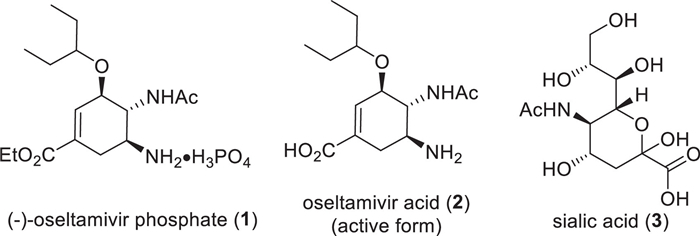
Figure 1.
Structures of (–)-oseltamivir phosphate (1), its active form oseltamivir acid (2) and sialic acid (3).

Seasonal influenza (the flu) is an acute respiratory infection caused by influenza viruses and remains a major worldwide health problem. Annually, the influenza has resulted in approximately 1 billion diagnosed cases of infection and >290 thousand related deaths [1]. (–)-Oseltamivir phosphate (1, Tamiflu, Fig. 1) is the water-soluble and orally bioavailable prodrug of the corresponding potent and selective inhibitor oseltamivir acid (2) of influenza neuraminidases A and B [2]. The acid mimics the transition state of the reaction catalyzed by the neuraminidase of the influenza virus, preventing the cleavage of sialic acid (3) from the glycoprotein during the viral replication process and inhibiting the spread of viruses to neighboring cells [3]. (–)-Oseltamivir phosphate (1) was developed by Gilead Science[4] and Hoffmann Roche [5] and approved by the US Food and Drug Administration (FDA) in 1999 for the treatment and prevention of both type A and type B human influenza. It has subsequently been included on the World Health Organization (WHO) Model List of Essential Medicines [6].
Nowadays, the industrial manufacture of (–)-oseltamivir phosphate (1) relies on the Roche’s semisynthetic approach utilizing naturally occurring (–)-shikimic acid as the starting material [5,7] which is obtained either by extraction of star anise or by a fermentation of genetically engineered Escherichia coli strain [8,9]. Numerous efforts have been devoted to improve the synthesis [10,11], resulting in some highly efficient and innovative routes [12–21]. Several different strategies have been developed for synthesizing the carbocyclic core structure of (–)-oseltamivir phosphate (1). The substituted cyclohexene can be obtained through selective functionalization of the carbocycle from commercial supplies [16,17,20], or prepared by bottom-up synthesis, including Diels-Alder reaction [12,15,19,21], intramolecular condensation reactions (Fig. S1 in Supporting information) [13,14,18]. Herein we present a concise and new asymmetric synthesis of (–)-oseltamivir phosphate (1) by employing a biphasic Pd-catalyzed 6-endo Heck reaction to form the carbocycle.
Our retrosynthetic analysis of (–)-oseltamivir phosphate (1) is depicted in Scheme 1. We envisioned that the target molecule 1 could be accessed from the advanced bicyclic oxazine 4 through a three-step sequence including Cu(OTf)2-mediated ring-opening etherification, deprotection and salt formation. The bicyclic core of 4 would be available by a regio-and diasteroselective hetero-Diels-Alder reaction of the chiral cyclohexadienyl amine 5 with the acyl nitroso dienophile. To access chiral cyclohexadienyl amine 5, a challenging Pd-catalyzed regioselective intramolecular Heck-type cyclization of (Z)-β-iodo homoallylic amine 6 would need to be addressed. (Z)-β-iodo homoallylic amine 6 would be derived from the asymmetric allylation of the chiral (Z)-N-sulfinyl imine 7 and Zn reagent, in situ derived from Zn and allyl bromide derivative. Chiral imine 7 could be prepared through a series of functional group transformations from the commercially available ethyl propiolate.
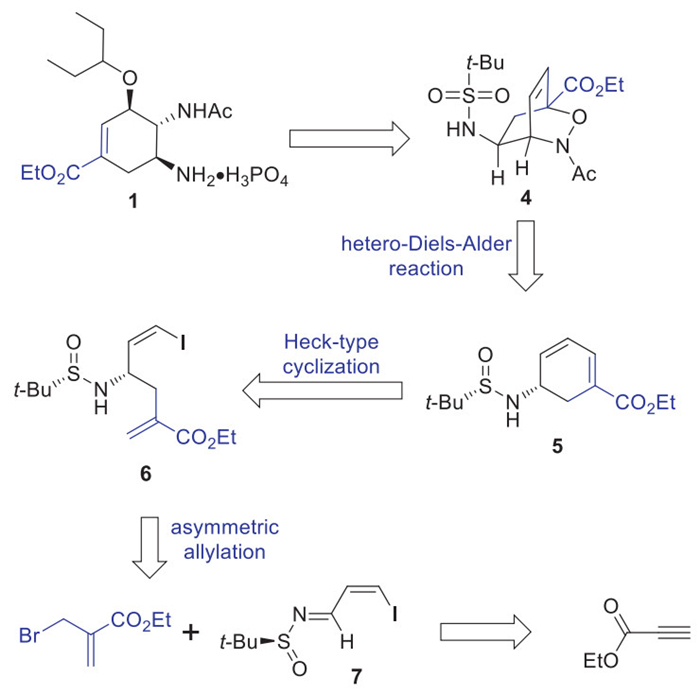
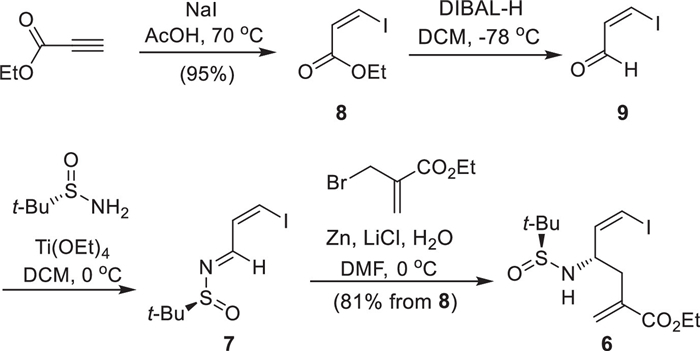
Our synthesis efforts commenced with the preparation of (Z)-β-iodo homoallylic amine 6 as shown in Scheme 2. Hydrohalogenation of commercially available ethyl propiolate with NaI under Ma’s conditions (AcOH, 70 ℃, 15 h) afforded exclusively the thermodynamically unfavorable ethyl (Z)-β-iodoacrylate (8) in excellent yield [22], which was readily converted into the (Z)-β-iodoacrolein (9) through DIBAL-H reduction of 8 followed by the Ti(O-i-Pr)4–mediated condensation with (S)-tert-butylsulfinamide at 0 ℃ within 9 h gave (Z)-N-sulfinyl imine 7 with a Z/E ratio of 9:1. The geometrically pure (Z)-N-sulfinyl imine 7 was obtained through the replacement of Ti(O-i-Pr)4 with Ti(OEt)4. Without the isolation of the required 7, the direct Barbier allylation with the Zn reagent, in situ derived from Zn powder and allyl bromide derivative in the presence of LiCl with H2O as additive furnished the desired (Z)-β-iodo homoallylic amine 6 in an overall yield of 81% with the excellent diastereoselectivity (dr > 95:5) over the three steps. It is worth mentioning that in the absence of water, only 59% of the desired product 6 was formed, indicating the addition of water is crucial for this asymmetric allylation reaction [23,24]. This is consistent with our previous observation of similar transformations [23].
With the chiral amine 6 in hand, we next turned our attention to the key Pd-catalyzed regioselective intramolecular Heck-type cyclization reaction for the synthesis of 5 along the projected pathway (Table 1) [25]. Initial efforts to directly form the desired 5 under Genêt’s conditions [26] led to the formation of complex and inseparable mixtures (entries 1 and 2). Attempts to run the cyclization process using Pd(P(tBu)3)2 and PdCl2dtbpf instead of PdCl2/TPPTS and Pd(OAc)2/TPPTS as catalysts were also unsuccessful, yielding an extremely low isolated yield of the desired product 5, along with significant formation of side product 10, which was proposed to be formed through a competing 5-exo cyclization followed by dealkoxycarbonylation (entries 3 and 4). Changing the solvent from MeCN/H2O to H2O as the sole solvent led to the predominant product 5 albeit no decrease in yield for 10 under the same reaction conditions (entry 5). To our delight, when performed in the presence of the 5 wt% TPGS-1000 (surfactant) under the reaction conditions of entry 6, the cyclization reaction produced 5 and 10 in 51% and 15% yields, indicating that the formation of exo-product 10 can be reduced. Decreasing reaction temperature from 70 ℃ to 50 ℃, the reaction delivered 71% of product 5 and 20% 5-exo product 10 (entry 7). However, the reaction at room temperature resulted in no reaction (entry 8). Reducing PdCl2dtbpf catalyst loading from 10 mol% into 5 mol% did not seem to affect this transformation (entry 9), retaining a similar result of entry 7 in Table 1. Unfortunately, a very poor outcome of 5 was obtained when the PdCl2dtbpf catalyst loading was diminished to 2 mol% (entry 10). Variation of the amount of TPGS-1000 used in the cyclization was next explored. It is important to note that the amount of TPGS-1000 has a significant effect on the regioselectivity of the Heck-type cyclization process (entries 11–13). The optimized reaction conditions were found to be the best results to generate 5 in 73% when the amount of TPGS-1000 was decreased to 3 wt% (entry 11). Screening of other surfactants including TPGS-750-M [27], Triton X-100 and Tween 80 did not improve the reaction results (entries 14–16).
 DownLoad:
CSV
DownLoad:
CSV
 |
||||||
| Entry | Catalyst (x mol%) | Surfactant (y wt%) | Solvent | Temp (℃) | Regioselectivity | |
| 6-endo product 5 (%)b | 5-exo product 10 (%)b | |||||
| 1 | PdCl2/TPPTS (10) | MeCN/H2O (6:1) | 70 | Mess | Mess | |
| 2 | Pd(OAc)2/TPPS (10) | MeCN/H2O (6:1) | 70 | Mess | Mess | |
| 3 | Pd(PtBu3)2 (10) | MeCN/H2O (6:1) | 70 | 12 | 33 | |
| 4 | PdCl2dtbpf (10) | MeCN/H2O (6:1) | 70 | 16 | 35 | |
| 5 | PdCl2dtbpf (10) | H2O | 70 | 47 | 36 | |
| 6 | PdCl2dtbpf (10) | TPGS-1000 (5) | H2O | 70 | 51 | 15 |
| 7 | PdCl2dtbpf (10) | TPGS-1000 (5) | H2O | 50 | 71 | 20 |
| 8 | PdCl2dtbpf (10) | TPGS-1000 (5) | H2O | r.t. | NR | NR |
| 9 | PdCl2dtbpf (5) | TPGS-1000 (5) | H2O | 50 | 70 | 19 |
| 10 | PdCl2dtbpf (2) | TPGS-1000 (5) | H2O | 50 | 40 | 11 |
| 11 | PdCl2dtbpf (5) | TPGS-1000 (3) | H2O | 50 | 73 | 18 |
| 12 | PdCl2dtbpf (5) | TPGS-1000 (2) | H2O | 50 | 59 | 25 |
| 13 | PdCl2dtbpf (5) | TPGS-1000 (1) | H2O | 50 | 50 | 37 |
| 14 | PdCl2dtbpf (5) | TPGS-750-M (3) | H2O | 50 | 45 | 46 |
| 15 | PdCl2dtbpf (5) | Triton X-100 (3) | H2O | 50 | 49 | 21 |
| 16 | PdCl2dtbpf (5) | Tween 80 (3) | H2O | 50 | Trace | Trace |
| a The reactions were carried out on a 0.2 mmol scale: 6 (79.8 mg, 0.2 mmol) was added catalyst (x mol%), Et3N (0.6 mmol) and surfactant (y wt%) in solvent (1.0 mL) under nitrogen conditions, and the mixture was stirring overnight. b Isolated yield. |
||||||
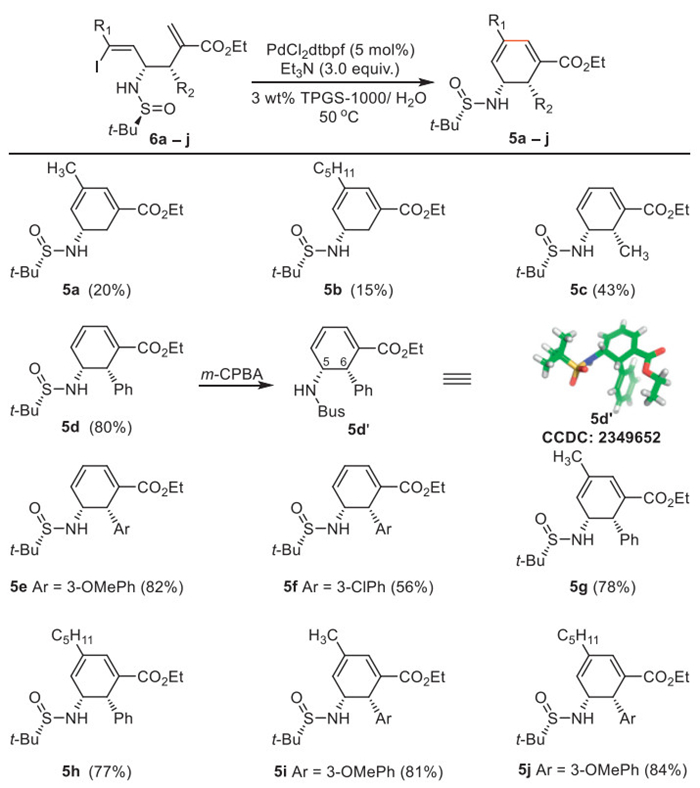
With the optimal reaction conditions established (Table 1, entry 11), we next proceeded to explore the substrate scope of the biphasic Pd-catalyzed intramolecular Heck-type cyclization shown in Scheme 3. For (Z)-β-iodo homoallylic amines with alkyl group (methyl and pentyl) at C3 position in 6 gave poor yield of desired products accompanied by a significant amount of 5-exo byproducts (details see Supporting information). The (Z)-β-iodo homoallylic amines with methyl and substituted aryl groups in 6 afforded the expected endo-Heck cyclization products 5c-5f in moderate to good yields. A similar trend was observed with disubstituted (Z)-β-iodo homoallylic amines delivering 5g-5j in good yields. The stereochemistry of products 5c-5j was elucidated by NOESY analysis (details see Supporting information), and the absolute configuration of 5d was further determined as (5R,6S) by X-ray crystallographic analysis of its oxidation product 5d’.
After successfully constructing the required six-membered ring core with the biphasic Pd-catalyzed intramolecular Heck-type cyclization methodology newly developed, we then turned our attention to complete the synthesis of 1 (Scheme 4). Our initial attempts to directly convert cyclohexadienyl amine 5 to the corresponding bromodiamide by following Corey's conditions (BrNHAc, SnBr4) proved to be unsuccessful [28], with only the decomposition of the starting material being observed. This failure was attributed to the lability of the tert-butyl sulfinamide group under the Lewis acidic conditions employed. Compound 5 was instead subjected to a controlled oxidation with 3-chloroperoxybenzoic acid (m-CPBA) (1.2 equiv.) in dilute ethyl acetate (EtOAc) to produce tert-butyl sulfonamide (Bus) 11 in good yield. The bromoamidation of compound 11 successfully yielded bromodiamide 12. Consistent with Corey's findings [21,29], the bromide reacted with the olefin from the counter steric face, which was confirmed by the X-ray structure of compound 12. Unfortunately, the intramolecular cyclization of 12 under basic conditions did not generate the desired N-alkylation product aziridine 14, but only the O-alkylation product oxazoline 13, which was a dead end for the synthesis.
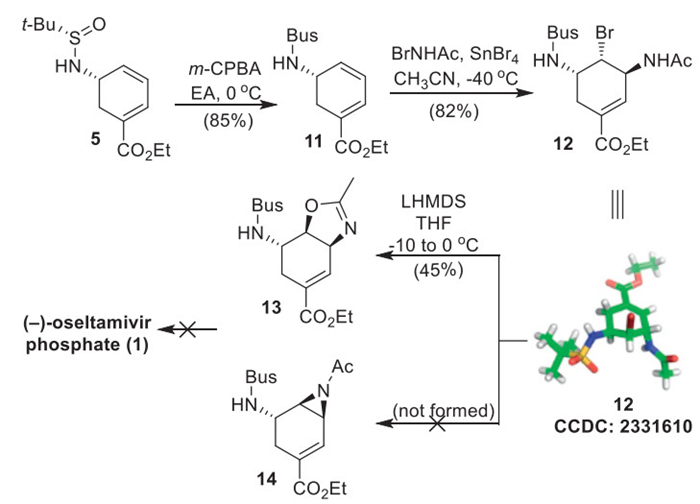
To address the challenges associated with aziridine formation, an alternative approach toward (–)-oseltamivir phosphate (1) was explored (Scheme 5). A hetero-Diels-Alder reaction between cyclohexadienyl amine 5 and the in situ generated acyl nitroso readily occurred in a highly regio- and diastereoselective fashion to provide bicyclic oxazine 15 as the major product [30]. The structure of 15 was elucidated by NMR spectroscopic analysis, and was further confirmed by X-ray crystallographic analysis of a single crystal. Subsequent oxidation of 15 with m-CPBA furnished sulfonamide 4 in 96% yield.
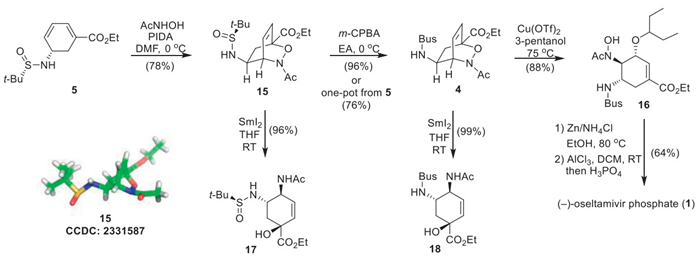
The installation of the 3-pentylether group was firstly pursued by adopting a SN’-type transformation. Accordingly, reductive cleavage of the labile N–O bond of 15 and 4 with samarium diiodide (SmI2) gave allylic alcohols 17 and 18, respectively, in excellent yields. Unfortunately, attempts to introduce the 3-pentylether group to 17 and 18 all failed despite trying a variety of conditions. In order to circumvent this obstacle, we decided to install the 3-pentylether group earlier in the route. First, it was found that the hetero-Diels-Alder reaction and the subsequent sulfinamide oxidation reaction could be carried out in a one-pot fashion, compound 4 was obtained from diene 5 directly in 76% yield. Ring opening of 4 with 3-pentanol was then investigated. Upon screening various conditions, it was revealed that Cu(OTf)2 could effectively mediate the regio- and diastereoselective ring-opening of 4 with 3-pentanol, affording trans-1,2-substituted diamino cyclohexyl amyl ether 16 in 88% yield, which already contained the skeleton and all three chiral centres of the final target. The end game of this synthesis included zinc-mediated reductive cleavage of the N–O bond under acidic conditions, AlCl3 enabled removal of the Bus group [31], and salt formation with H3PO4, providing (–)-oseltamivir phosphate (1) in 64% yield. All the spectra data of thus synthesized 1 matched well with that previously reported in the literature [12,20,32].
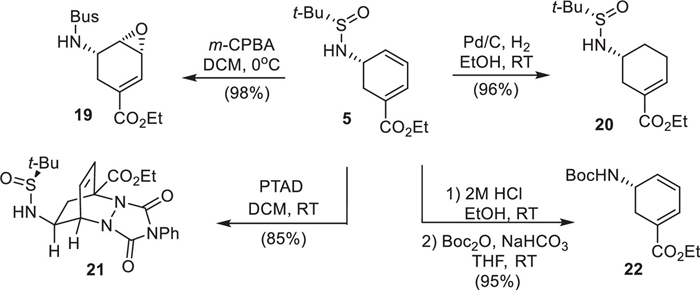
Furthermore, these chiral cyclohexadienyl amines can serve as useful synthons, as demonstrated by the selective transformations of chiral cyclohexadienyl amine 5 (Scheme 6). Treatment of 5 with excess m-CPBA first induced sulfur oxidation, converting the tert-butyl sulfinamide into a sulfonamide and subsequent directed olefin epoxidation yielded epoxide 19 stereoselectively as a single isomer [33]. Selective catalytic hydrogenation produced the cyclohexene derivative 20, while a Diels-Alder reaction with 4-phenyl-1,2,4-triazoline-3,5-dione (PTAD) yielded the adduct 21. Removal of the chiral auxiliary and protection of the amine with Boc furnished Corey’s intermediate 22 [21], thereby completing a formal synthesis of (–)-oseltamivir phosphate (1).
In summary, a concise asymmetric synthesis of (–)-oseltamivir phosphate (1) has been successfully developed, which proceeds with a longest linear sequence of 7 steps from the known aldehyde 9 (or 9 steps from ethyl propiolate, 24% overall yield). The key bond disconnections employed in our synthesis of 1 are unprecedented in published routes. Notable transformations include a diastereoselective allylation reaction of cis-α,β-unsaturated chiral imines, a Pd-catalyzed regioselective intramolecular Heck-type cyclization for the synthesis of chiral cyclohexadienyl amines, a regioselective and diastereoselective nitroso hetero-Diels-Alder reaction and a Cu(OTf)2-mediated regioselective and diastereoselective nucleophilic substitution reaction. Moreover, the chiral cyclohexadienyl amines are versatile synthons for further selective transformations, as exemplified in a formal synthesis of (–)-oseltamivir phosphate (1). Particularly, the convergence and modularity nature of our synthesis of 1 render it amenable to derivative synthesis. Further progress in this direction will be reported in due course.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Qi Wang: Writing – original draft, Investigation, Data curation. Bichu Cheng: Writing – review & editing, Writing – original draft, Funding acquisition, Formal analysis. Minjie Liu: Investigation. Fen-Er Chen: Writing – review & editing, Writing – original draft, Investigation, Formal analysis.
Financial support from the National Natural Science Foundation of China (No. 22461023) is gratefully acknowledged.
Supplementary material associated with this article can be found, in the online version, at doi:
Influenza (Seasonal), 2024. n.d. accessed June 24
C.U. Kim, W. Lew, M.A. Williams, et al., J. Med. Chem. 41 (1998) 2451–2460. doi: 10.1021/jm980162u
C.U. Kim, W. Lew, M.A. Williams, et al., J. Am. Chem. Soc. 119 (1997) 681–690. doi: 10.1021/ja963036t
J.C. Rohloff, K.M. Kent, M.J. Postich, et al., J. Org. Chem. 63 (1998) 4545–4550. doi: 10.1021/jo980330q
M. Federspiel, R. Fischer, M. Hennig, et al., Org. Proc. Res. Dev. 3 (1999) 266–274. doi: 10.1021/op9900176
WHO endorses landmark public health decisions on Essential Medicines for Multiple Sclerosis, 2024. n.d. accessed July 23.
S. Abrecht, P. Harrington, H. Iding, et al., Chimia 58 (2004) 621. doi: 10.2533/000942904777677605
D.R. Knop, K.M. Draths, S.S. Chandran, et al., J. Am. Chem. Soc. 123 (2001) 10173–10182. doi: 10.1021/ja0109444
S.S. Chandran, J. Yi, K.M. Draths, et al., Biotech. Prog. 19 (2003) 808–814. doi: 10.1021/bp025769p
B. Limbani, S. Bera, D. Mondal, Chemistry Select 5 (2020) 6083–6122. doi: 10.1002/slct.202000675
C.R. Sagandira, F.M. Mathe, U. Guyo, P. Watts, Tetrahedron 76 (2020) 131440.
H. Li, S.J. Shen, C.L. Zhu, H. Xu, J. Am. Chem. Soc. 140 (2018) 10619–10626. doi: 10.1021/jacs.8b06900
S. Zhu, S. Yu, Y. Wang, D. Ma, Angew. Chem. Int. Ed. 49 (2010) 4656–4660. doi: 10.1002/anie.201001644
H. Ishikawa, T. Suzuki, Y. Hayashi, Angew. Chem. Int. Ed. 48 (2009) 1304–1307. doi: 10.1002/anie.200804883
K. Yamatsugu, L. Yin, S. Kamijo, et al., Angew. Chem. Int. Ed. 48 (2009) 1070–1076. doi: 10.1002/anie.200804777
B. Sullivan, I. Carrera, M. Drouin, T. Hudlicky, Angew. Chem. Int. Ed. 48 (2009) 4229–4231. doi: 10.1002/anie.200901345
B.M. Trost, T. Zhang, Angew. Chem. Int. Ed. 47 (2008) 3759–3761. doi: 10.1002/anie.200800282
J.J. Shie, J.M. Fang, S.Y. Wang, et al., J. Am. Chem. Soc. 129 (2007) 11892–11893. doi: 10.1021/ja073992i
N. Satoh, T. Akiba, S. Yokoshima, T. Fukuyama, Angew. Chem. Int. Ed. 46 (2007) 5734–5736. doi: 10.1002/anie.200701754
Y. Fukuta, T. Mita, N. Fukuda, M. Kanai, M. Shibasaki, J. Am. Chem. Soc. 128 (2006) 6312–6313. doi: 10.1021/ja061696k
Y.Y. Yeung, S. Hong, E.J. Corey, J. Am. Chem. Soc. 128 (2006) 6310–6311. doi: 10.1021/ja0616433
S. Ma, X. Lu, Z. Li, J. Org. Chem. 57 (1992) 709–713. doi: 10.1021/jo00028a055
S. Bonazzi, B. Cheng, J.S. Wzorek, D.A. Evans, J. Am. Chem. Soc. 135 (2013) 9338–9341. doi: 10.1021/ja404673s
A. Shen, M. Liu, Z.S. Jia, M.H. Xu, G.Q. Lin, Org. Lett. 12 (2010) 5154–5157. doi: 10.1021/ol102148b
H. Zhang, B. Xu, L. Zhou, Z.M. Zhang, J. Zhang, Green Synth. Catal. 5 (2024) 102–107.
J.H. Rigby, R.C. Hughes, M.J. Heeg, J. Am. Chem. Soc. 117 (1995) 7834–7835. doi: 10.1021/ja00134a040
B.H. Lipshutz, S. Ghorai, A.R. Abela, et al., J. Org. Chem. 76 (2011) 4379–4391. doi: 10.1021/jo101974u
S.Hong Yeung, E.J. Corey, J. Am. Chem. Soc. 128 (2006) 6310–6311.
X.Gao Yeung, E.J. Corey, J. Am. Chem. Soc. 128 (2006) 9644–9645. doi: 10.1021/ja063675w
B.S. Bodnar, M.J. Miller, Angew. Chem. Int. Ed. 50 (2011) 5630–5647. doi: 10.1002/anie.201005764
D. Enders, M. Seppelt, T. Beck, Adv. Synth. Catal. 352 (2010) 1413–1418. doi: 10.1002/adsc.201000143
J.C. Rohloff, K.M. Kent, M.J. Postich, et al., J. Org. Chem. 63 (1998) 4545–4550.
A.H. Hoveyda, D.A. Evans, G.C. Fu, Chem. Rev. 93 (1993) 1307–1370. doi: 10.1021/cr00020a002

Figure 1 Structures of (–)-oseltamivir phosphate (1), its active form oseltamivir acid (2) and sialic acid (3).

Scheme 3 Scope of six-membered chiral amine via Pd-catalyzed intramolecular Heck cyclization.
Table 1. Optimization of the biphasic Pd-catalyzed intramolecular Heck-type cyclization conditions of (Z)-β-iodo homoallylic amine 6.
|
|
||||||
| Entry | Catalyst (x mol%) | Surfactant (y wt%) | Solvent | Temp (℃) | Regioselectivity | |
| 6-endo product 5 (%)b | 5-exo product 10 (%)b | |||||
| 1 | PdCl2/TPPTS (10) | MeCN/H2O (6:1) | 70 | Mess | Mess | |
| 2 | Pd(OAc)2/TPPS (10) | MeCN/H2O (6:1) | 70 | Mess | Mess | |
| 3 | Pd(PtBu3)2 (10) | MeCN/H2O (6:1) | 70 | 12 | 33 | |
| 4 | PdCl2dtbpf (10) | MeCN/H2O (6:1) | 70 | 16 | 35 | |
| 5 | PdCl2dtbpf (10) | H2O | 70 | 47 | 36 | |
| 6 | PdCl2dtbpf (10) | TPGS-1000 (5) | H2O | 70 | 51 | 15 |
| 7 | PdCl2dtbpf (10) | TPGS-1000 (5) | H2O | 50 | 71 | 20 |
| 8 | PdCl2dtbpf (10) | TPGS-1000 (5) | H2O | r.t. | NR | NR |
| 9 | PdCl2dtbpf (5) | TPGS-1000 (5) | H2O | 50 | 70 | 19 |
| 10 | PdCl2dtbpf (2) | TPGS-1000 (5) | H2O | 50 | 40 | 11 |
| 11 | PdCl2dtbpf (5) | TPGS-1000 (3) | H2O | 50 | 73 | 18 |
| 12 | PdCl2dtbpf (5) | TPGS-1000 (2) | H2O | 50 | 59 | 25 |
| 13 | PdCl2dtbpf (5) | TPGS-1000 (1) | H2O | 50 | 50 | 37 |
| 14 | PdCl2dtbpf (5) | TPGS-750-M (3) | H2O | 50 | 45 | 46 |
| 15 | PdCl2dtbpf (5) | Triton X-100 (3) | H2O | 50 | 49 | 21 |
| 16 | PdCl2dtbpf (5) | Tween 80 (3) | H2O | 50 | Trace | Trace |
| a The reactions were carried out on a 0.2 mmol scale: 6 (79.8 mg, 0.2 mmol) was added catalyst (x mol%), Et3N (0.6 mmol) and surfactant (y wt%) in solvent (1.0 mL) under nitrogen conditions, and the mixture was stirring overnight. b Isolated yield. |
||||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们





 下载:
下载: