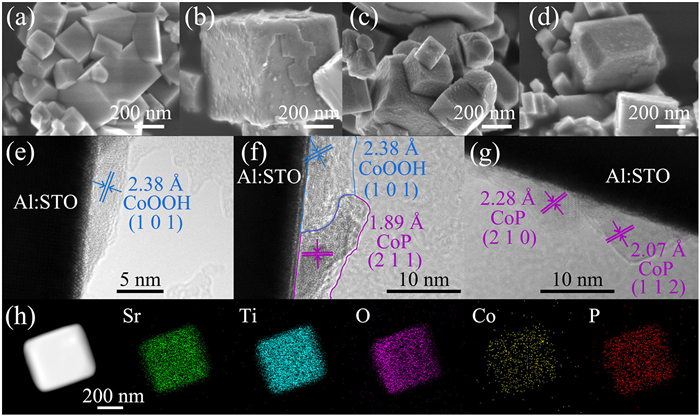
Figure 1.
SEM images of (a) Al: STO, (b) CoOOH/Al: STO, (c) CoP-CoOOH/Al: STO, (d) CoP/Al: STO. TEM images of (e) CoOOH/Al: STO, (f) CoP-CoOOH/Al: STO, (g) CoP/Al: STO. (h) STEM and element mapping images of CoP-CoOOH/Al: STO.
Boosting photocatalytic overall water splitting efficiency and stability with adjacent cobalt-based dual cocatalysts
Li Tian , Xiangjiu Guan , Jingkuo Qu , Anna Dai , Jiaye Cai , Zheng Zhang , Shichao Zong , Liejin Guo

Photocatalytic overall water splitting (POWS), with solar irradiation and water as the only energy and substance input, has emerged as a promising scenario for green H2 production [1-6]. Upon the excitation and separation of charge carriers, the subsequent redox reaction on the surface of photocatalyst represents as one of the essential issues for the efficient water splitting process, with cocatalyst lowering the overpotentials and serving reactive sites [7-9]. Although some excellent examples have been reported, the high efficiency on POWS currently relies on the utilization of noble metal cocatalyst, such as Rh and Pt, which hinders the potential large-scale application [8,10-12].
Cobalt-based cocatalysts, with merits of high electrical conductivity and diverse compositional characteristics, have become ideal choice for low-cost photocatalytic water splitting [13-16]. Specifically, cobalt phosphide (CoP), owing to the metallic properties and potential Schottky effect when combining with semiconductor, has been widely investigated as H2 evolution reaction (HER) cocatalysts [17-19]. Typically, CoP/CdS hybrid photocatalytic system with CoP as cocatalyst exhibited excellent activity and stability for over 100 h in the presence of sacrificial reagents [20]. Our group have demonstrated POWS from Al-doped SrTiO3 (Al: STO) with CoP cocatalyst tightly attached through a photodeposition-phosphorization method [21], yet further improvements in efficiency are still desired from developing novel cocatalytic structure beyond the exclusive role of cocatalyst solely in H2 evolution.
Employing dual cocatalysts respectively for hydrogen and oxygen evolution has emerged as a pivotal approach to enhancing the efficiency of POWS by the spatial separation of charge carriers and active sites [22-26]. As a cost-effective candidate, cobalt oxyhydroxide (CoOOH) has garnered significant attention due to its immense potential to enhance electrocatalytic O2 production [27,28]. In recent years, CoOOH has been introduced into photocatalytic systems [29-31]. For instance, Al: STO co-loaded with CoOOH and Rh/CrOx has achieved near 100% carrier separation efficiency while demonstrating stability over 1000 h [32,33], demonstrating great potential in dual-cocatalyst modified photocatalytic system in POWS when efficient low-cost metals are incorporated.
Even though given the advantages of spatial charge-carrier separation, the traditional dual cocatalysts might encounter some limitations when taken the water molecule adsorption and splitting process on the surface of the cocatalyst into consideration. With spatial separated active sites on photocatalyst, once the H—O bond in water molecule adsorbed on the oxygen evolution reaction (OER) cocatalyst is broken during water oxidation, the produced hydrogen radicals will take a relatively long journey to reach the HER cocatalyst. It could be proposed that a configuration of adjacent dual cocatalysts would be more efficient since it inherits the advantages the charge-carrier separation while providing rapid consumption of reactant species with reduced distance between reduction and oxidation sites [34,35]. However, it is challenging to realize the facile loading of adjacent dual cocatalysts while simultaneously ensuring the well dispersion and intimate contact of these cocatalysts on the surface of photocatalyst.
In this contribution, using Al: STO as probe photocatalyst, we report on modification of adjacent cobalt-based dual cocatalysts for efficient POWS without noble metal. By controlling the phosphorization process of CoOOH deposited on Al: STO, highly dispersed CoP-CoOOH could be obtained as adjacent dual cocatalysts, which contribute to the boosted efficiency and stability of POWS. Through structural characterizations combined with spectra technologies, it is revealed that the adjacent CoP-CoOOH dual cocatalysts both facilitate charge separation and inhibit potential photocorrosion of CoP during POWS process. This work not only provides novel strategy for efficient low-cost cocatalysts but also lays a solid foundation for advancing the development and application of photocatalysis technology.
Fig. S1 (Supporting information) briefly illustrates the synthesis process of CoP-CoOOH/Al: STO. With the assistance of oxidative environment provided by NaIO4 under irradiation of ultraviolet light, Co2+ ions in the solution were oxidized into Co3+ species (characterized as CoOOH in the following discussion) deposited on the surface of Al: STO, and were then partially converted into cobalt phosphide by controlling the duration of the following phosphorization process. The detailed synthesis procedure and characterization methods were introduced in Texts S1-S4 (Supporting information).
Fig. S2a (Supporting information) and Fig. 1 exhibit the crystal structure and the morphological characteristics of as-prepared samples. As shown in the X-ray diffraction (XRD) patterns in Fig. S2 (Supporting information), the crystal structure of Al: STO remained unchanged after loading Co species. The Co species, due to their low loading amount, were not observed in XRD patterns. Scanning electron microscope (SEM) images in Figs. 1a and b show the microstructures of the samples before and after loading Co species. It could be seen that Al: STO presented a cubic structure with smooth surface. After Co was loaded, a large number of nanoparticles were uniformly distributed on the surface of Al: STO, indicating the successful loading of cobalt species. The high-resolution transmission electron microscopy (HRTEM) image in Fig. 1e demonstrates that the Co species were in close contact with the surface of Al: STO, with the lattice (2.38 Å) of the Co species identified as the (101) plane of CoOOH [36,37], confirming that the Co species loaded onto the Al: STO surface were in the form of CoOOH, designated as CoOOH/Al: STO.
Then, CoOOH/Al: STO was phosphorized for 2 h in a tubular furnace. As shown in Fig. S2a, the phosphorization process did not change the crystal structure of Al: STO. As observed from the SEM image in Fig. 1c, the morphology of the sample after phosphorization was consistent with that before phosphorization, indicating that the phosphorization process did not alter the uniformity of the size and distribution of the cocatalyst nanoparticles. From the HRTEM image in Fig. 1f, a new lattice corresponding to the (211) plane of CoP (1.89 Å) could be observed [38,39], while the lattice of CoOOH was also presented, which indicated that CoOOH was partially converted to CoP during the phosphorization process. Furthermore, CoP was in close contact with both CoOOH and Al: STO. Thus, the as-prepared sample was confirmed as adjacent CoP and CoOOH co-loaded on Al: STO, designated as CoP-CoOOH/Al: STO. Fig. 1h shows the STEM image of CoP-CoOOH/Al: STO and its elemental mapping, explicitly demonstrating the uniform loading of Co and P elements on the surface of Al: STO.
When phosphorization time was increased to 5 h, it could be observed from the SEM image (Fig. 1d) that the cocatalyst maintained dispersed nanoparticle morphology on the surface of Al: STO photocatalyst. In the HRTEM image (Fig. 1g), only lattice fringes corresponding to the (201) and (112) crystal planes of CoP were observed [40,41], while the lattice fringes of CoOOH could not be observed, indicating that CoOOH had been completely converted to CoP after phosphorization for 5 h. Therefore, the sample was confirmed CoP-modified Al: STO, designated as CoP/Al: STO.
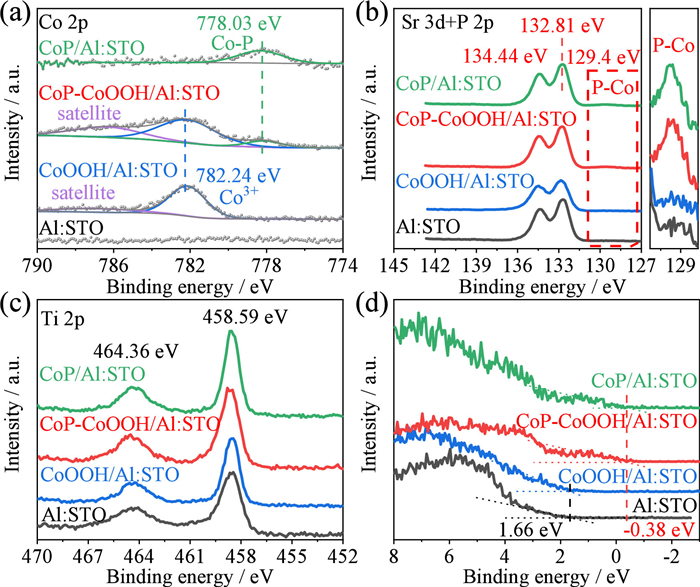
The elemental chemical states of as-prepared samples were characterized by X-ray photoelectron spectroscopy (XPS) to further determine the structure and composition. As shown in the survey scan XPS in Fig. S3 (Supporting information), Al: STO exhibited typical peaks attributed to Sr, Ti, and O elements in SrTiO3, additional peaks ascribed to Co and P were distinctly observed in CoP-CoOOH/Al: STO, conclusively demonstrating the successful introduction of both Co and P. The high-resolution XPS of Co, P, Sr, and Ti were further characterized. As shown in Co 2p spectra in Fig. 2a, a binding energy of 782.24 eV observed both in CoOOH/Al: STO and CoP-CoOOH/Al: STO indicated the presence of Co3+ in CoOOH [42,43], while the Co 2p orbital of CoP-CoOOH/Al: STO exhibited a new peak at 778.03 eV attributed to Co-P in CoP, which is further demonstrated by the Co 2p orbital of CoP/Al: STO [21]. It has been reported that Co3+ could serve as active sites for the OER [43], suggesting that CoP-CoOOH dual cocatalyst could facilitate both HER and OER process in POWS. In further analysis, the Co 2p orbital of CoP/Al: STO only exhibited a major peak of Co-P, without detecting any characteristic peaks attributed to Co3+ of CoOOH, unequivocally indicated that CoOOH had been completely converted into CoP. Subsequently, the high-resolution Sr 3d and P 2p spectra of as-prepared samples were presented in Fig. 2b. It was evident that two peaks attributed to Sr 3d were located at 134.44 and 132.81 eV. Additionally, a peak at 129.4 eV attributed to the P-Co bond in CoP was observed in CoP-CoOOH/Al: STO and CoP/Al: STO, while was absent in CoOOH/Al: STO [21,44]. Accordingly, Ti 2p spectra of the as-prepared samples (Fig. 2c) showed two main peaks of Ti 2p3/2 and Ti 2p1/2 components at 464.36 and 458.59 eV, respectively. Energy dispersive X-ray spectroscopy (EDS) of SEM was used to characterize the atomic ratio of P and Co in the samples with different phosphorization time. The results in Table S1 (Supporting information). further indicated that the unphosphorized sample of CoOOH/Al: STO does not contain phosphorus. With the increment of phosphorization time, the atomic ratio of phosphorus to cobalt gradually increased, indicating that CoOOH was gradually converted to CoP. When the phosphorization time reached 5 h, the atomic ratio of phosphorus to cobalt was approximately 1, demonstrating the complete conversion of CoOOH into CoP. This phenomenon is further supported by the XPS valence band (VB) analysis, as depicted in Fig. 2d. Al: STO and CoOOH/Al: STO exhibited a distinct VB edge positioned at approximately 1.66 eV (vs. the Fermi level), corresponding to the valence band maximum of Al: STO [21]. Notably, the CoP-CoOOH/Al: STO and CoP/Al: STO exhibited a VB edge located at −0.38 eV below the Fermi level, attributed to the unique metallic character of CoP.
As such, successful deposition of adjacent CoP-CoOOH dual cocatalysts was demonstrated, and composition of cocatalyst could be facilely tailored by controlling of phosphorization process. This characteristic was further corroborated by the high absorption coefficient of CoP-CoOOH/Al: STO in the ultraviolet-to-visible light region (Fig. S4 in Supporting information). Upon a detailed analysis of the UV–vis spectra, Al: STO exhibited an absorption edge of approximately 390 nm, corresponding to a bandgap energy of approximately 3.19 eV for SrTiO3, while the weak absorption in visible light region was ascribed to the formation of oxygen vacancy by Al-doping. Upon the loading of CoOOH and CoP, the absorption intensity beyond the absorption edge significantly increased (Fig. S4a), and was observed to be elevated with further increment of CoP-CoOOH content (Fig. S4b). It was noteworthy that the absorption edge remained almost identical to that of pure Al: STO, suggesting that CoP-CoOOH adjacent dual cocatalysts were primarily loaded on the surface of Al: STO without penetrating deeply into its crystal lattice, hence maintaining the inherent high crystallinity and excellent bulk charge separation properties of Al: STO for potentially efficient POWS.
The POWS performance was evaluated and shown in Fig. 3. As could be seen from Fig. 3a, the loading of CoP-CoOOH had a significant impact on the POWS performance of Al: STO. Neither pristine Al: STO nor CoOOH/Al: STO exhibited activity for POWS since no HER sites were provided. In contrast, Al: STO loaded with CoP-CoOOH displayed the highest activity for POWS, with H2 and O2 evolution rates of 4.86 and 2.30 mmol h-1 g-1, respectively. The AQY of 12.7% at 350 ± 10 nm surpasses most of recently reported results on POWS without noble metal. Detailed information regarding the calculation of AQY was supplied in Table S2 (Supporting information), and the recently reported results were summarized in Table S3 (Supporting information). However, inferior activity was observed when CoOOH was completely converted into CoP, with no CoOOH left as OER sites. The synergistic loading of CoP and CoOOH as dual cocatalysts could significantly enhance the activity, thereby achieving efficient POWS. Comparison activity (Fig. S5 in Supporting information) further demonstrated the advantage of adjacent dual-cocatalysts.
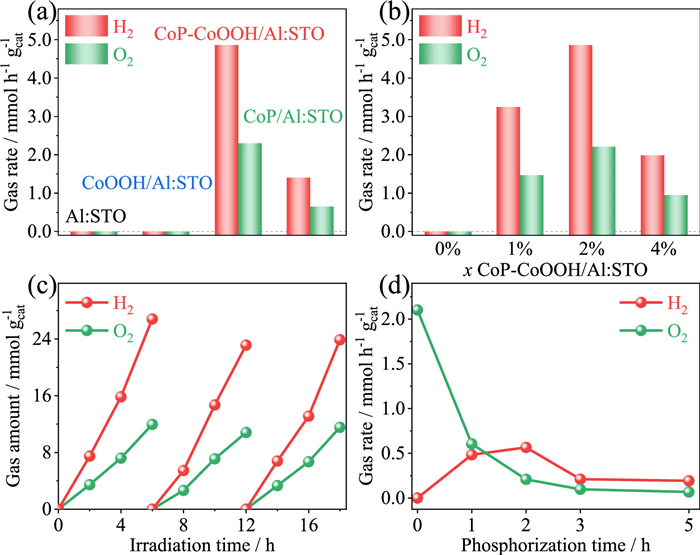
The impact of cocatalyst loading amounts on the performance of POWS was investigated (Fig. 3b). Initially, pristine Al: STO was unable to split water due to the lack of active sites. However, upon loading with CoP-CoOOH, the POWS performance of Al: STO was noticeably enhanced, which can be attributed to CoP-CoOOH serving as a cocatalyst that provides sufficient reactive sites and effectively promotes the redox reactions. Optimized POWS performance was observed when loading adjacent CoP-CoOOH dual-cocatalysts with 2 wt% Co, while further increasing the loading amount of CoP-CoOOH led to a decrease in the POWS performance of CoP-CoOOH/Al: STO, which could be ascribed to excessive surface loading of CoP shielding the light absorption of Al: STO [21]. The stability of CoP-CoOOH/Al: STO was tested, and the results are shown in Fig. 3c, demonstrating excellent stability with no significant decrease in POWS activity during 18 h test.
To identify the distinct roles of CoP and CoOOH in POWS, the photocatalytic H2 and O2 evolution tests were conducted on cobalt-based cocatalysts modified Al: STO with different phosphorization time (Fig. 3d). The O2-evolution rate exhibited a downward trend with the extension of phosphorization time, while the H2-evolution performance experienced a process of initially increasing and then decreasing. From the perspective of O2-evolution performance, the unphosphorized sample, which was Al: STO loaded with CoOOH, exhibited the highest O2-evolution performance. This suggested that CoOOH played a crucial role of OER cocatalyst. However, with increased phosphorization time, the O2-evolution performance started to decrease since CoOOH was gradually converted into CoP upon phosphorization. For the trend of H2-evolution performance, the unphosphorized sample did not exhibit significant H2 evolution ability. With increased phosphorization time, the H2-evolution performance gradually improved, indicating that the as-converted CoP during phosphorization process served as effective HER cocatalyst. When the phosphorization time exceeded 3 h, the H2 and O2 evolution performances remained stable and no significant changes were observed. This may be due to the nearly complete conversion of CoOOH to CoP.
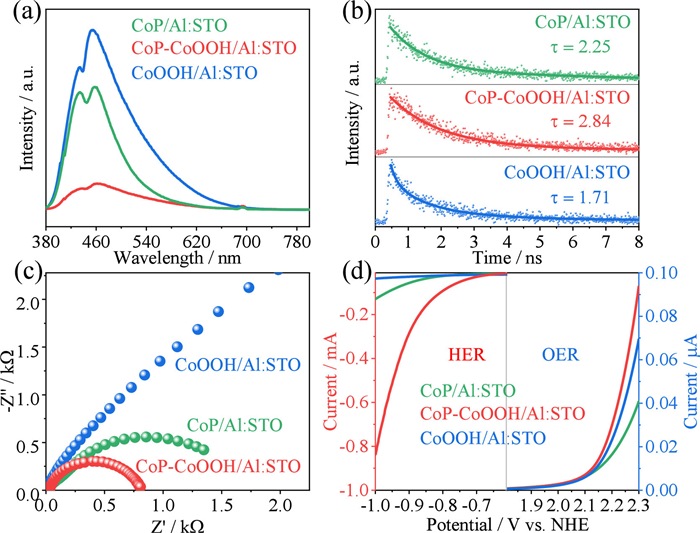
Detailed characterizations of charge carrier transport performance for the CoOOH/Al: STO, CoP-CoOOH/Al: STO and CoP/Al: STO were conducted (Fig. 4), so as to highlight the significant advantages of the dual cocatalyst on enhancing the photocatalytic activity of Al: STO. Through steady-state photoluminescence (PL) and time-resolved PL (TR-PL) spectral analysis (Figs. 4a and b), it was observed that all as-prepared samples exhibited distinct emission peaks at approximately 450 nm. Specifically, the emission peak intensity of CoP-CoOOH/Al: STO was significantly lower than that of CoOOH/Al: STO and CoP/Al: STO, directly reflecting that dual-cocatalyst loading could significantly reduce the carrier recombination rate [45]. Furthermore, through TR-PL spectral fitting, the average carrier lifetime (τav = 2.84 ns) of the CoP-CoOOH/Al: STO photocatalyst was longer than that of both CoOOH/Al: STO and CoP/Al: STO, promising sufficient redox reaction of water splitting on the surface of photocatalyst. Electrochemical impedance spectroscopy (EIS) analysis further revealed the advantages of dual cocatalysts in charge transportation (Fig. 4c). Compared to CoOOH/Al: STO and CoP/Al: STO, CoP-CoOOH/Al: STO exhibited the smallest semicircle in EIS spectra, indicating that dual-cocatalyst loading can substantially diminish carrier transport resistance, facilitate the migration of photogenerated carriers, and further contribute to the enhanced photocatalytic efficiency [46,47]. Furthermore, as shown in Fig. 4d, the linear sweep voltammetry (LSV) curves of HER and OER indicated that the overpotentials for H2 and O2 evolution of CoP-CoOOH/Al: STO were lower than those of CoOOH/Al: STO and CoP/Al: STO. This could be attributed to both accelerated HER and OER processes on the surface of photocatalyst with the assistance of dual cocatalysts, which in turn facilitated separation and migration of charge carriers.
Further analysis was conducted to delve into the role of the interfacial contact between CoP and CoOOH in the CoP-CoOOH dual-cocatalyst system for enhancing the performance of OWS. As depicted in Fig. 5a, the work function of metallic CoP (5.58 eV) was larger than that of CoOOH (5.2 eV) [48]. This difference drove the flow of electrons from CoOOH to CoP upon contact (Fig. 5b), thereby constructing a Schottky junction at the interface [49,50]. The unique characteristic of this Schottky junction was its ability to unilaterally facilitate electron transfer from CoOOH to CoP, effectively preventing reverse migration. This phenomenon inhibited the recombination of photogenerated carriers, thus enhancing the separation efficiency.
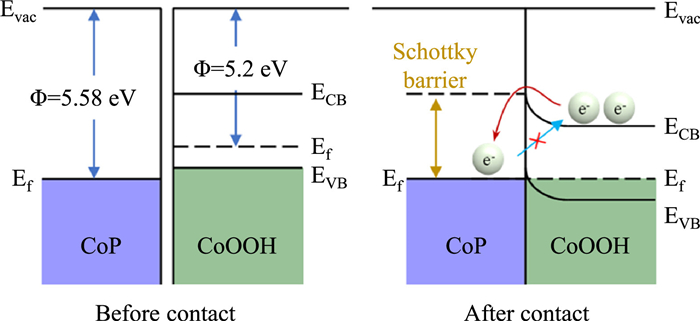
Furthermore, it was noteworthy that phosphorus elements were susceptible to photocorrosion in photocatalytic reactions [51], resulting in the relatively poor stability of CoP cocatalysts without hole scavengers. As shown in Fig. S6 (Supporting information), the activity of CoP/Al: STO decreased significantly during a 9-h test. Interestingly, when CoP was combined with CoOOH to form the CoP-CoOOH adjacent dual-cocatalyst structure, the as-established Schottky junction facilitated the holes flowing to CoOOH rather than CoP, thus effectively preventing the photocorrosion of CoP caused by photogenerated holes. Consequently, the activity of CoP-CoOOH/Al: STO remained unchanged during a continuous 18-h test (Fig. 3c). The synergistic effect of CoP and CoOOH enabled the POWS reaction to proceed efficiently, providing new insights for the design of more efficient photocatalytic systems.
Different loading methods of cocatalysts can significantly affect their distribution [26,52]. To deeply explore the superiority of in-situ photo-assisted chemical oxidation technology, a systematic comparison was made of CoP-CoOOH loaded by various methods and the results were displayed in Figs. S7 and S8 (Supporting information). As shown in Fig. S7, the loading of cocatalysts did not alter the crystal structure of Al: STO and neither Co nor P species were detected due to their low loading amounts. Fig. S8a presents the distribution of CoP-CoOOH/Al: STO cocatalyst synthesized using in-situ photo-assisted chemical oxidation technology with NaIO4 as an oxidization sacrificial agent, showing a uniform distribution of nanoparticles. Figs. S8b and c show the distribution of the 2 wt% cocatalysts loaded by a photodeposition method without a sacrificial agent and an impregnation method, respectively. Obviously, both of them exhibited significant agglomeration, sometimes even covering the entire surface of Al: STO. This distribution not only increased the light-shielding effect on the photocatalyst and reduced the utilization efficiency of light energy, but also reduced utilization efficiency of the cocatalyst.
In addition, the uniform distribution of cocatalysts loaded via NaIO4-assisted in-situ photo-assisted chemical oxidation method (Fig. S8a) effectively reduced the light-shielding effect on the photocatalyst and Optimized utilization efficiency of the cocatalyst and elevated density of reaction sites. Based on the above advantages, it was not difficult to conclude that the POWS H2 production performance of the sample loaded by the in-situ photo-assisted chemical oxidation method was higher than that of the samples loaded with other methods, as shown in Fig. S8d.
Based on previous characterization and analysis, the possible mechanism of the excellent POWS performance on CoP-CoOOH/Al: STO was proposed and depicted in Fig. 6. By utilizing in-situ photo-assisted oxidation technology, CoOOH nanoparticles were uniformly deposited, and adjacent CoP-CoOOH dual cocatalysts modified Al: STO was achieved by the controlled phosphorization process. Dual cocatalysts help to efficiently separate charge carriers and promote water splitting reaction on the surface of photocatalyst by promoting both HER and OER process. Particularly, in comparison with conventional separated dual-cocatalyst, the special structure of adjacent CoP-CoOOH dual cocatalysts could shorten the migration distance of carriers, hence reducing the possibility of carrier recombination during migration. On the other hand, the adjacent dual-cocatalyst structure could help to shorten the transfer pathway of proton when compared to conventional separated dual cocatalysts. Besides, during POWS process, the Schottky junction constructed at the CoP-CoOOH interface allows electron transfer from CoOOH to CoP, which guarantee the gathering of electrons and holes respectively on CoP and CoOOH, hence significantly inhibiting the recombination and prolonging the lifetime of charge carriers as well as preventing CoP from being corroded by photogenerated holes. Consequently, the CoP-CoOOH/Al: STO system exhibits exceptional POWS activity and stability.
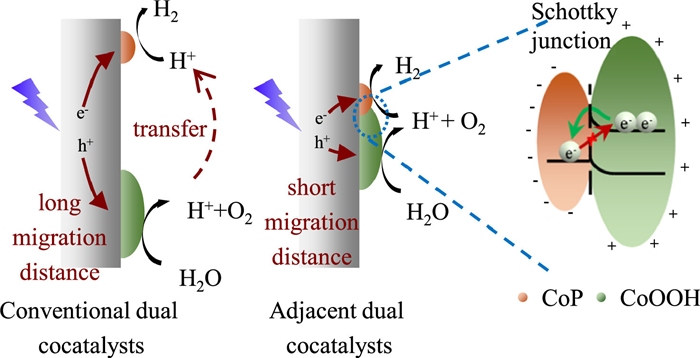
In summary, adjacent CoP-CoOOH dual cocatalysts are loaded via in-situ photo-assisted oxidation deposition method followed by controlled phosphorization, which enables the uniform deposition of cocatalyst nanoparticles on the surface of Al: STO photocatalyst. Besides inheriting the conventional merits of dual cocatalysts that simultaneously promote HER and OER, the unique adjacent structure of CoP-CoOOH dual cocatalysts significantly reduces the distance of photogenerated charge carriers migrating from bulk to the surface and the distance of proton transfer from oxidation sites to reduction sites. In addition, the formation of Schottky junction at the CoP-CoOOH interface further enhances charge-separation efficiency, prolongs carrier lifetime, and inhibits the photocorrosion of CoP. As a result, the CoP-CoOOH/Al: STO photocatalyst demonstrates exceptional performance in POWS, achieving an H₂ and O2 production rate of 4.86 and 2.30 mmol h-1 g-1, with an AQY of 12.7% at 350 ± 10 nm. This study not only provides a cost-effective and highly efficient strategy for solar energy conversion into chemical energy but also advances the understanding of cocatalysts in photocatalytic systems.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Li Tian: Writing – original draft, Investigation, Formal analysis, Data curation, Conceptualization. Xiangjiu Guan: Writing – review & editing, Supervision, Project administration, Funding acquisition, Formal analysis. Jingkuo Qu: Formal analysis. Anna Dai: Formal analysis. Jiaye Cai: Funding acquisition. Zheng Zhang: Formal analysis. Shichao Zong: Writing – review & editing, Formal analysis. Liejin Guo: Supervision, Resources, Project administration, Funding acquisition.
This work is supported by the National Natural Science Foundation of China (Nos. 52488201, 52376209), the Natural Science Basic Research Program of Shaanxi (No. 2023-JC-QN-0618), the Natural Science Foundation of Sichuan Province (No. 2025ZNSFSC1250), the Shaanxi Postdoctoral Science Foundation (No. 2023BSHYDZZ126), and China Fundamental Research Funds for the Central Universities.
Supplementary material associated with this article can be found, in the online version, at doi:
L. Wang, Y. Zhang, L. Chen, et al., Adv. Mater. 30 (2018) 1801955. doi: 10.1002/adma.201801955
S. Chen, T. Takata, K. Domen, Nat. Rev. Mater. 2 (2017) 17050. doi: 10.1038/natrevmats.2017.50
Z. Wang, Z. Sun, H. Yin, et al., eScience 3 (2023) 100136. doi: 10.1016/j.esci.2023.100136
Z. Wang, C. Li, K. Domen, Chem. Soc. Rev. 48 (2019) 2109–2125. doi: 10.1039/C8CS00542G
L. Tian, X. Guan, Y. Dong, et al., Environ. Chem. Lett. 21 (2023) 1257–1264. doi: 10.1007/s10311-023-01580-8
Y. Ma, Z. Feng, Y. Dong, et al., Chin. Chem. Lett. 36 (2025) 110922. doi: 10.1016/j.cclet.2025.110922
G. Zhao, X. Xu, Nanoscale 13 (2021) 10649–10667. doi: 10.1039/D1NR02464G
H. Zhao, Q. Mao, L. Jian, et al., Chin. J. Catal. 43 (2022) 1774–1804. doi: 10.1016/S1872-2067(22)64105-6
J. Yang, D. Wang, H. Han, C. Li, Acc. Chem. Res. 46 (2013) 1900–1909. doi: 10.1021/ar300227e
J. Ran, J. Zhang, J. Yu, et al., Chem. Soc. Rev. 43 (2014) 7787–7812. doi: 10.1039/C3CS60425J
Z. Wang, J. Fan, B. Cheng, et al., Mater. Today Phys. 15 (2020) 100279. doi: 10.1016/j.mtphys.2020.100279
B. Zhou, S. Sun, Front. Energy 18 (2024) 122–124. doi: 10.1007/s11708-023-0870-z
V. Soni, C. Xia, C.K. Cheng, et al., Appl. Mater. Today 24 (2021) 101074. doi: 10.1016/j.apmt.2021.101074
X. Deng, H. Tüysüz, ACS Catal. 4 (2014) 3701–3714. doi: 10.1021/cs500713d
V. Artero, M. Chavarot-Kerlidou, M. Fontecave, Angew. Chem. Int. Ed. 50 (2011) 7238–7266. doi: 10.1002/anie.201007987
L. Dong, J. Qu, T. Zhang, et al., Chin. Chem. Lett. 36 (2025) 110397. doi: 10.1016/j.cclet.2024.110397
S. Cao, Y. Chen, H. Wang, et al., Joule 2 (2018) 549–557. doi: 10.1016/j.joule.2018.01.007
Y. Li, Y. Li, C. Yang, et al., Appl. Surf. Sci. 639 (2023) 158180. doi: 10.1016/j.apsusc.2023.158180
Q. Liu, J. Huang, H. Tang, et al., J. Mater. Sci. Technol. 56 (2020) 196–205. doi: 10.1016/j.jmst.2020.04.026
S. Cao, Y. Chen, C.J. Wang, et al., Chem. Commun. 51 (2015) 8708–8711. doi: 10.1039/C5CC01799H
S. Zong, L. Tian, X. Guan, et al., J. Colloid Interface Sci. 606 (2022) 491–499. doi: 10.1016/j.jcis.2021.08.049
A. Meng, L. Zhang, B. Cheng, J. Yu, Adv. Mater. 31 (2019) 1807660. doi: 10.1002/adma.201807660
T. Ohno, L. Bai, T. Hisatomi, et al., J. Am. Chem. Soc. 134 (2012) 8254–8259. doi: 10.1021/ja302479f
J. Seo, D. Ishizuka, T. Hisatomi, et al., J. Mater. Chem. A 9 (2021) 8655–8662. doi: 10.1039/D1TA00164G
G. Xue, S. Ping, H. Ce, et al., Energy Mater. 3 (2023) 300016.
L. Tian, X. Guan, S. Zong, et al., Catalysts 13 (2023) 355. doi: 10.3390/catal13020355
Z. Jia, Y. Yuan, Y. Zhang, et al., Carbon Energy 6 (2024) e418. doi: 10.1002/cey2.418
H. Jia, N. Yao, C. Yu, et al., Angew. Chem. Int. Ed. 62 (2023) e202313886. doi: 10.1002/anie.202313886
Y. Yang, Q. Zhang, X. Yang, et al., Energy Fuels 36 (2022) 11457–11466. doi: 10.1021/acs.energyfuels.2c01392
B. Zeng, Q. Zhou, N. Ta, et al., ChemPhotoChem 8 (2024) e202400107. doi: 10.1002/cptc.202400107
L. Zhang, B. Wang, W. Yang, et al., Chin. Chem. Lett. 37 (2026) 111142. doi: 10.1016/j.cclet.2025.111142
T. Takata, J. Jiang, Y. Sakata, et al., Nature 581 (2020) 411–414. doi: 10.1038/s41586-020-2278-9
H. Lyu, T. Hisatomi, Y. Goto, et al., Chem. Sci. 10 (2019) 3196–3201. doi: 10.1039/C8SC05757E
B. Ma, Y. Dang, D. Li, et al., Appl. Catal. B 298 (2021) 120491. doi: 10.1016/j.apcatb.2021.120491
X. Yan, Z. Chen, Y. Yue, et al., Appl. Catal. B 360 (2025) 124527. doi: 10.1016/j.apcatb.2024.124527
W. Wen, D. Liang, J.P. Cheng, J.M. Wu, RSC Adv. 6 (2016) 70947–70951. doi: 10.1039/C6RA14347D
W. Guo, C. Yu, S. Li, et al., Adv. Mater. 31 (2019) 1901241. doi: 10.1002/adma.201901241
R. Beltrán-Suito, P.W. Menezes, M. Driess, J. Mater. Chem. A 7 (2019) 15749–15756. doi: 10.1039/C9TA04583J
C.C. Zhang, S. Wei, L.X. Sun, et al., Rare Met. 43 (2024) 1095–1107. doi: 10.1007/s12598-023-02494-8
S. Wei, W. Yongjing, X. Kun, et al., Acta Phys. Chim. Sin. 40 (2024) 2308015. doi: 10.3866/PKU.WHXB202308015
W. Guo, Z. Liang, J. Zhao, et al., Small Methods 2 (2018) 1800204. doi: 10.1002/smtd.201800204
K. Zhang, T.H. Lee, J.H. Cha, et al., ACS Omega 4 (2019) 21410–21416. doi: 10.1021/acsomega.9b03104
S. Song, H. Bao, X. Lin, et al., J. Energy Chem. 42 (2020) 5–10. doi: 10.1016/j.jechem.2019.05.021
H. Luo, X. Zhang, H. Zhu, et al., J. Mater. Sci. Technol. 166 (2023) 164–172. doi: 10.1016/j.jmst.2023.05.028
C. Cao, J. Li, L. Zhang, et al., J. Mater. Sci. Technol. 214 (2025) 180–193. doi: 10.1016/j.jmst.2024.06.038
B. Li, W. Guo, X.F. Lu, et al., Mater. Rep. : Energy 3 (2023) 100230.
B. Wang, B. An, X. Li, S. Shen, Front. Energy 18 (2024) 101–109. doi: 10.1007/s11708-023-0894-4
H. Wang, Y. Zhou, S. Tao, Appl. Catal. B 315 (2022) 121588. doi: 10.1016/j.apcatb.2022.121588
J. Zhang, J. Liu, Z. Meng, et al., J. Mater. Sci. Technol. 159 (2023) 1–9.
M. Zhang, Y. Zhang, L. Tang, et al., Appl. Catal. B 295 (2021) 120278. doi: 10.1016/j.apcatb.2021.120278
X. Ren, D. Philo, Y. Li, et al., Coord. Chem. Rev. 424 (2020) 213516. doi: 10.1016/j.ccr.2020.213516
T. Kawawaki, Y. Kataoka, S. Ozaki, et al., Chem. Commun. 57 (2021) 417–440. doi: 10.1039/D0CC06809H

Figure 1 SEM images of (a) Al: STO, (b) CoOOH/Al: STO, (c) CoP-CoOOH/Al: STO, (d) CoP/Al: STO. TEM images of (e) CoOOH/Al: STO, (f) CoP-CoOOH/Al: STO, (g) CoP/Al: STO. (h) STEM and element mapping images of CoP-CoOOH/Al: STO.

Figure 2 High-resolution XPS spectra of (a) Co 2p, (b) Sr 3d and P 2p, (c) Ti 2p orbitals, and (d) VB edge of as-prepared samples.

Figure 3 (a) POWS of Al: STO with different cocatalysts. (b) POWS on xCoP-CoOOH/Al: STO (x refer the mass ratio of Co to Al: STO). (c) Time course of POWS on CoP-CoOOH/Al: STO. (d) H2 and O2 evolution rate on cobalt-based cocatalysts modified Al: STO with different phosphorized time (sacrificial agent: 20 vol% CH3OH for H2 evolution, 20 mmol/L NaIO3 for O2 evolution).

Figure 4 (a) PL spectra, (b) TR-PL spectra, (c) EIS curves, and (d) LSV curves of HER and OER on as-prepared samples.

Figure 5 Energy diagrams of CoP and CoOOH. The Evac, Ef, ECB, EVB and Φ represent potentials of vacuum level, Fermi level, conduction band, valence band and work function, respectively.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:

 下载:
下载: