State Key Laboratory of Flexible Electronics (KLoFE) & Institute of Advanced Materials (IAM), Nanjing University of Posts & Telecommunications, Nanjing 210023, China
b.
State Key Laboratory of Flexible Electronics (KLoFE) & Institute of Advanced Materials (IAM), Nanjing Tech University (NanjingTech), Nanjing 211816, China
c.
Institute of Quantum and Sustainable Technology (IQST), School of Chemistry and Chemical Engineering, Jiangsu University, Zhenjiang 212013, China
Received Date:
06 November 2024 Accepted Date:
06 July 2025 Revised Date:
03 July 2025 Available Online:
15 January 2026
Abstract:
To precisely control intrachain π-electron delocalization and interchain interaction simultaneously is the prerequisite to obtain stable and efficient deep-blue light-emitting p-n polymer semiconductors for the polymer light-emitting diodes (PLEDs). Herein, we introduced the steric carbazole-fluorene nanogrid into light-emitting diphenyl sulfone-based p-n polymer semiconductors (PG and PDG) via metal-free CN coupling polymerization for the fabrication of deep-blue PLEDs. The steric, rigid and twisted configuration between nanogrid and diphenyl sulfone in PG and PDG present the unique characteristic of large steric hindrance interaction to suppress interchain aggregation in solid state. Due to the different length of electron-deficient diphenyl sulfone monomers, PG showed a deep-blue emission with a maximum peak at 428 nm but red-shifted to 480 nm for the PDG films. Interestingly, similar deep-blue emission behavior of PG in diluted non-polar solution and films suggested the extremely weak interchain aggregation. Finally, PLEDs based on PG are fabricated with a stable deep-blue emission of CIE (0.15, 0.10), and corresponding EL spectral profile is also completely identical to PL ones of diluted solution, revealed the intrachain emission without obvious interchain excited state, confirmed effectiveness of the steric hindrance functionalization of nanogrid in p-n polymer semiconductor for deep-blue light-emitting organic optoelectronics.
Since the first polymer semiconductor based on polyacetylenes is discovered in 1970s [1], emerging polymer semiconductors are widely applied in the polymer light-emitting diodes (PLEDs), polymer field-effect transistor (PFETs) and polymer solar cell (PSCs) [2]. In general, the electronic structure of polymer semiconductors is the fundamental factor to dominate a variety of optoelectronic processes spanning from charge transport to the photophysics of neutral excited species [3-5]. As the key parameter of electronic structures, the band-gap of organic materials, especial for the polymer semiconductors, can be precisely tuned by the p-n molecular design principle [4,6,7], as displayed in Scheme 1a. Bandgap of organic molecules linked by simple σ-bond are very large, estimated about > 4.5 eV, resulted into extremely low conductivity (insulated property), but reduced to ~3.5 eV via introducing p- and n-type aromatic units for the excellent intramolecular π-electron delocalization [6,7]. Up to date, the deep-blue emission (< 430 nm) is mostly observed from the hydrocarbon polymer semiconductors without strong electron-deficient heteroatomic aromatic units (Scheme 1b) [8,9]. Besides, controlling the intrachain π-electron delocalization between the p- and n-type aromatic segments is a precondition to obtain wide-bandgap polymer semiconductors with narrowband deep-blue emission [9,10]. However, ubiquitous interchain π-π interactions in light-emitting p-n polymer semiconductor easily resulted into the formation of interchain excited state, lead to low emission efficiency and narrow bandgap (Scheme 1c) [5,9,11-15]. These are very undesirable for the deep-blue PLEDs [16]. More important, up to date, deep-blue light-emitting hydrocarbon and p-n π-conjugated polymers are mostly manufactured via metallic-catalyst polymerization, such as Suzuki, Yamamoto and Stille reaction [12,17,18]. Therefore, introducing an active monomer to prepare the deep-blue light-emitting p-n polymer semiconductors via metal-free polymerization is essential, and inevitably, the intrachain π-electron delocalization and interchain interactions in solid states that govern these bandgap and excited behavior need to be taken into consideration.
Scheme 1
Scheme 1.
Design principle of the light-emitting p-n polymer semiconductors for deep-blue PLEDs. (a) Band-gap engineering of organic materials toward light-emitting optoelectronics. (b) Jablonski diagram of organic light-emitting materials. (c) To achieved deep-blue light-emitting polymer via controlling intrachain charge-transfer behavior. (d) Design and preparation of the steric carbazole-fluorene nanogrid and diphenyl sulfone p-n polymers semiconductors prepared via metal-free C—N polymerization for deep-blue PLEDs.
In general, to isolate the wide-bandgap organic chromophore with extremely weak interchain interaction is an effective strategy to obtain stable and efficient deep-blue emission [11,16,19]. In this regard, there are two factors need to consider for the realization of the preparation of the stable and efficient deep-blue light-emitting p-n polymers: (ⅰ) To establish organic chromophore with the electronic structure of wide bandgap (> 3.0 eV) via controlling the intrachain π-electron delocalization [8,19-22]. (ⅱ) To suppress interchain π-π interactions and ensure single-chromophore emission behavior without the formation of interchain excited or charge transfer states [9,11,13,14,23]. In fact, π-conjugation-interrupted strategy is a convenient and simple method to precisely tune intrachain π-electron delocalization via controlling the π-electron capacity of p and n monomers, and their copolymerization ratio, space distance and topological structures [24-29]. However, as we discussion above, due to the freedom σ-bond, π-conjugation-interrupted p-n polymers present a diverse chain morphology and conformational behavior, which may induce complicated interchain aggregations and space charge-transfer behavior [24,28,30]. These may result into low color purity and the emission efficiency [28]. More interestingly, recently, our group developed a series of outstanding nano-grids with steric, stereoisomeric and three (3)-dimensional topological structures, which provided a fantastic molecular segmental monomer to design and prepare wide bandgap deep-blue p-n polymer semiconductors [31-35]. Herein, we demonstrated a steric carbazole-fluorene nano-grid to prepare diphenyl sulfone-based p-n polymers (PG and PDG) via metal-free C—N polymerization for the deep-blue PLEDs (Scheme 1d) [36-39]. Two n-type monomers are selected to explore the effect of intrachain π-electron delocalization on the emission behavior of p-n polymers [39]. Steric carbazole-fluorene nanorgid is introduced to suppress interchain aggregation to ensure single-chromophore deep-blue emission behavior. Finally, stable deep-blue PLEDs are further fabricated with a CIE of (015, 0.10).
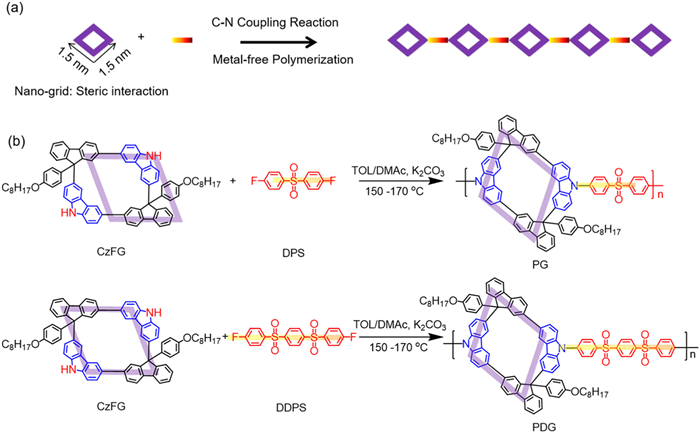
In general, residual metal catalyst is a key topic for the wide application of polymer semiconductors in organic electronics [17,18]. However, up to date, most p-n polymer semiconductors are prepared via metallic-catalyst polymerization, which result into a serious problem of residual metal catalyst [17]. Therefore, to explore and introduce the metal-free polymerization is essential for the preparation of p-n polymer semiconductors. In the last decades, the C—N coupling polymerizations are widely applied to achieve the functional polymers with an excellent high temperature thermoplastic [37-39]. Herein, we designed a series of novel carbazole-fluorene nanogrid (rod)-coil p-n polymer prepared via N—C coupling polymerizations for the deep-blue PLEDs. The steric nanogrid can easily suppress interchain aggregation to reduce the formation of interchain excited states (Fig. 1a). Therefore, steric carbazole-fluorene nano-grids is obtained with a 3D-topological structures [36], and two diphenyl sulfones, G and DG, are introduced as the secondary monomer to check the effect of intrachain π-electron delocalization on the emission behavior. Firstly, two polymers, PG and PDG, are prepared via metal-free C—N polymerization. Firstly, the nanogrids prepared via Friedel-Crafts (F-C) cycling reactions, according to previous work [36]. Preparation of PG and PDG were obtained according to previous works [37-39], as depicted in Fig. 1b. The 1H and 13C NMR spectral analyses are introduced here to confirm their chemical structures (Fig. 1, Figs. S1 and S2 in Supporting information). GPC measurements revealed the number-average molecular weight (Mn) and polydispersity index (PDI) of PG and PDG are calculated about 2.33 × 104 and 1.42, 2.02 × 104 and 2.00, respectively (Fig. S3 in Supporting information). Meanwhile, PG and PDG showed a high decomposition temperature (Td) of up to 380 ℃ and 350 ℃ (Fig. S4 in Supporting information), and presented a glass transition temperature (Tg) of ~130 ℃ and 175 ℃ (Fig. S5 in Supporting information), which indicated their excellent thermal stability. Meanwhile, two polymers showed excellent solubility in common solvents, such as polar/non-polar solvent, toluene (TOL), chloroform (CHCl3), tetrahydrofuran (THF), and dichloromethane (DCM). Subsequently, atomic force microscopy (AFM) is also explored to evaluate the surface morphology. According to the AFM analysis (Fig. S6 in Supporting information), the roughness-mean-square (RMS) of PG and PDG pristine spin-coated films was ~1.86 and 1.34 nm, which are useful to fabricate high-performance PLEDs. In addition, cyclic voltammetry (CV) study shows the highest occupied molecular orbital (HUMO) and low occupied molecular orbital (LUMO) energy levels of PG are −5.15, −1.67 eV and PDG are −5.15, −2.15 eV, respectively (Fig. S7 in Supporting information). The optical band gap (Eg) of the two materials is calculated about 3.48 and 3.05 eV, respectively, which confirmed their excellent ultra-wide bandgap for the realization of deep-blue emission behaviour.
Figure 1
Figure 1.
Steric-nanogridization of conjugated molecules for p-n polymer semiconductors. (a) Steric nanogrid-based p-n polymer prepared via metal-free C—N polymerization. (b) Synthesis route of carbazole-fluorene nanogrid and diphenyl sulfone-based light-emitting p-n polymer, PG and PDG.
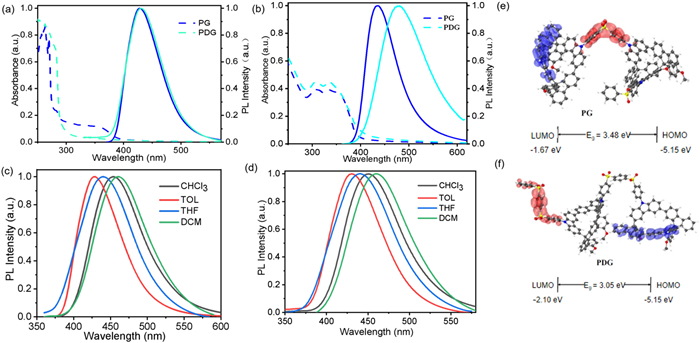
Beyond the intrinsically electronic structures, intrachain conformation and interchain interactions of polymer semiconductors will determine their optoelectronic property. Meanwhile, strong intrachain charge transfers states also may increase the Huang-Rhys factor (S factor) to result into a red-shifted and broaden emission spectra in organic p-n fluorescent materials. These intrachain charge transfers are seriously enhanced in the polar solvents. Therefore, we further explored the optical property of PG and PDG in various states, as showed Fig. 2. Firstly, PG and PDG in diluted toluene solution presented the similar broaden absorption spectra, with a large region range from 300 nm and 380 nm (Fig. 2a). These broaden absorption region are also revealed the formation weak charge-transfer states in our model materials, respectively. Compared to the steric nanogrid monomers [36], these red-shifted absorption spectra of PG and PDG in diluted solution revealed the formation of intrachain charge transfer from nanogrid to aryl sulfone segments. PDG with bis-benzene sulfonyl units have a smaller Eg than those of PG with diphenyl sulfone (DPS) via the optical band gap calculated by the absorption spectral edges of the PG and PDG. Consider the same backbone structure, it can be seen that the change of receptor unit (bis-benzene sulfonyl/diphenyl sulfone) has no significant effect on the luminescence behavior in toluene solution, and showed deep-blue emission. In addition, strong absorption band from 300 nm to 400 nm are easily observed for PG and PDG spin-coated films, indicated the strong intrachain charge transfer (Fig. 2b). As we expected, both PG and PDG diluted solution had a completely similar broaden emission spectral profile with a maximum peak at 428 nm. In general, significantly different absorption and fluorescence spectral profile between diluted solution and film is easily observed for light-emitting polymer semiconductors, due to the ubiquitous interchain π-π interactions and aggregations. Interestingly, the PL spectra of PG film is quite similar to those of diluted solution but slight red-shifts about ~5 nm, suggested their extremely weak interchain interaction (Fig. 2) [40]. These results indicated that the nanogrids may be suppressed the π-π stacking between chain aromatic units. However, PDG spin-coated films displayed a PL spectrum with a maximum peak at 480 nm, serious red-shifted about 50 nm, compared to the diluted solution. These serious red-shifted emission behaviors in solid states also indicated the formation of strong intrachain charge-transfer states induced by the restriction of the intrachain conformational motion. Therefore, to precisely control the intrachain charge-transfer is the precondition to ensure efficient deep-blue emission in p-n fluorescent materials.
Figure 2
Figure 2.
Optical property and electronic structure of PG and PDG. The absorption and emission spectra of PG and PDG (a) in diluted toluene solution (10–3 mg/mL) and (b) films. Fluorescence spectra of PG (c) and PDG (d) in various solvents. Theoretical calculation of electronic structure of PG (e) and PDG (f).
In order to check the charge transfer behavior, emission spectra of PG and PDG in various solvents are recorded, as displayed in Figs. 2c and d. It is easily observed that the maximum emission peak of PG diluted solution is about 428, 442, 456 and 463 nm in toluene, THF, chloroform and DCM diluted solution, which indicated the strong intrachain charge transfer with increasing the solvent polarity. And there is a similar change tendency for the PDG, revealed the formation of strong intrachain charge transfer states. Therefore, relatively strong intrachain charge transfer induced the restriction of the intrachain conformational motion in PDG spin-coated films that those of PG, can reasonably explain their large red-shifted PL spectra between diluted solution and solid states. The main reason is that the no-polar toluene solvent has intramolecular charge transfer interaction with PG and PDG molecular chains, and the interaction with polar solvent is easy to produce intramolecular charge transfer and intermolecular charge transfer which increased interchain conjugation and leads to chain-solvent interaction (solvation effect) [15,41]. To further explore the electron structure and charge transfer properties of the PG and PDG, DFT simulations were explored for their chain segments, as shown in Figs. 2e and f. Similar to previous work [39], the completely separated HOMO and LUMO of PG and PDG chain segments are mostly distributed along carbazole-fluorene nanogrid and diphenyl sulfone, respectively, with no obvious overlap on the phenyl bridges and nitrogen atoms. And the calculated wide bandgap is about 3.48 and 3.05 eV for the PG and PDG, similar to the experimental value. In addition, PG and PDG spin-coated films showed PL decay lifetime of 1.0 and 3.5 ns (Fig. S8 in Supporting information), much longer than those of tradition deep-blue polyfluorenes, induced by the intrachain charge transfer states. Moreover, the fluorescence quantum yields (PLQY, Φ) of PG and PDG in solution and pristine film are 56% and 20%, 24% and 10%, respectively. Relatively low emission efficiency resulted from the heavy atoms effect of S atom, formation of intrachain charge-transfer and interchain excited states. Compared to PDG, relatively high efficiency of PG is useful to enhance the efficiency of deep-blue PLEDs. In this regard, introduce of steric nano-grid into p-n polymer semiconductors can effective suppress interchain aggregation to obtain single-chromophore emission behavior in solid state.
Due to the wide bandgap, both PG and PDG may be possible act as the deep-blue emission layer in PLEDs. Therefore, preliminary PLEDs with a sandwich structure of ITO/PEDOT: PSS/PG and PDG films/TPBi (30 nm)/LiF (0.5 nm)/Al (100 nm) were fabricated without systematically optimized (Fig. 3a). The similar HOMO energy level of PG and PDG are beneficial for the improvement of hole injection from PEDOT: PSS layer to emission layers. Interestingly, EL spectra of PG-based on PLEDs consisted of only one peak at 428 nm, completely similar to PL one (Fig. 3b), confirmed their extremely weak interchain aggregation. Corresponding CIE value of PG-based PLEDs is estimated about (0.15, 0.10) (Fig. 3c). More important, with increasing applied voltage from 10 V to 12 V (Fig. S9 in Supporting information), similar EL spectral profiles are suggested the excellent stable deep-blue emission behaviour. However, there is broaden EL spectral profile with long tail to 700 nm but red-shifted to 455 nm observed for the PDG-based PLEDs, revealed the emission from intrachain charge-transfer states, that the CIE is about 0.20, 0.16, revealed the sky-blue colour emission. Besides, turn on voltage (Von) of PG and PDG-based PLED are estimated about 6.0 and 6.5 V, respectively. And the maximum brightness is calculated about 247 and 101 cd/m2, with a current efficiency (C. E.) of 0.52 and 0.03 cd/A for the PG and PDG device, respectively (Figs. 3d-f). Therefore, PG showed stable deep-blue intrachain electroluminescent behaviour in solid states.
Figure 3
Figure 3.
Performance of deep-blue Pled based on PG and PDG. (a) Device structure of PG and PDG-based PLEDs. (b) EL spectra of PG and PDG-based PLEDs, corresponding CIE value (c). Inset showed the photograph of PG-based PLEDs. Luminance (d) and current density (e) vs. voltage curves of PG and PDG-based PLEDs, and corresponding current efficiency vs. current density curves (f).
In summary, two light-emitting p-n polymer semiconductors are successfully synthesized from carbazole-fluorene nanogrid and bis(4-fluorophenyl)-sulfone monomers via metal-free C—N polymerization coupling reactions for the deep-blue PLEDs. The carbazole-fluorene nanogrids are introduced as steric monomer segments to suppress the formation of interchain excited and charge transfer states, which is useful to obtain single-chromophore deep-blue emission. More interestingly, PG chain presents a completely similar deep-blue emission in diluted and film states without obvious red-shifting, confirmed the extremely weak interchain aggregation under solid states. However, compared to diluted solution, PDG films displayed a red-shifted emission spectra (~50 nm), indicated the strong intrachain charge-transfer induced by the restriction of the intrachain conformational motion in film. Finally, deep-blue PLEDs-based on PG showed a reasonably similar and stable EL spectral profile to PL one, revealed the intrachain electroluminescent behavior in solid states, suggested the effectiveness of our nanogrid p-n polymer prepared via metal-free C—N polymerization for deep-blue PLEDs.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
J. Lin acknowledges the support from the Jiangsu Provincial Senior Talent Program (Dengfeng, Jiangsu University). L. Xie and J. Lin acknowledge the support from the National Key R & D Program of China (No. 2024YFB3612600), the National Natural Science Foundation of China (Nos. 22275098, 62288102), Basic Research Program of Jiangsu (No. BK20243057), the Natural Science Research Start-up Foundation of Recruiting Talents of Nanjing University of Posts and Telecommunications (No. NY222097), the National Natural Science Foundation of China (No. 62205035).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.111551.
[1]
H. Shirakawa, E.J. Louis, A.G. MacDiarmid, C.K. Chiang, A.J. Heeger, J. Chem. Soc. Chem. Commun. (1977) 578–580.
J. Lin, B. Liu, M. Yu, et al., Adv. Mater. 31 (2019) 1804811. doi: 10.1002/adma.201804811
[41]
Z.R. Grabowski, K. Rotkiewicz, W. Rettig, Chem. Rev. 103 (2003) 3899–4032. doi: 10.1021/cr940745l
Scheme 1
Design principle of the light-emitting p-n polymer semiconductors for deep-blue PLEDs. (a) Band-gap engineering of organic materials toward light-emitting optoelectronics. (b) Jablonski diagram of organic light-emitting materials. (c) To achieved deep-blue light-emitting polymer via controlling intrachain charge-transfer behavior. (d) Design and preparation of the steric carbazole-fluorene nanogrid and diphenyl sulfone p-n polymers semiconductors prepared via metal-free C—N polymerization for deep-blue PLEDs.
Figure 2
Optical property and electronic structure of PG and PDG. The absorption and emission spectra of PG and PDG (a) in diluted toluene solution (10–3 mg/mL) and (b) films. Fluorescence spectra of PG (c) and PDG (d) in various solvents. Theoretical calculation of electronic structure of PG (e) and PDG (f).
Figure 3
Performance of deep-blue Pled based on PG and PDG. (a) Device structure of PG and PDG-based PLEDs. (b) EL spectra of PG and PDG-based PLEDs, corresponding CIE value (c). Inset showed the photograph of PG-based PLEDs. Luminance (d) and current density (e) vs. voltage curves of PG and PDG-based PLEDs, and corresponding current efficiency vs. current density curves (f).


 DownLoad:
DownLoad:


 下载:
下载:





 下载:
下载:
