermal decomposition temperatures (Td, 5% weight loss) higher than 300 ℃ (Fig. S7 in Supporting information). The differential scanning calorimetry (DSC) profile of FA-poly-120-Upy showed glass transition (Tg, 98 ℃) and melting (Tm, 228 ℃) temperatures (Fig. S8 in Supporting information) higher than those of the FA-poly-120-Pd(Ⅱ) precursor (Tg = 95 ℃; Tm = 224 ℃).
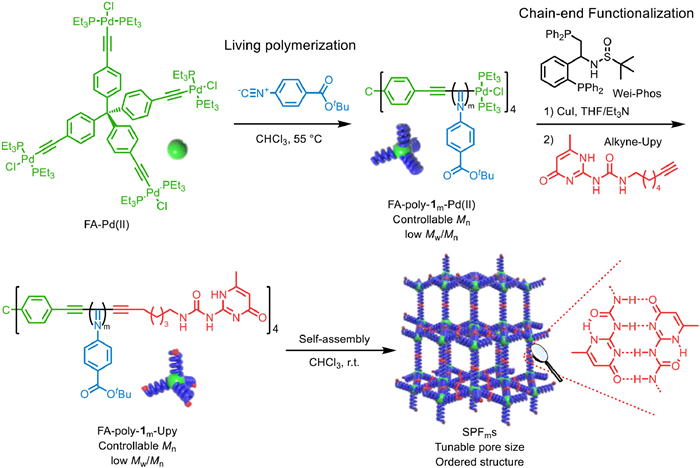
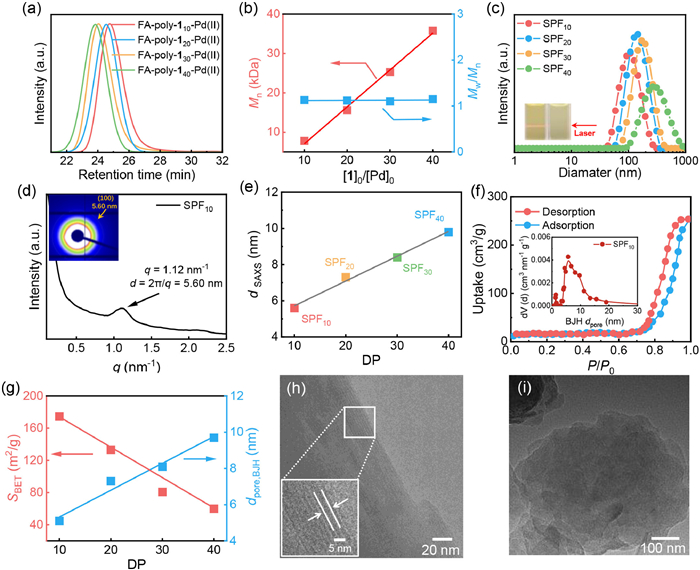
The isolated FA-poly-1m-Upy polymers with different DPs were self-assembly into porous SPFms (m = DP) in non-polar solvents (CHCl3) via intermolecular hydrogen bonding between the terminal Upy units. Notably, the CHCl3 solution of FA-poly-1m-Pd(Ⅱ) was clear and transparent, lacking of any Tyndall effect, regardless of DPs. In contrast, a pronounced Tyndall effect was observed in the CHCl3 solutions of all the synthesized FA-poly-1m-Upy bearing Upy unit on the terminals, suggesting they were self-assembled into porous SPFm, confirming the formation of the expected frameworks (Fig. 2c). Dynamic light scattering (DLS) analysis indicated that FA-poly-110-Pd(Ⅱ) had a hydrodynamic diameter of 7.8 nm in CHCl3, suggesting its molecular dissolution (Fig. S9 in Supporting information). Meanwhile, the hydrodynamic diameters of SPF10, SPF20, SPF30, and SPF40 were 100.5, 139.2, 174.5, and 277.2 nm, respectively, even when diluted to 0.1 mg/mL (Fig. 2c and Fig. S10 in Supporting information). This result indicated the FA-poly-1m-Upy with different molecular weights were self-assembly into large-sized frameworks. The ordering of the porous SPFms was further verified by synchrotron small-angle X-ray scattering (SAXS) analysis. The SAXS curve of the CHCl3 solution of SPF10 showed a clear peak at q ≈ 1.12 nm−1, which corresponded to a simulated pore aperture of 5.60 nm (Fig. 2d). Similarly, the SAXS curves of SPF20, SPF30, and SPF40, showed peaks at q ≈ 0.86, 0.74, and 0.64 nm−1, respectively (Fig. S11 in Supporting information), which corresponded to pore apertures of 7.30, 8.40, and 9.80 nm. These results further confirmed the formation of frameworks with different pore apertures. Interestingly, the SAXS analyses suggested that the aperture size of the SPFms was linearly correlated to the DP of the polyisocyanide links (Fig. 2e), due to the rigid rodlike backbone of helical polyisocydnie. The linear correlation between the pore-aperture with the DP of polyisocyanide backbone supports high controllability of pore size. That means control over the DP of polyisocyanide via the living polymerization technique could readily tune the pore size of the SPFms. Based on the slop of the plot in Fig. 2e, it can be concluded that the every one increment in the DP of polyisocyanide backbone, the pore size increases by ca. 0.15 nm.
The porosities of the generated frameworks were further investigated by nitrogen sorption isotherm measurements. All the SPFms exhibited typical type Ⅳ isotherms, indicating the formation of mesoporous frameworks. As summarized in Table S2 (Supporting information), the Brunauer–Emmett–Teller (BET) specific surface area (SBET) of SPF10 was 98.34 m2/g, and the Barrett–Joyner–Halenda (BJH) pore size distribution was centered at ca. 5.06 nm (Fig. 2f). The pore volume (Vpore) of SPF10 was calculated to be 0.44 g/mL. As the DP of the polyisocyanide arms increased, the SBET values of SPF20, SPF30, and SPF40 decreased to 87.26, 53.28, and 32.15 m2/g, respectively, while their pore apertures increased to 7.27, 8.12, and 9.72 nm, respectively. The pore apertures obtained from nitrogen sorption isotherm measurements was linearly correlated to the DP of the polyisocyanide arms (Fig. 2g and Table S2), agreeing well with the SAXS analyses. In addition, the BJH pore volumes (Vpore) of SPF20, SPF30, and SPF40 were 0.30, 0.21, and 0.18 cm3/g, respectively (Fig. S12 and Table S2 in Supporting information). SBET and Vpore were also correlated to the DP of the polyisocyanide links. As plotted in Fig. 2g, the SBET values linearly decreased with increasing DP, while the pore aperture and Vpore linearly increased with increasing DP (Fig. S13 and Table S2 in Supporting information). Notably, owing to the dense hydrophobic pendants of the polyisocyanide links, the SPFs maintained their porous structures after being treated with water, as confirmed by nitrogen adsorption-desorption analysis (Fig. S14 in Supporting information). This was expected to facilitate the encapsulation of enzymes in water. As displayed in Fig. 2h, the transmission electron microscope (TEM) image of SPF10 clearly shows clear lattice fringe with the spacing of 5.32 nm and it is consistent with this estimated through the SAXS and BET analyses. Similarly, SPF20, SPF30 and SPF40 are also analyzed by TEM, but it is difficult to detect their lattice fringes due to the large pore size (Fig. 2i and Fig. S15 in Supporting information). Therefore, the pore structures were visualized by scanning electron microscopy (SEM). The SPFms with different pore sizes exhibited different morphologies [48]. For instance, SPF10 and SPF20, synthesized with relatively short polyisocyanide blocks, exhibited rough surfaces decorated with pores (Figs. 3a and b). As the length of the polyisocyanide increased, SPF30 and SPF40 showed fleshy fluffy surfaces (Figs. 3c and d). These observations suggest that the morphology of these porous polymers can be controlled by adjusting the length of the polyisocyanide links. This result can be attributed to the formation of pores by intermolecular hydrogen bonding between the terminal Upy units, which means that the pore aperture size directly depends on the length of the rigid polyisocyanide links.

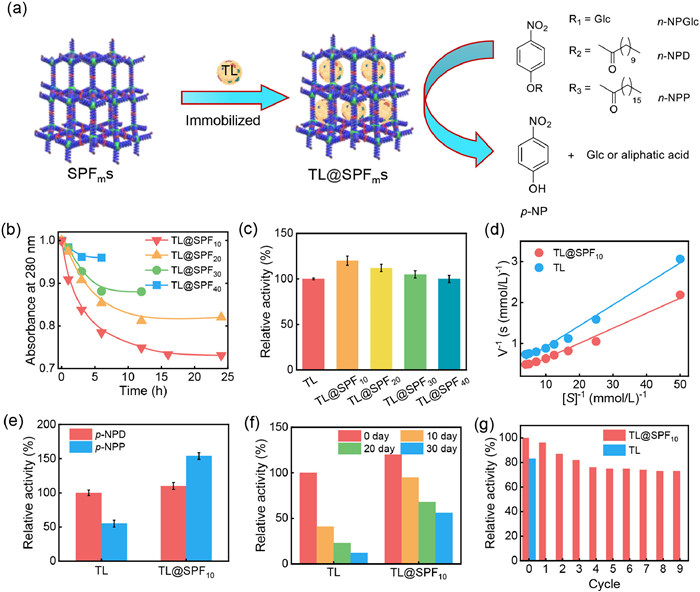
The porous SPFms with controllable pore sizes show great promise for enzyme encapsulation. Therefore, to investigate their enzyme encapsulation capability, the encapsulation of lipase from Thermophila lanuginosa (TL, size: 3.5 × 4.5 × 5.0 nm3) was first explored. SPFms with different sizes were immersed into a phosphate-buffered saline solution containing TL, and the UV–vis absorbance of the supernatant was measured. Based on the change in the characteristic absorbance at 280 nm, approximately 26, 19, and 12 wt% of the TL was encapsulated into SPF10, SPF20, and SPF30, respectively. In contrast, only a smaller amount of TL (less than ca. 6 wt%) was encapsulated in the larger pores of SPF40, which was because the pore size of this sample was too large to encapsulate TL (Table S3 in Supporting information). SPF10 showed the highest encapsulation capacity because its pore size matched the size of TL (Figs. 4a and b). To confirm the encapsulation of TL in the pores of SPFms, sodium dodecyl sulfate (SDS) was used to remove the physically adsorbed enzymes from the surface of the SPFms [27]. It was revealed that only 2%–10% of the TL removed from the solution was physically adsorbed as free enzymes on the SPF surface, meaning that > 90% of the enzymes were encapsulated into the SPFms (Fig. S16 in Supporting information). The nitrogen sorption isotherms of the TL-encapsulated SPFms (TL@SPFms) showed a reduction in BET surface area (Fig. S17 in Supporting information). The FT-IR spectrum of TL@SPF10 displayed characteristic adsorption peaks at 3296, 2871, and 1654 cm−1 that were ascribed to TL (Fig. S18 in Supporting information). Furthermore, the circular dichroism (CD) spectrum of TL@SPF10 was almost the same to that of the free TL (Fig. S19 in Supporting information), indicating that the encapsulated TL maintained its pristine conformation. To assess the catalytic performance of TL@SPFms, the hydrolysis reaction of p-nitrophenyl β-D-glucopyranoside (p-NPGlc) catalyzed by TL and TL@SPF10 was studied in phosphate-buffered saline. The production of p-nitrophenol (p-NP) was detected by measuring the UV–vis absorbance at a wavelength of 405 nm to determine the activity of TL and TL@SPF10. Both free TL and encapsulated TL@SPF10 catalyzed the hydrolysis of p-NPGlc and produced targeted p-NP. However, the catalytic activity of TL@SPF10 was 1.2 times higher than that of the free TL due to the confinement effect of the encapsulated enzymes in TL@SPF10 (Figs. 4c and d, and Table S4 in Supporting information). The porosity of the SPF10 was beneficial for the enrichment of p-NPGlc and strengthening the interaction between the encapsulated TL and p-NPGlc, leading to enhanced catalytic activity. Note that pure SPF10 did not show any catalytic activity, confirming that the catalytic of the TL@SPF10 was entirely derived from the encapsulated TL. Additionally, the catalytic activities of TL@SPF20, TL@SPF30, and TL@SPF40 were also investigated. TL@SPF20 and TL@SPF30 showed catalytic activities that were 1.12 times and 1.05 times higher than that of free TL, respectively. The catalytic activity of TL@SPF40 was comparable to that of free TL. These results further demonstrated that SPFs with pore sizes matching the size of TL were favorable for improving catalytic activity. Furthermore, the catalytic activity of TL@SPF10 toward substrates with different sizes (p-nitrophenyl decanoate (p-NPD) and p-nitrophenyl palmitate (p-NPP)) was also investigated. When free TL was used as a catalyst, the hydrolysis of p-NPD, which has shorter alkyl chains, proceeded more rapidly than that of p-NPP. The p-NPD/p-NPP reaction ratio of this catalyst was 1.8. Surprisingly, when TL@SPF10 was used as the catalyst, the hydrolysis of p-NPP, which has longer alkyl chains, was much more rapid than that of p-NPD, and the p-NPP/p-NPD reaction rate ratio was 1.4 (Fig. 4e). This was because the longer alkyl chains of p-NPP meant that this reactant was retained in the pores of SPF10 for a longer period, increasing the chance of contact with the enzyme. TL@SPF10 also showed high stability, retaining 56% of its catalytic activity after 30 days of storage. In contrast, the free TL only maintained 12% of its catalytic activity (Fig. 4f). Furthermore, the TL@SPFms were easily recovered from the reaction mixture by centrifugation for reuse in the hydrolysis process. Based on the product yield, the catalytic activity remained above 75% even after 5 cycles. Under the same conditions, the catalytic activity of free TL was completely lost after the first recycle (Fig. 4g).
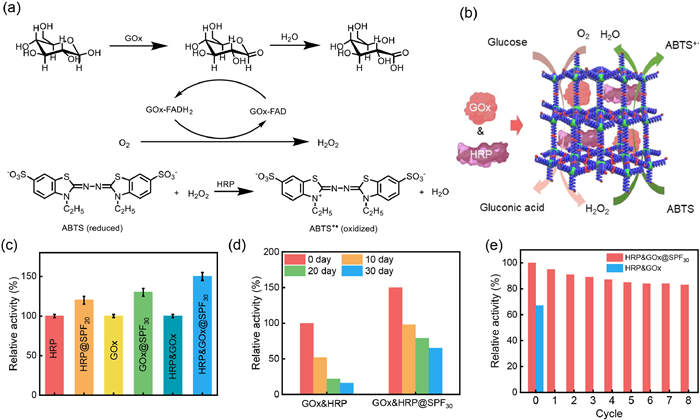
To demonstrate that SPFs with controllable pore size could serve as versatile nanoplatforms for enzyme encapsulation, the encapsulation of glucose oxidase (GOx, 6.0 × 5.2 × 7.7 nm3) and horseradish peroxidase (HRP, 4.0 × 4.4 × 6.8 nm3) into the SPFms was attempted using the same procedure described above. Given the different sizes of HRP and GOx, different encapsulation efficiencies were observed. HRP was most effectively encapsulated within SPF20 (21 wt%), followed by SPF30 (18 wt%), SPF40 (9 wt%), and SPF10 (6 wt%) (Table S3). In contrast, GOx was most effectively encapsulated within SPF30 (24 wt%), followed by SPF40 (16 wt%), SPF20 (10 wt%), and SPF10 (8 wt%) (Table S3). These results further confirm the importance of matchable pore size for the effective immobilization of biomacromolecules (Table S2). The HRP-encapsulated SPFms (HRP@SPFms) and GOx-encapsulated SPFms (GOx@SPFms) were further characterized by N2 adsorption, FT-IR measurements, and CD spectroscopy (Figs. S20-S22 in Supporting information), which confirmed that the enzymes were encapsulated into the pores and maintained their pristine conformations. Next, the catalytic activities of the HRP@SPF20, free HRP, GOx@SPF30, and free GOx were investigated. HRP@SPF20 showed catalytic activity that was 1.2 times higher than that of free HRP. Similarly, the catalytic activity of GOx@SPF30 was 1.3 times higher than that of free GOx. These results indicate that the SPFs with matched pore sizes are favorable for biocatalysis. Furthermore, large-sized frameworks could also be applied for the encapsulation of dual enzymes to perform cascade reactions. As shown in Fig. 5a, two enzymes (GOx and HRP) were encapsulated into SPF30. FT-IR and N2 adsorption analyses indicated that GOx and HRP were both encapsulated into SPF30 (15 wt% GOx and 13 wt% HRP), forming GOx & HRP-encapsulated SPF30 (GOx & HRP@SPF30) (Figs. S23 and S24 in Supporting information). When GOx & HRP@SPF30 used as a dual-enzyme catalyst, GOx in GOx & HRP@SPF30 specifically catalyzes the conversion of β-D-glucose to gluconic acid and H2O2 in the presence of O2 [49]. The catalytic reaction involves a reduction step and an oxidation step (Figs. 5a and b). The reduction step is as follows: β-D-glucose is catalytically oxidized by GOx in the presence of one molecule of oxygen to D-glucono-δ-lactone, which is then non-enzymatically hydrolyzed with one molecule of water to gluconic acid, and the flavin adenine dinucleotide (FAD) cofactor on the GOx is reduced to FADH2. In the oxidation step, the oxygen molecules oxidize the FADH2 to FAD and H2O2 is produced. Next, HRP in GOx & HRP@SPF30 oxidizes the substrate (diammonium 2,2′-azino-bis(3-ethyl-benzothiazoline-6-sulphonate (ABTS)) in conjunction with H2O2, converting it to the oxidized state (ABTS+•), while producing H2O. Compared with a homogeneous mixture of free enzymes in solution, GOx & HRP@SPF30 showed a 1.5-fold enhancement in catalytic activity for the cascade reactions (Fig. 5c). This improvement was attributed to the reduced distance between GOx and HRP after encapsulation, which minimized the diffusion limitations of intermediates. The encapsulated GOx & HRP@SPF30 remained highly active after 30 days of storage (65% activity retention), while the free GOx & HRP showed poor activity (only 16% activity retention) (Fig. 5d). Moreover, the encapsulated GOx & HRP@SPF30 was easily recovered by centrifugation, retaining > 85% of its activity after 5 cycles (Fig. 5e). These findings further emphasize the importance of matchable aperture encapsulation strategies in cascade reactions.
In summary, a series of tetrahedral four-arm polyisocyanides with controlled Mn and low Mw/Mn was successfully synthesized. Though the terminal the Pd(Ⅱ)-catalyzed Sonogashira cross coupling reaction, Upy was installed onto the chain ends of the four-arm polyisoycnaides. Driven by intermolecular hydrogen bonding between the terminal Upy, well-ordered porous frameworks were successfully obtained (SPFs). SAXS, SEM, and nitrogen adsorption-desorption analysis demonstrated that the prepared SPFs possess ordered crystalline frameworks. The pore size of the SPFs is directly rely on the DP of the polyisocyanide arms. Therefore, by adjusting the DP, it is readily to precisely control the pore size of the SPFs, enabling the preparation of porous frameworks with highly tunable pore sizes. By designing frameworks with pore sizes matching the dimensions of encapsulated enzymes, the presence of these SPFms as exoskeletons significantly enhances the catalytic activity and stability of the enzymes. Furthermore, encapsulating two enzymes within an SPFm with a large pore size enables dual-enzyme cascade reactions. This study offers a method for precisely constructing porous frameworks with highly tunable pore-size and provides a novel exoskeleton material for the encapsulation of enzymes in practical applications.
Runtan Gao: Writing – review & editing, Writing – original draft. Yang Zong: Data curation. Tingting Li: Data curation. Na Liu: Writing – review & editing. Zongquan Wu: Writing – review & editing, Writing – original draft, Funding acquisition.
The National Natural Science Foundation of China (NSFC, Nos. 92256201, 52273006, 22071041, 92356302, and 21971052) and Natural Science Foundation of Jilin Province (No. 20240101181JC) are gratefully appreciated for financial the supports. This work was also supported by the User Experiment Assist System of Shanghai Synchrotron Radiation Facility (SSRF).
Supplementary material associated with this article can be found, in the online version, at doi:
J. Wei, Z. Sun, W. Luo, et al., J. Am. Chem. Soc. 139 (2017) 1706–1713. doi: 10.1021/jacs.6b11411
J. Han, J. Feng, J. Kang, et al., Science 383 (2024) 1014–1019. doi: 10.1126/science.adk8680
J. Dong, Y. Liu, Y. Cui, Acc. Chem. Res. 54 (2021) 194–206. doi: 10.1021/acs.accounts.0c00604
X. Zhao, P. Pachfule, S. Li, et al., J. Am. Chem. Soc. 141 (2019) 6623–6630. doi: 10.1021/jacs.9b01226
E. Altay, D. Nykypanchuk, J. Rzayev, ACS Nano 11 (2017) 8207–8214. doi: 10.1021/acsnano.7b03214
X. Cao, Y. Jin, H. Wang, et al., Chin. Chem. Lett. 35 (2024) 109201. doi: 10.1016/j.cclet.2023.109201
C. Yao, J. Tian, H. Wang, et al., Chin. Chem. Lett. 28 (2017) 893–899. doi: 10.1016/j.cclet.2017.01.005
S.E. Neumann, J. Kwon, C. Gropp, et al., Science 383 (2024) 1337–1343. doi: 10.1126/science.adf2573
P.H.J. Kouwer, P. de Almeida, O. ven den Boomen, et al., Chin. Chem. Lett. 29 (2018) 281–284. doi: 10.1016/j.cclet.2017.11.002
N. Liu, L. Zhou, Z.Q. Wu, Acc. Chem. Res. 54 (2021) 3953–3967. doi: 10.1021/acs.accounts.1c00489
E. Yashima, K. Maeda, H. Iida, Y. Furusho, K. Nagai, Chem. Rev. 109 (2009) 6102–6211. doi: 10.1021/cr900162q
C. Du, X. Zhu, C. Yang, M. Liu, Angew. Chem. Int. Ed. 61 (2022) e202113979. doi: 10.1002/anie.202113979
L. Zhou, K. He, S.M. Kang, et al., Angew. Chem. Int. Ed. 62 (2023) e202310105. doi: 10.1002/anie.202310105
H. He, W. Tan, J. Guo, et al., Chem. Rev. 120 (2020) 9994–10078. doi: 10.1021/acs.chemrev.0c00306
S. Chakrabarty, E.O. Romero, J.B. Pyser, J.A. Yazarians, A.R.H. Narayan, Acc. Chem. Res. 54 (2021) 1374–1384. doi: 10.1021/acs.accounts.0c00810
S. Tang, A.T. Beattie, L. Kafkova, et al., Nature 602 (2022) 701–707. doi: 10.1038/s41586-022-04414-9
J. Shi, Y. Wu, S. Zhang, et al., Chem. Soc. Rev. 47 (2018) 4295–4313. doi: 10.1039/c7cs00914c
S. Qiao, H. Jin, A. Zuo, Y. Chen, Acc. Chem. Res. 57 (2024) 93–105. doi: 10.1021/acs.accounts.3c00565
W. Xu, Y. Wu, W. Gu, et al., Chem. Soc. Rev. 53 (2024) 137–162. doi: 10.1039/D3CS00767G
K.Y. Wang, J. Zhang, Y.C. Hsu, et al., Zhou, Chem. Rev. 123 (2023) 5347–5420. doi: 10.1021/acs.chemrev.2c00879
J. Qiao, L. Liu, J. Shen, L. Qi, Chin. Chem. Lett. 32 (2021) 3195–3198. doi: 10.1016/j.cclet.2021.03.021
M. Wang, S. Fa, G. Zhang, J. Yu, Q. Zhang, Small 19 (2023) 2304957. doi: 10.1002/smll.202304957
J. Zhou, Z. Su, M. Wang, et al., Chem. Eng. J. 399 (2020) 125767. doi: 10.1016/j.cej.2020.125767
S.D. Diwakara, W.S.Y. Ong, Y.H. Wijesundara, et al., J. Am. Chem. Soc. 144 (2022) 2468–2473. doi: 10.1021/jacs.1c12020
R.T. Gao, S.Y. Li, Y. Zong, et al., Angew. Chem. Int. Ed. 63 (2024) e202410010. doi: 10.1002/anie.202410010
Y. Zhang, C. Xing, Z. Mu, et al., J. Am. Chem. Soc. 145 (2023) 13469–13475. doi: 10.1021/jacs.3c04183
R. Gao, X. Kou, L. Tong, et al., Angew. Chem. Int. Ed. 63 (2024) e202319876. doi: 10.1002/anie.202319876
W. Liang, F. Carraro, M.B. Solomon, et al., J. Am. Chem. Soc. 141 (2019) 14298–14305. doi: 10.1021/jacs.9b06589
C. Xing, P. Mei, Z. Mu, et al., Angew. Chem. Int. Ed. 61 (2022) e202201378. doi: 10.1002/anie.202201378
A. Kumar Mahato, S. Pal, K. Dey, et al., J. Am. Chem. Soc. 145 (2023) 12793–12801. doi: 10.1021/jacs.3c03562
Q. Sun, C.W. Fu, B. Aguila, et al., J. Am. Chem. Soc. 140 (2018) 984–992. doi: 10.1021/jacs.7b10642
H. Liu, Y. Zhou, J. Guo, et al., J. Am. Chem. Soc. 145 (2023) 23227–23237. doi: 10.1021/jacs.3c07904
S. Paul, M. Gupta, K. Dey, et al., Chem. Sci. 14 (2023) 6643–6653. doi: 10.1039/d3sc01367g
J.L. Lin, Z.K. Wang, Z.Y. Xu, et al., J. Am. Chem. Soc. 142 (2020) 3577–3582. doi: 10.1021/jacs.9b13263
G. Lin, H. Ding, R. Chen, et al., J. Am. Chem. Soc. 139 (2017) 8705–8709. doi: 10.1021/jacs.7b04141
E. Elacqua, K.B. Manning, D.S. Lye, et al., J. Am. Chem. Soc. 139 (2017) 12240–12250. doi: 10.1021/jacs.7b06201
P. Sun, B. Qin, J.F. Xu, X. Zhang, Prog. Polym. Sci. 124 (2022) 101486. doi: 10.1016/j.progpolymsci.2021.101486
E.R. Draper, D.J. Adams, Nat. Mater. 23 (2024) 13–15. doi: 10.1038/s41563-023-01765-0
Y. Liu, L. Wang, L. Zhao, et al., Chem. Soc. Rev. 53 (2024) 1592–1623. doi: 10.1039/d3cs00705g
Y. Zhou, L. Kan, J.F. Eubank, et al., Chem. Sci. 10 (2019) 6565–6571. doi: 10.1039/c9sc00290a
F. Lin, S.B. Yu, Y.Y. Liu, et al., Adv. Mater. 34 (2022) 2200549. doi: 10.1002/adma.202200549
Q. Li, J.D. Sun, B. Yang, et al., Chin. Chem. Lett. 33 (2022) 1988–1992. doi: 10.1016/j.cclet.2021.10.017
K. Yang, Q. Li, S. Tian, et al., J. Am. Chem. Soc. 146 (2024) 10699–10707. doi: 10.1021/jacs.4c00541
J. Verjans, R. Hoogenboom, Prog. Polym. Sci. 142 (2023) 101689. doi: 10.1016/j.progpolymsci.2023.101689
A.J.P. Teunissen, T.F.E. Paffen, G. Ercolani, T.F.A. de Greef, E.W. Meijer, J. Am. Chem. Soc. 138 (2016) 6852–6860. doi: 10.1021/jacs.6b03421
Z. Zhang, Y. Ye, S. Xiang, B. Chen, Acc. Chem. Res. 55 (2022) 3752–3766. doi: 10.1021/acs.accounts.2c00686
L. Yue, S. Wang, D. Zhou, et al., Nat. Commun. 7 (2016) 10742. doi: 10.1038/ncomms10742
X.H. Xu, Y.X. Li, L. Zhou, N. Liu, Z.Q. Wu, Chem. Sci. 13 (2022) 1111–1118. doi: 10.1039/D1SC05361B
S.B. Bankar, M.V. Bule, R.S. Singhal, L. Ananthanarayan, Biotechnol. Adv. 27 (2009) 489–501. doi: 10.1016/j.biotechadv.2009.04.003

Figure 2 (a) SEC curves of FA-poly-1m-Pd(Ⅱ)s. (b) Plot of Mn and Mw/Mn of FA-poly-1m-Pd(Ⅱ)s against the DP of polyisocyanide link. (c) DLS curves of SPFms (0.2 mg/mL, CHCl3). Insets: Tyndell effects of SPF10 (left side) and FA-poly-110-Pd(Ⅱ) (right side) in CHCl3 (2.0 mg/mL). (d) Solution-phase synchrotron SAXS profiles of SPF10 in CHCl3. (e) Plot of d-spacing (SAXS) vs. the DP of polyisocyanide link. (f) Nitrogen sorption isotherms of SPF10 (insets: pore size distribution). (g) Plots of the SBET and dpore, BJH of SPFms vs. the DP of the polyisocyanide backbone. TEM image of SPF10 (h) and SPF20 (i) casted from the CHCl3 solutions (insets: partial enlarged image).

Figure 3 SEM images of SPF10 (a) (insets: partial enlarged image), SPF20 (b), SPF30 (c) and SPF40 (d) casted from the CHCl3 solutions.

Figure 4 (a) Schematic diagram for encapsulating enzyme into SPFms for catalysis. (b) Encapsulations of TL into SPFms with different apertures. (c) The catalytic activities of pNPGlc catalyzed by TL and TL@SPFms. (d) Lineweaver−Burk plot of n-NPGlc concentrations vs. reaction velocity for TL and TL@SPF10. (e) The relative hydrolysis activity of TL and TL@SPFms towards p-NPD and p-NPP. (f) Relative stability of TL and TL@SPF10 in storage at room temperature. (g) The catalytic cycles of pNPGlc catalyzed by TL and TL@SPFms.

Figure 5 (a) Mechanism diagram of the catalytic activity for the cascade reactions. (b) Encapsulation of HRP and GOx into the SPFms for catalyzed glucose and ABTS cascade reaction. (c) Relative activity of HRP, GOx, HRP & GOx, and encapsulated HRP@SPF20, GOx@SPF30, and HRP & GOx@SPF30. (d) Relative stability of HRP & GOx and HRP & GOx@SPF30 after storage for catalyzed glucose and ABTS cascade reaction. (e) Catalytic cycles of HRP@GOx and HRP & GOx@SPF30 for catalyzed glucose and ABTS cascade reaction.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:

 下载:
下载: