Citation:
Giulia Brufani, Edoardo Bazzica, Yanlong Gu, Francesco Mauriello, Luigi Vaccaro. Csp2–H functionalization as an efficient catalytic route to carbazoles[J]. Chinese Chemical Letters,
2026, 37(1): 111545.
doi:
10.1016/j.cclet.2025.111545
Csp2–H functionalization as an efficient catalytic route to carbazoles
English
Csp2–H functionalization as an efficient catalytic route to carbazoles
Laboratory of Green S.O.C. – Dipartimento di Chimica, Biologia e Biotecnologie, Università degli Studi di Perugia, Via Elce di Sotto 8, 06123, Perugia, Italy
b.
Department of Civil, Energy, Environmental and Material Engineering (DICEAM), Università degli Studi Mediterranea di Reggio Calabria, via Graziella, Feo di Vito, 89122 Reggio Calabria, Italy
c.
Key Laboratory of Material Chemistry for Energy Conversion and Storage, Ministry of Education, Hubei Key Laboratory of Material Chemistry and Service Failure, School of Chemistry and Chemical Engineering, Huazhong University of Science and Technology, Wuhan 430074, China
* Corresponding author. E-mail address: luigi.vaccaro@unipg.it (L. Vaccaro). 1 These authors contributed equally to this work.
Received Date:
21 April 2025 Accepted Date:
03 July 2025 Revised Date:
17 June 2025 Available Online:
15 January 2026
Abstract:
Given the broad applicability of carbazole structural moieties in materials science and medicinal chemistry, significant efforts have been devoted to developing efficient synthetic catalytic methodologies to access this valuable scaffold. Catalyzed direct Csp2–H functionalization provides an effective and cost-efficient approach to synthesizing carbazoles from simple and readily available starting materials, ensuring a promising path characterized by excellent atom and step economy. This review highlights the substantial progress made in the last 10 years in advancing catalytic Csp2–H functionalization techniques for synthesizing carbazoles.
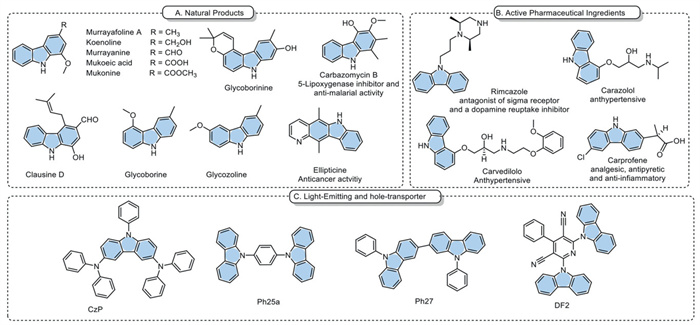
The carbazole scaffold emerges prominently within N-heterocyclic compounds, serving as a pivotal structural motif in various biologically relevant natural products characterized by diverse biological applications, such as antimalarial [1], antibacterial [2,3], and antimicrobial properties (Fig. 1a) [4]. They find extensive application as active pharmaceutical ingredients (APIs) across multiple applications such as anticancer [5], and antihypertensive as Carazolol and Carvedilol (Coreg tablets) [6-8], antiviral [9], and anti-inflammatory [10] as Carprofen, which is a COX-1 and COX-2 inhibitor used in veterinary medicine (Fig. 1b) [11]. Beyond their biological significance, carbazole derivatives exhibit remarkable properties, including effective photoconductivity [12], photorefractivity [13], low ionization potentials [14], and luminescence [15], rendering them indispensable as building blocks for materials capable of carrying charges and emitting light (Fig. 1c) [16-18].
Figure 1
Figure 1.
Bioactive and synthetically relevant molecules incorporating the carbazole unit: Representative examples of natural products (A), pharmaceutically and biologically active compounds (B) and hole-transporting and light-emitting molecules (C).
Given the wide applicability of these pivotal structural moieties in materials science and medicinal chemistry, considerable efforts have been devoted to developing effective synthetic methodologies to access this privileged scaffold in an atom- and step-economical fashion [19-21].
Traditional methods relied on the Fisher-Borsche synthesis [22,23], the Graebe-Ullmann synthesis [24,25], or the Cadogan cyclization [26,27]. These protocols have proven to be highly effective. However, they often require harsh reaction conditions, high temperatures, and complex starting materials, which frequently result in the formation of potentially toxic by-products. These factors negatively impact efficiency and sustainability. Additionally, the regioselective installation of various substituents on different positions of the aromatic ring remains challenging. For example, the presence of electron-withdrawing nitro groups or electron-donating methoxy substituents significantly reduces the reaction yield.
As a result, there is a continued need for the development of novel, effective synthetic routes, such as annulation from indole derivatives [28,29], ring-closing metathesis [30], oxidative Heck coupling reactions (Fujiwara-Moritani reaction) [31], and cycloaddition [32,33].
The growing interest in developing atom-economy-driven methodologies has the potential to revolutionize the synthesis of heterocycles. This trend has significantly advanced synthetic strategies that enable the formation of C–C or C–X (X = N, O, S) bonds via direct C–H functionalization, providing an effective and cost-efficient approach to constructing crucial structural motifs [34-37]. Such methodologies facilitate the synthesis of natural products and APIs from simple, readily available starting materials [38-40]. This approach ensures a promising, direct pathway characterized by excellent atom and step economy, which has proven to be highly valuable in accessing carbazoles over the past decade [41-43].
This review highlights the substantial efforts invested between 2015 and 2024 in advancing catalytic Csp2–H functionalization for synthesizing the privileged carbazole scaffold. In this review, synthetic methodologies are categorized into multi-component intermolecular and intramolecular Csp2–H functionalization.
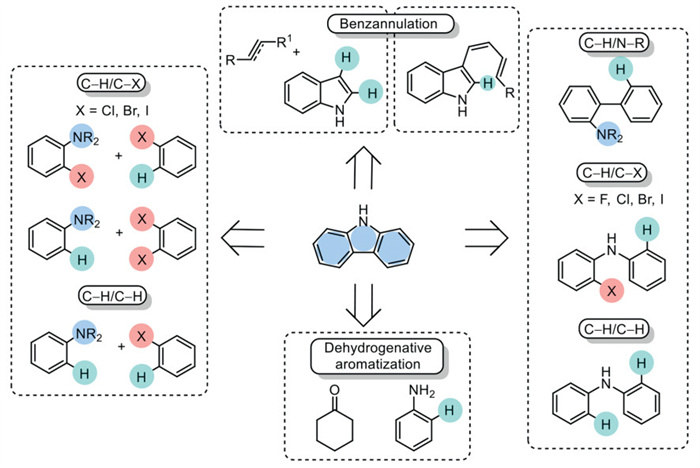
Among multi-component intermolecular catalytic Csp2–H functionalization, both Csp2–Csp2 and Csp2–N bonds are to be synthesized in the same step via a Buchwald-Hartwig domino amination starting from 2-halo aniline and aryl halide or anilines and aryl dihalide, or even via a Suzuki-Miyaura tandem cross-coupling followed by SNAr reaction of aniline-derived boronic esters with aryl dihalide (Fig. 2). Intramolecular coupling allows the formation of Csp2–N bonds via the coupling of Csp2–H and N–H bonds of a [1,1′-biphenyl]−2-amine or Csp2–H and Csp2–X (X = F, Cl, Br, I) bond starting from halogenated diarylamines. An intriguing alternative involves the direct activation of Csp2–H/Csp2–H bonds within the diarylamines scaffold, improving the atom economy by circumventing the need for derivatization. The distinctions between the functionalized partners of the Csp2–H bond are explored, classifying them into Csp2–X (X = F, Cl, Br, I), Csp2–H, and N–R (Fig. 2).
Figure 2
Figure 2.
An overview of carbazole production: common synthetic pathways involving the synthesis of carbazoles via Csp2–H functionalization.
The dehydrogenative aromatization process offers a promising approach for carbazole synthesis, allowing for the use of non-aromatic substrates as starting materials. The last route that can be considered is benzannulation, which is a well-established method for synthesizing aromatic compounds such as carbazoles, benzocarbazoles, indolocarbazoles, and carbolines. This strategy encompasses several types, including electrocyclization, formal cycloaddition, transition-metal-catalyzed Csp2–H bond functionalization, and Brønsted or Lewis acid-catalyzed electrophilic annulation. Given the extensive application of this method, intramolecular and intermolecular benzannulation strategies have been thoroughly covered in recent reviews. For comprehensive coverage of developments, we suggest referring to the following review [44,45].
The final approach examined in this review is the derivatization of simple, unfunctionalized carbazoles through direct Csp2–H functionalization, a highly effective strategy for enhancing scaffold complexity. This methodology circumvents the need for pre-installed functional groups and eliminates cumbersome protection and deprotection steps. Leveraging the electron-rich nature of the C3 position, direct Csp2–H functionalization through electrophilic aromatic substitution (EAS) processes is well-established. However, selective functionalization at other positions, particularly the C1 position, remains a significant challenge. The concluding section of the review highlights more recent advancements that enable targeted functionalization of carbazoles, expanding the scope of this synthetic strategy.
2.
Intramolecular Csp2–H functionalization
The most efficient and well-documented approach to synthesizing carbazoles is intramolecular Csp2–H functionalization. This method can be broadly categorized into two main types based on the formation of either a Csp2–C or a Csp2–N bond. The first category encompasses the Csp2–H activation of 2-halo-N-arylanilines and the double Csp2–H/Csp2–H functionalization of diarylamines. The second category involves Csp2–H/N–R functionalization of 2-aminobiphenyl.
2.1
Csp2–H/C–X coupling
The first category to be considered involves the Csp2–H activation of 2-halo-N-arylanilines, enabling the closure of the carbazole ring through the formation of a new Csp2–Csp2 bond. This methodology is well-documented and typically utilizes a Pd-catalyst under basic conditions. Giving a brief examination of examples from periods prior to those considered for this review, Bedford and Cazin developed a one-pot, sequential Pd-catalyzed N-arylation and Csp2–H activation reaction starting from aryl bromides and 2–chloro-N-alkylated anilines, using NaOtBu as the base and PtBu3 as a ligand [46]. Later, Fagnou et al. used the combination of Pd(OAc)2 and N-heterocyclic carbene (NHC) or phosphine ligand in the presence of K2CO3 [47-49]. Bedford et al. used a combination of Pd(OAc)2 as a catalyst, PtBu3 as a ligand, and NaOtBu as a base [50]; and Ackermann et al. employed a combination of Pd(OAc)2 as a catalyst, PCy3 as a ligand, and K3PO4 as a base [51]; while Buchwald et al. used Pd2(dba)3, TrixiePhos, and NaOtBu [52]. Ligand-free Pd-catalyzed intramolecular Csp2–H arylation has also been reported. Sakamoto et al. prepared carbolines via a Pd-catalyzed intramolecular Csp2–H arylation of ortho–bromo-substituted anilinopyridines in the presence of Na2CO3 [53], and Fagnou et al. prepared carbazoles using the heterogeneous Pd(OH)2/C in the presence of KOAc [54]. Recently, Das et al. synthesized naturally occurring carbazoles via a Pd-catalyzed intramolecular Csp2–H arylation of 2-iodo-N-arylanilines using K2CO3 [55]. However, many of these methods have drawbacks, such as high catalyst loading, reliance on sensitive or expensive ligands, use of inorganic bases, and reaction conditions that are incompatible with base-sensitive functional groups. Consequently, improved protocols that utilize inexpensive reagents, catalysts, and additives under milder reaction conditions are still needed. Recent advancements in this area are presented.
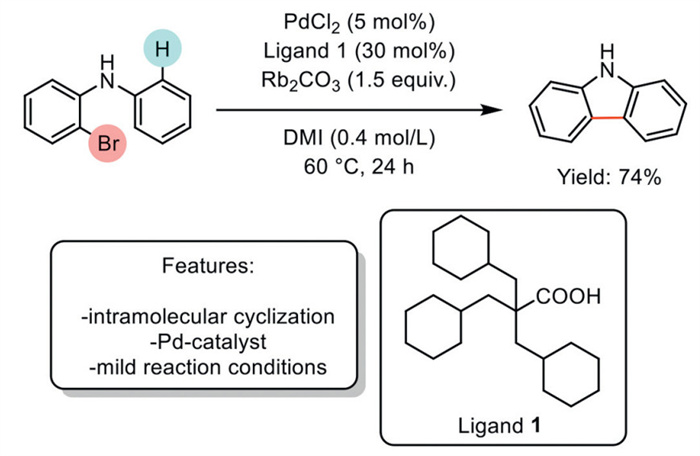
In 2018, Fujihara, Tsuji et al. reported the use of a bulky carboxylic acid, tri(cyclohexylmethyl)acetic acid, as an efficient ligand in Pd-catalyzed intramolecular Csp2–H arylation reactions. This methodology was applied in the synthesis of carbazole. Typically, such reactions require high temperatures; however, this reaction proceeds smoothly at moderate temperatures due to the steric bulk of the carboxylate ligand, which accelerates the rate-determining Csp2–H bond activation step in the catalytic cycle. The optimized protocol requires the use of Rb2CO3 as a base to facilitate the effective deprotonation of the hydrogen involved in the functionalization. Although this methodology is effective for synthesizing various heterocycles and carbocycles, it is limited to a single example of carbazole synthesis (Scheme 1) [56].
Scheme 1
Scheme 1.
Bulky carboxylic acid in Pd-catalyzed intramolecular Csp2–H arylation reactions to carbazoles.
In 2019, Dhimne, Alam, et al. developed a two-step consecutive Cu-catalyzed Chan-Lam N-arylation of various o-iodoanilines and boronic acids followed by the intramolecular Pd-catalyzed aryl Csp2–H activation of 2-iodo-N-arylanilines. The first step involves the use of Cu(OAc)2·H2O as a catalyst, and the second step involves Pd(OAc)2. In both steps, 1,8-diazabicyclo[5.4.0]undec–7-ene (DBU) was employed as an effective base, and toluene was used as the solvent. The developed methodologies provide access to a diverse array of 30 carbazoles, from moderate to excellent yield, by incorporating various electron-donating and electron-withdrawing substituents, including halogens and other reactive functional groups. Despite their wide applicability, a general trend observed is a reduction in yield when electron-withdrawing substituents are present on the second aromatic ring. Conversely, higher yields are obtained when the 2-iodoaniline ring contains an electron-withdrawing group and/or the second aromatic ring features an electron-donating substituent. Furthermore, the presence of an electron-withdrawing N-tosyl group significantly reduced conversions and yields, even after 24 h, with no cyclization occurring through the Csp2–H bond of the p-tolyl group. This optimized protocol was successfully utilized in the synthesis of two natural products: Clausine L and Clausine H. Related to the second step of the consecutive protocols, the authors proposed a plausible mechanism in which, after the in situ formation of a Pd(0) species, an initial oxidative addition occurred, generating the Pd-δ-complex. The Csp2–H activation step occurred via a δ-bond metathesis or through an SEAr path, in the case of electron-rich aryl. DBU promotes the formation of the palladacycle, and the final reductive elimination leads to carbazole formation (Scheme 2) [57].
Scheme 2
Scheme 2.
Consecutive Cu-catalyzed Chan-Lam N-arylation of o-iodoanilines and boronic acids followed by the intramolecular Pd-catalyzed aryl Csp2–H activation.
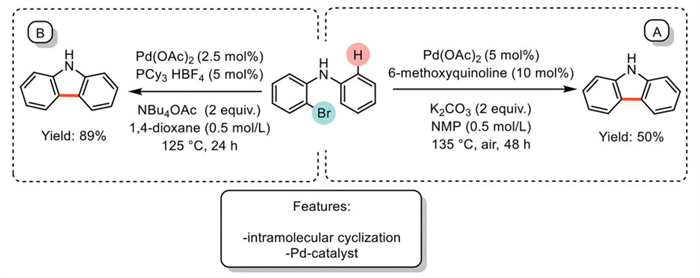
In 2021, McGlacken et al. introduced a similar approach, using a quinoline ligand in Pd-catalyzed intramolecular protocols for carbazole synthesis under basic conditions provided by K2CO3. The optimized methodology was applied in the synthesis of benzofurans. However, the authors suggest that this method could be potentially extended to obtain carbazoles. Notably, carbazole was successfully synthesized with a yield of 50% (Scheme 3a) [58]. The same authors reported an NBu4OAc-mediated Pd-catalyzed intramolecular protocol for dibenzofuran synthesis, which was applied to the synthesis of carbazoles using PCy3·HBF4 as the ligand, achieving an 89% yield (Scheme 3b) [59]. Although both methodologies are effective for synthesizing various benzofurans, they are limited to a single example of carbazole synthesis.
Scheme 3
Scheme 3.
Pd-catalyzed intramolecular protocols for carbazole synthesis.
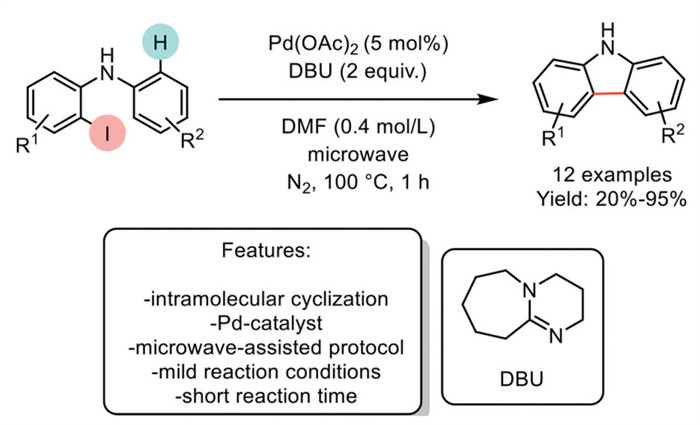
In 2022, Ghosh et al. presented a Pd-catalyzed microwave-assisted synthesis of functionalized carbazoles from 2-iodo-N-arylanilines under mild reaction conditions. This method demonstrated the potential benefits of DBU as a base in the synthesis of carbazoles. The use of microwave irradiation led to significant improvements, including shortened reaction times, enhanced yields ranging from 20% to 95%, and reduced requirements for base and solvent. Furthermore, the authors conducted a docking study to investigate the interaction between the synthesized carbazoles and calf-thymus DNA, highlighting the potential biological applications of their work (Scheme 4) [60].
Scheme 4
Scheme 4.
Pd-catalyzed microwave-assisted synthesis of functionalized carbazoles from 2-iodo-N-arylanilines.
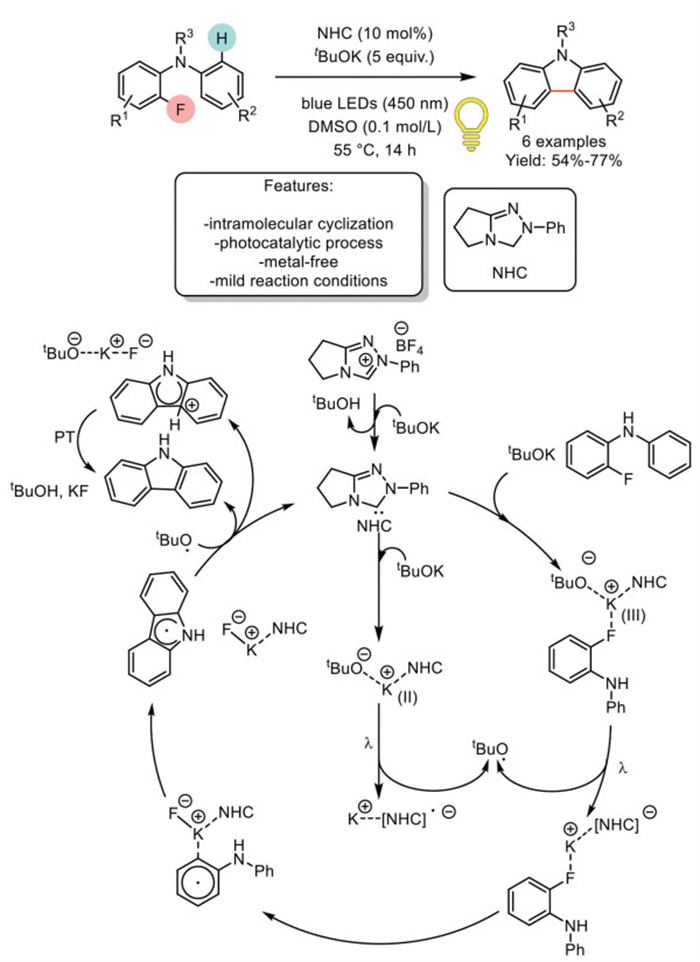
In 2023, Chen et al. made significant progress in the synthesis of carbazoles by developing a transition metal-free, blue light-induced method. This approach utilized NHC as a catalyst for the single-electron reduction of monofluoroarenes to carbazoles. The combination of NHC and the base tBuOK formed a highly effective photoactive complex under blue LEDs irradiation in DMSO, which was successfully utilized to synthesize 6 different carbazoles via intramolecular cross-coupling. This method achieved moderate to good yields under mild reaction conditions. However, the authors did not investigate the influence of electron-rich or electron-poor substituents. The method was applied exclusively to simple carbazoles, as well as methyl and N-methyl-substituted carbazoles, dimeric carbazoles, and phenyl-substituted carbazoles. A plausible reaction mechanism was proposed, and the corresponding mechanism for fluorocarbazoles, consistent with these findings, is presented here. The mechanism involves the in situ formation of a free NHC species, either as a binary or ternary complex with tBuOK and fluorophenyl species. Both complexes are photoactive under irradiation with blue light. Photoactivation promotes a single-electron transfer (SET) process from the tBuO- anion to the NHC, generating an NHC radical anion. Subsequently, the complex undergoes a further SET event, resulting in the formation of a phenyl radical. Through a concerted mechanism, the SET induces Caryl−F cleavage, and KF bond formation occurs simultaneously. In the following steps, the radical reacts intramolecularly via a sequence of radical addition, single-electron oxidation, and deprotonation, yielding the final product. As an alternative to the single-electron oxidation/deprotonation sequence, hydrogen atom transfer (HAT) pathways via tert–butoxyl/methyl radicals could also lead to the product (Scheme 5) [61].
Scheme 5
Scheme 5.
Blue light-induced NHC-catalyzed single electron reduction of monofluoroarenes to carbazoles.
Aiming to maximize atom economy and approaching the ideal case of Csp2–H activation, where both partners are unfunctionalized, developing double Csp2–H/Csp2–H functionalization to form novel Csp2–Csp2 bonds and close the carbazole ring is highly desirable. However, there are limited examples documenting the direct activation of Csp2–H/Csp2–H bonds in diarylamine scaffolds. These scaffolds are commercially and synthetically useful starting materials with a wide range of applications. They are commonly prepared from aniline precursors through metal-catalyzed Csp2–N coupling reactions, such as the Ullmann and Buchwald-Hartwig reactions [62-64]. Generally, these procedures involve the use of a Pd-catalyst in combination with stoichiometric inorganic oxidants [65,66]. Fagnou et al. developed Pd(Ⅱ)-catalyzed oxidative intramolecular synthesis of carbazoles under air as a terminal oxidant [67]. Tokunaga et al. developed supported palladium hydroxide-catalyzed intramolecular double Csp2–H/Csp2–H bond functionalization for the synthesis of carbazoles using O2 under pressure as the terminal oxidant [68]. Recent progress in this direction is presented.
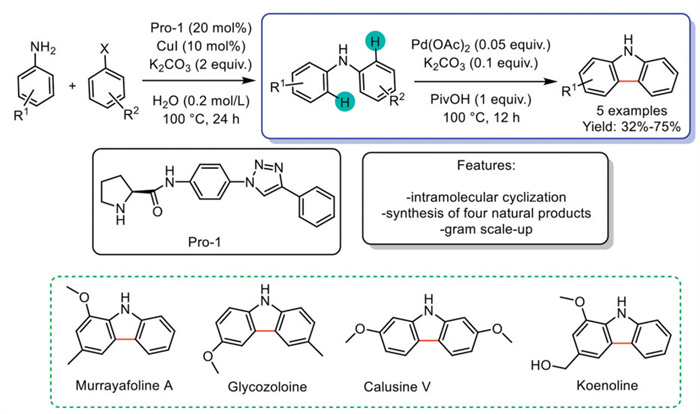
In 2018, Dash et al. introduced a consecutive two-step process for synthesizing carbazoles from anilines. First, the protocol was optimized for the synthesis of diarylamines via a Cu-catalyzed Ullmann-type cross-coupling. The reaction employed CuI as the catalyst and a prolinamide ligand and was performed in water using K2CO3 as the base. Subsequently, carbazole formation was achieved in the next step through Pd-catalysis along with pivalic acid, and resorting to the use of a catalytic amount of K2CO3. This approach enabled direct access to naturally occurring carbazoles, eliminating the need for additional pre-functionalization steps (Scheme 6) [69].
Scheme 6
Scheme 6.
Consecutive two-step Cu-catalyzed Ullmann-type cross-coupling and Pd-catalyzed intramolecular Csp2–H activation to carbazoles.
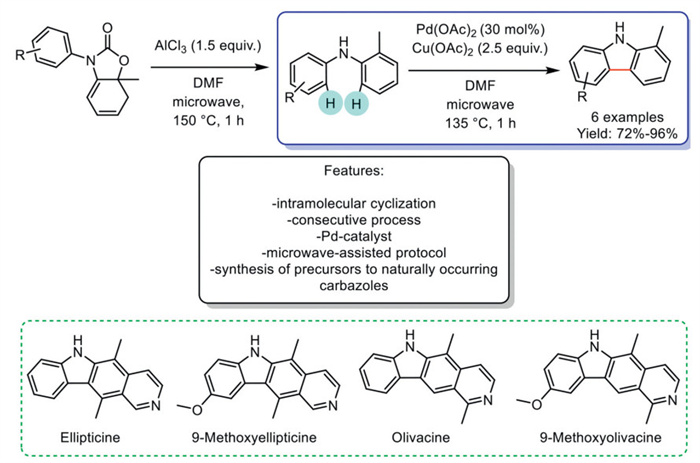
In 2022, Tomariz et al. reported an innovative microwave-promoted one-pot cascade reaction of 4-oxazolin-2-ones, yielding methylated diarylamines that were subsequently cyclized into a series of carbazoles. The process began with the derivation of diarylamines from 4-oxazolin-2-ones, followed by hydrolysis and aromatization using AlCl3 under microwave irradiation. Carbazoles were then synthesized from these diarylamines under Pd(Ⅱ) catalysis, employing an excess of Cu(OAc)2 as an oxidant. This Pd(Ⅱ)-catalyzed method, based on oxidative Csp2–H/Csp2–H functionalization, enabled the formal total synthesis of four naturally occurring pyrido[4,3-b]carbazole alkaloids (ellipticine, 9-methoxyellipticine, olivacine, and 9-methoxyolivacine) by synthesizing key precursor compounds. Additionally, a three-step conversion with the Vilsmeier-Haack formylation species enabled the synthesis of a series of 1-methyl and 1,4-dimethyl-3-formylcarbazoles. The limitation of the developed methodology is its restricted applicability, as it is mainly effective for methoxy and methyl-substituted carbazoles (Scheme 7) [70].
Scheme 7
Scheme 7.
Pd-catalyzed synthesis of carbazoles with Cu(OAc)2 as an oxidant under microwave irradiation.
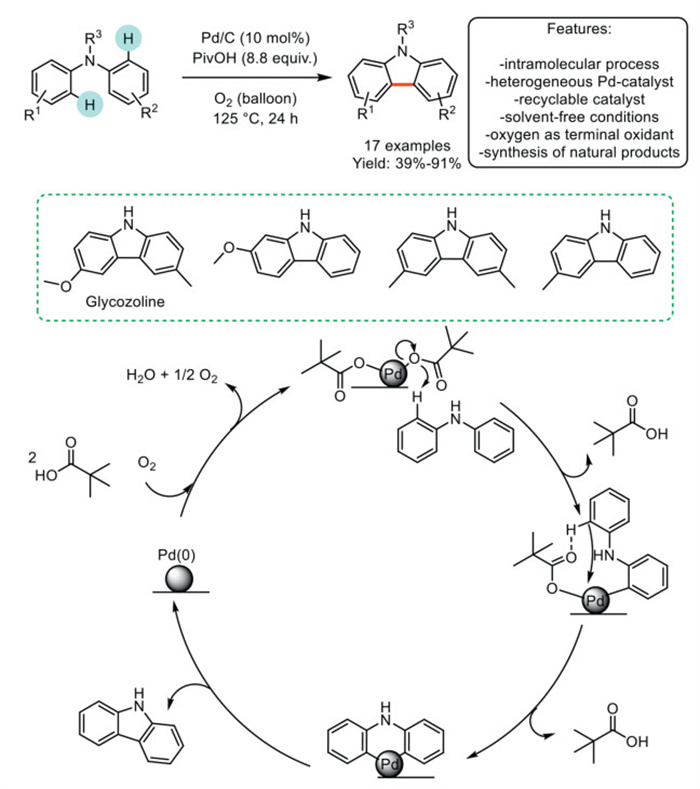
In 2024, Vaccaro et al. developed a sustainable access route to carbazoles via Csp2–H/Csp2–H oxidative functionalization/cyclization of diarylamines and triarylamines. The authors utilized the readily available, commercial, and recyclable Pd/C as a heterogeneous catalyst under mild conditions, thereby eliminating the need for stoichiometric or excessive amounts of organic or inorganic oxidants. These methodologies relied on low-pressure molecular oxygen to ensure effective waste reduction and were conducted under solvent-free conditions, utilizing only pivalic acid as a ligand and acidic additive. The versatility of these protocols was demonstrated in the synthesis of 17 structurally diverse carbazoles, encompassing both natural products and light-emitting N-aryl-carbazole derivatives. The established protocol necessitates a precise range of reactivity, as an increased electron density in diarylamines negatively impacts the reaction, promoting a side reaction in which substrates react with pivalic acid to form C–O bonds. Moreover, the use of diarylamines containing electron-withdrawing groups leads to a significant decrease in reactivity. The authors proposed a plausible mechanism involving Pd(Ⅱ)–pivalate species undergoing two electrophilic aromatic substitutions to form a palladate species. Following reductive elimination, carbazole formation ensued, with Pd(0) undergoing oxidative regeneration facilitated by the oxygen atmosphere (Scheme 8) [71].
Scheme 8
Scheme 8.
Pd/C catalyzed the synthesis of carbazoles from diarylmines with O2 as a terminal oxidant.
One of the most straightforward methods for constructing carbazoles is the dehydrogenative Csp2−H/N−H coupling of 2-aminobiphenyls. Amino groups substituted with carbonyl, sulfonyl, alkyl, and heteroaryl groups have proven to be effective directing groups for Csp2−H bond cleavage at the 2′-position. Under Pd or Cu catalysis, these reactions yield N-substituted carbazoles [72-77]. Herein, recent examples of this methodology are reported.
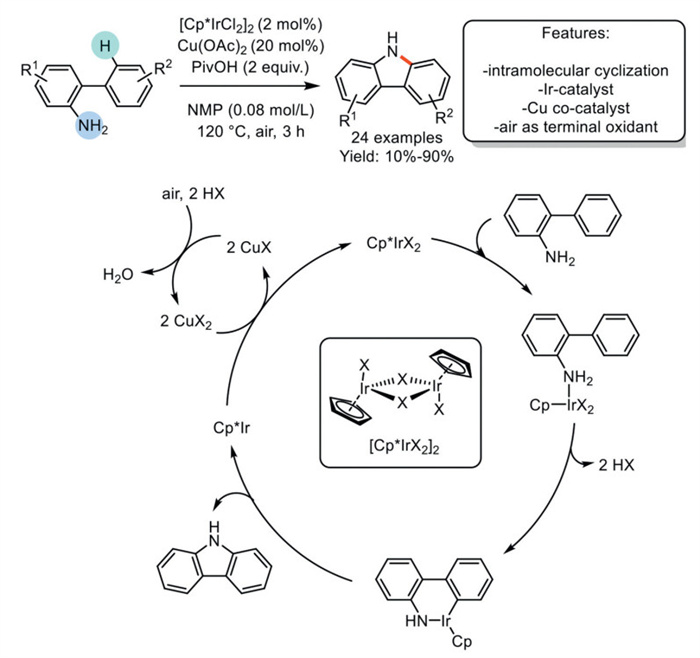
In 2015, Satoh and Miura et al. developed an intramolecular direct Csp2–H/N–H Ir-catalyzed dehydrogenative cyclization of 2-aminobiphenyls in the presence of Cu-cocatalyst and under air as a terminal oxidant to yield carbazoles. The optimized protocol requires the use of [Cp*IrCl2]2 as the catalyst and Cu(OAc)2 as the co-catalyst, with pivalic acid (PivOH) as the acid. This approach facilitated the synthesis of 24 examples with yields ranging from moderate to excellent. A plausible mechanism involves the coordination of the nitrogen to Cp*-Ir(Ⅲ). The amino-directed Csp2–H bond cleavage of this complex takes place, forming an iridacycle-intermediate, which undergoes Csp2−N reductive elimination to afford the desired carbazole and Cp*-Ir(Ⅰ). The latter is re-oxidized by the Cu(Ⅱ)-cocatalyst, forming Cu(Ⅰ), which is then re-oxidized by air as a terminal oxidant (Scheme 9). This reaction can lead to the formation of dimerization by-products, which may reduce the overall yield of the desired carbazole. Additionally, the reaction mechanism for the formation of this by-product is unclear, and further tests are necessary to elucidate the reaction pathway [78].
Scheme 9
Scheme 9.
Ir-catalyzed dehydrogenative cyclization of 2-aminobiphenyls with Cu-cocatalyst and air as a terminal oxidant to carbazoles.
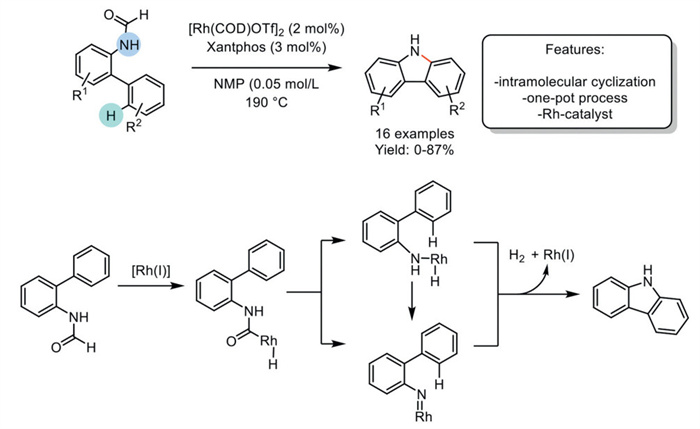
In 2015, Feng et al. developed a one-pot Rh(Ⅰ)-catalyzed synthesis of carbazoles via a direct Csp2–H amination starting from N-formyl 2-amino biaryls. The optimized protocol employed [Rh(COD)OTf]2 (COD = 1,5-cyclooctadiene) as the preferred catalyst and Xantphos as the ligand. It demonstrated broad applicability across a broad spectrum of 2-aminoacyl derivatives, yielding moderate to good results, irrespective of the presence of electron-donating or electron-withdrawing groups. However, the method results in reduced yields when applied to substrates with substantial steric hindrance. Additionally, it fails to produce products when N-substituted biaryl derivatives are used as reagents, highlighting a limitation in the substrate scope for certain compounds. Control experiments elucidated a plausible mechanism involving an initial oxidative addition of the Rh catalyst to the C–H bond of the formamide species, followed by a decarboxylation step, and concluding with a Csp2–H activation of the phenyl, leading to H2 and Rh(Ⅰ) species and ultimately yielding the desired product. (Scheme 10) [79].
Scheme 10
Scheme 10.
Rh-catalyzed synthesis of carbazoles via a direct Csp2–H amination from N-formyl 2-amino biaryls [79].
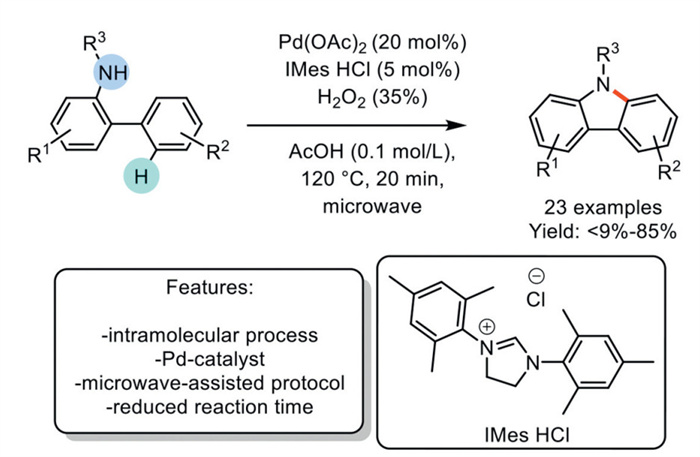
In 2016, Bjørsvik et al. developed a protocol for the synthesis of carbazoles from 2-aminobiphenyls and 2-N-acetylaminobiphenyls, involving a tandem Pd-catalyzed Csp2–H activation and intramolecular Csp2–N bond formation under microwave irradiation. The process was optimized using Pd(OAc)2 as the catalyst and IMes (1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene) as the ligand, with acetic acid (AcOH) serving as both the acid and the solvent. After ring closure, the reduced Pd-catalyst was oxidized to Pd(Ⅱ) by hydrogen peroxide. The developed methodology was effective with both electron-withdrawing and electron-donating groups, yielding 23 target carbazoles in a reduced reaction time. However, the method leads to low and zero isolated yields, respectively, with substrates replaced with methoxy substituent and free hydroxy groups; the latter must therefore be protected. Furthermore, the protocol showcased its capability by obtaining a precursor of carbazomycin (Scheme 11) [80].
Scheme 11
Scheme 11.
Pd-catalyzed synthesis of carbazoles via a direct Csp2–H amination from 2-N-acetylaminobiphenyls.
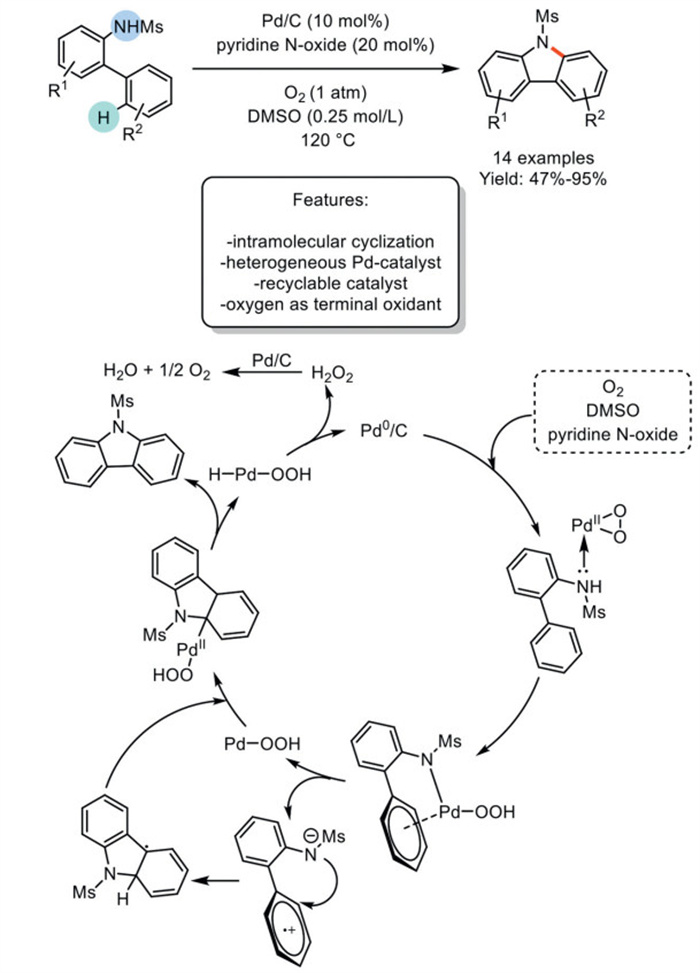
In 2016, Sajiki et al. demonstrated the efficacy of Pd/C in catalyzing the intramolecular Csp2–H amination of various N-mesylated 2-aminobiphenyls. This reaction, catalyzed by Pd and pyridine N-oxide and carried out in dimethyl sulfoxide (DMSO) under an oxygen atmosphere, afforded the corresponding N-mesylcarbazoles. The scope is restricted to N-protected aminobiphenyls, and the resulting carbazole derivatives require deprotection using Cs2CO3 in tetrahydrofuran (THF) and methanol (MeOH) solution. The authors proposed a plausible mechanism wherein Pd(0) species are oxidized to Pd(Ⅱ)-oxide through the oxidative cooperation of O2, DMSO, and pyridine N-oxide, leaching from Pd/C. According to the proposed mechanism, the sulfonamide moiety of N-Ms-2-aminobiphenyl undergoes a nucleophilic attack on the Pd-catalyst, forming a Pd(Ⅱ) amide complex. This complex leads to the formation of a charge-transfer complex. A single electron transfer from the aromatic ring to Pd(Ⅱ) generates a Pd(Ⅰ)-OOH species and a radical species. Subsequent intramolecular cyclization transforms the radical and Pd(Ⅰ)-OOH into a complex, facilitating β-hydrogen elimination to produce the desired carbazole and H-Pd(Ⅱ)-OOH. The ensuing reductive elimination yields Pd(0) species and H2O2. Furthermore, H2O2 formed during the reaction can be efficiently decomposed to water and O2 in the presence of 10% Pd/C (Scheme 12) [81].
Scheme 12
Scheme 12.
Pd/C catalyzes intramolecular Csp2–H amination of N-mesylated 2-aminobiphenyls to carbazoles.
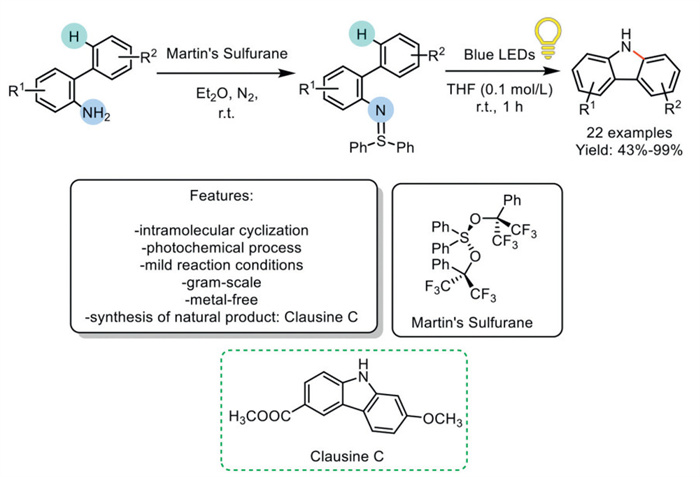
In 2020, Hashmi et al. developed a visible-light-induced intramolecular Csp2–H amination reaction using ortho-substituted aryl sulfilimines, rather than the corresponding hazardous azides, as a new generation of nitrene precursors. Blue LEDs promote the decomposition of sulfilimine, leading to the formation of the target product in THF. The protocol worked under mild reaction conditions, enabling the synthesis of a wide range of carbazoles with moderate to excellent yields. Despite the excellent features of this method, the reactivity of the second step may be limited to aryl sulfilimine substrates, requiring the use of Martin's sulfurane and consequently lowering the overall atom economy of the process. To highlight the protocol's efficiency, a gram-scale synthesis of the naturally occurring product Clausine C was performed (Scheme 13) [82].
Scheme 13
Scheme 13.
Visible-light-induced intramolecular Csp2–H amination of ortho-substituted aryl sulfilimines to carbazoles.
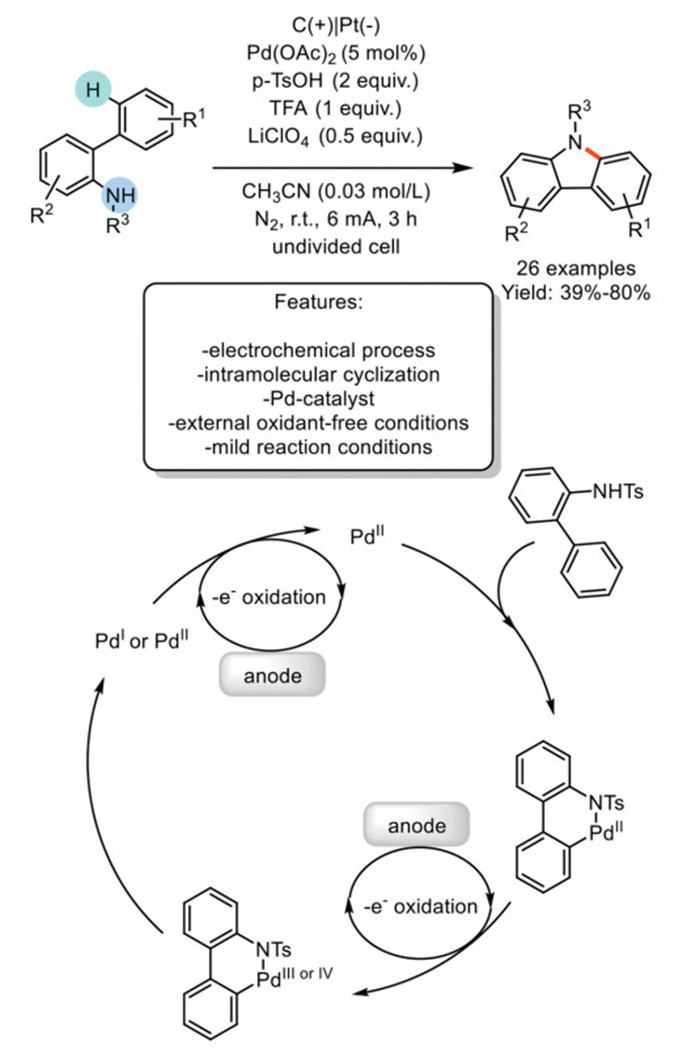
In 2021, Zhang and Lei et al. developed an electrochemical Pd-catalyzed intramolecular oxidative Csp2–H amination of 2-amidobiaryls to obtain carbazoles under mild reaction conditions, thereby avoiding the use of an external chemical oxidant. The reaction was carried out in an undivided cell under constant current electrolysis (CCE) with Pd(OAc)2 as a catalyst and LiClO4 as the supporting electrolyte. The methodology was adopted for the synthesis of a wide range of substrates, including halogen-containing carbazoles, which are useful for the introduction of additional functionalization. To further demonstrate the utility of this protocol, the scale-up reaction was carried out on a 7.0 mmol scale. The method is limited to using 2-amidobiaryls N-substituted with protective groups, resulting in the formation of N-protected carbazoles that require subsequent deprotection. Regarding the mechanism, the authors proposed that the coordination of the amido group of the 2-amido biaryls to Pd(OAc)2 facilitates cyclopalladation, resulting in the formation of a six-membered palladacycle. The palladacycle may be oxidized to a Pd(Ⅲ) or Pd(Ⅳ) complex. Then, the palladium complex undergoes a reductive elimination process to give the desired product and Pd(Ⅰ) or Pd(Ⅱ). Pd(Ⅰ) could be oxidized at the anode to give Pd(Ⅱ) to finish the catalytic cycle. The reductive half-reaction involves the formation of H2 at the cathode (Scheme 14) [83].
Scheme 14
Scheme 14.
Pd-catalyzed intramolecular oxidative Csp2–H amination of 2-amidobiaryls to obtain carbazoles.
Azides stand out as remarkable amino group sources in Csp2–H amination; indeed, they are readily prepared, exhibit efficiency under non-oxidative and non-basic conditions, and produce solely N2 gas as the sole by-product. Consequently, the intramolecular Csp2–H amination of 2-azido biphenyls has emerged as a pivotal approach to accessing N–H carbazoles.
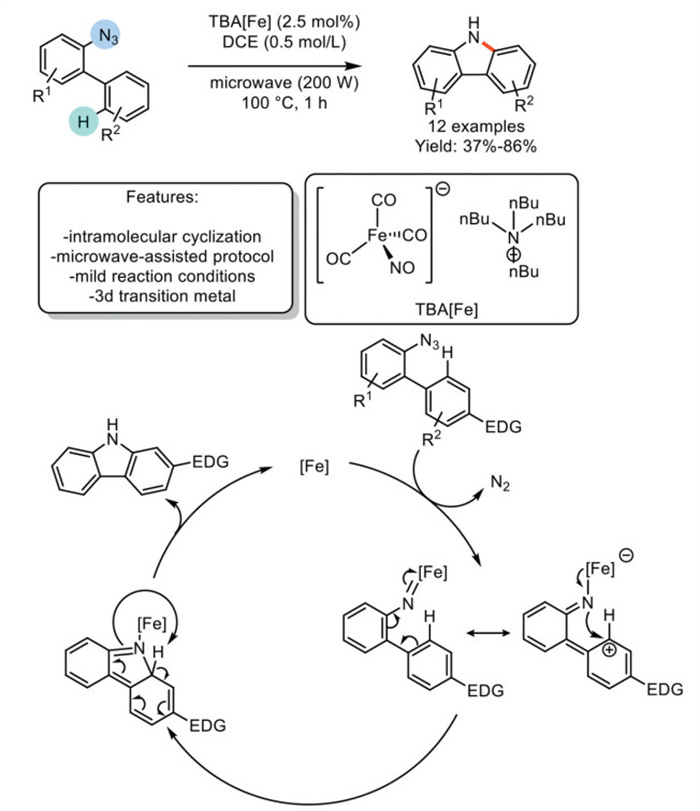
In 2016, Plietker et al. developed a direct intramolecular Csp2–H amination of 2-azido biphenyls into the corresponding carbazoles, catalyzed by the nucleophilic iron complex Bu4N[Fe(CO)3(NO)] (TBA[Fe]), resulting in N2 formation as the sole by-product. Various substituted carbazole derivatives were obtained in good to excellent yields under microwave irradiation, accelerating the reaction through more efficient energy transfer compared to the thermal version and significantly reducing the reaction time. Among these features, the primary drawbacks are the use of a toxic solvent, such as 1,2-dichloroethane, which has been identified as the only effective option, and the relatively narrow scope of the method. The authors proposed a reaction mechanism in which the [Fe(CO)3(NO)]- anion reacts with aryl azide, releasing N2 and forming the iron nitrene intermediate, which generates the zwitterionic mesomeric structure with a partial positive charge on the ortho carbon atom. Metal-to-ligand charge transfer sets the stage for the Csp2–N bond-forming process, yielding the intermediate, which is then transformed into carbazole through a 1,5-hydrogen shift (Scheme 15) [84].
Scheme 15
Scheme 15.
Iron-catalyzed intramolecular Csp2–H amination of α-azidobiaryls to carbazoles.
In 2017, Collins et al. reported that purple LEDs mediate photochemical intramolecular Csp2–H amination of 2-azido biphenyls, leading to the formation of carbazoles and polycyclic heterocycles within a continuous flow reactor. This approach offered a versatile route to various carbazoles and polycyclic heteroaromatic skeletons under mild reaction conditions. However, with electron-withdrawing groups, the optimal conditions were achieved under 394 nm irradiation. In contrast, higher yields were obtained for substrates with electron-donating groups when irradiated at 254 nm. Remarkably, the reaction was successfully applied to the synthesis of carprofen. The authors delineated a plausible mechanism for the photochemical generation of carbazoles. The reaction initiates with the formation of a singlet biphenyl nitrene, which subsequently undergoes addition to an adjacent aromatic ring. The resulting isocarbazole intermediate undergoes rearrangement to furnish the corresponding carbazoles (Scheme 16) [85].
Scheme 16
Scheme 16.
Purple LEDs-mediated photochemical intramolecular Csp2–H amination of 2-azido biphenyls to carbazoles.
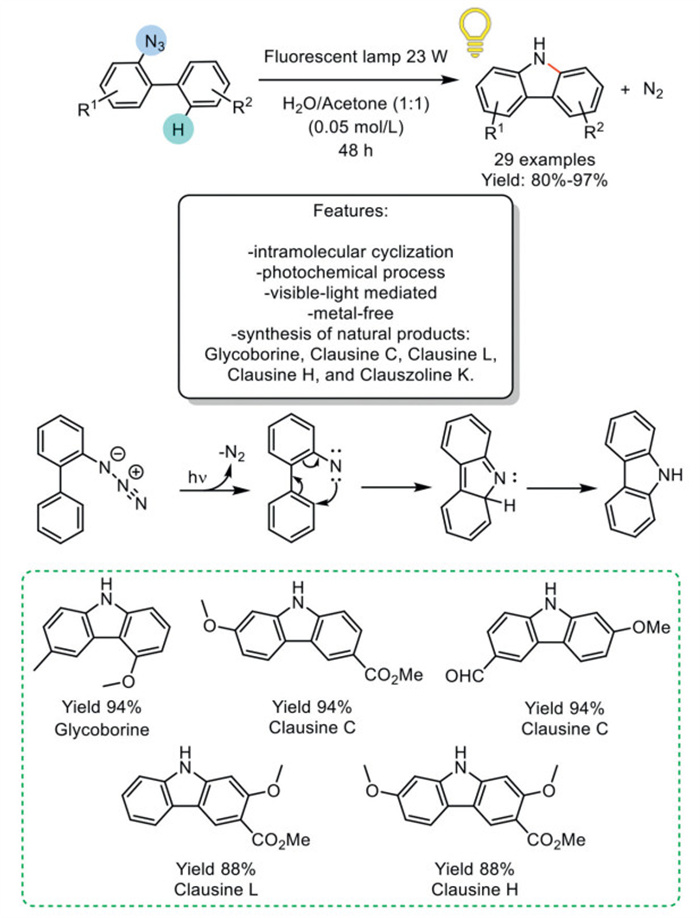
In 2018, Lu et al. reported a method for the visible-light-promoted intramolecular Csp2–H amination of 2-azido biphenyls, yielding carbazoles, utilizing water as a solvent and operating under mild reaction conditions. This protocol facilitated the synthesis of a diverse range of functionalized bioactive natural alkaloids, with N2 as the sole byproduct. Through optimized reaction conditions, a broad spectrum of variably functionalized carbazoles was synthesized, boasting yields ranging from good to excellent. Among the synthesized compounds were notable natural alkaloids, including glycoborine, clausine C, clausine L, clausine H, and clauszoline K. The authors delineated a plausible reaction mechanism for the photodecomposition of azides, wherein 2-azido biphenyls are transformed into a nitrene intermediate via the release of N2 under visible light. Subsequently, an electrocyclic ring closure enables the formation of the isocarbazole intermediate, which, via a [1,5] H-shift, undergoes conversion to the final product through tautomerization. Kinetic isotope effects analysis unveiled that the extrusion of N2 serves as the rate-determining step in this intricate process (Scheme 17) [86].
Scheme 17
Scheme 17.
Visible-light-promoted intramolecular Csp2–H amination of 2-azido biphenyls to yield carbazoles.
In 2021, Jana et al. introduced an intramolecular Csp2–H amination of 2-azido biphenyls, enabling the synthesis of N–H carbazoles. This approach was successfully employed to synthesize (±)-Mahanine and 12 other naturally occurring carbazole alkaloids. The Suzuki coupling reaction enables the synthesis of diarylamines, which are subsequently converted to the corresponding azides via diazotization with tert–butylnitrite and trimethylsilylazide. The key 2-azidobiphenyls were then converted, and metal-free Csp2–N coupling was performed to obtain the carbazole core, which is then converted to naturally occurring carbazole alkaloids. Despite the efficiency of the Csp2–N coupling step, the protocol's applicability has been tested on only a limited number of substrates. Additionally, since the hydroxyl groups are protected, a subsequent deprotection step is necessary. (Scheme 18) [87].
Scheme 18
Scheme 18.
Intramolecular catalyst-free Csp2–H amination of 2-azido biphenyls to access N–H carbazoles.
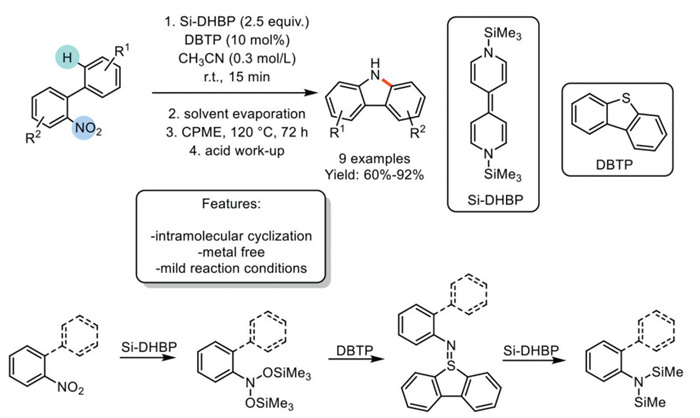
In 2018, Mashima and Tsurugi et al. developed a metal-free deoxygenation and reductive distillation method for nitroarenes using organosilicon-reducing reagents (Si-DHBP) and dibenzothiophene (DBTP) as a catalyst to synthesize carbazole structural moieties. This process, conducted under mild reaction conditions, demonstrated broad functional group tolerance and enabled the synthesis of 9 variously functionalized carbazoles with yields ranging from good to excellent. However, the process requires multiple synthetic steps, several additives, and an excess of the organosilicon reagent. The authors proposed a plausible reaction mechanism wherein the deoxygenation of nitrobenzene is promoted by an excess of Si-DHBP, resulting in the formation of N, O-bis(trimethylsilyl)phenylhydroxylamine and the release of (Me3Si)2O. Upon heating, the thermal decomposition of N, O-bis(trimethylsilyl)phenylhydroxylamine produces an unstable nitrene intermediate. DBTP acts as a catalytic stabilizer by trapping the nitrene species through the formation of the corresponding sulfimine. Finally, the N=S bond of the intermediate is reduced by Si-DHBP to form N, N-bis(trimethylsilyl)aniline, regenerating the DBTP catalyst. Additionally, 2-nitrobiphenyls were successfully converted to carbazoles in cyclopentyl methyl ether (CPME) through the in situ generation of arylnitrene species, which were inserted into the Csp2–H bond of the adjacent aryl ring (Scheme 19) [88].
Scheme 19
Scheme 19.
Metal-free deoxygenation and reductive distillation method for nitroarenes with organosilicon-reducing reagents to synthesize carbazole structural moieties.
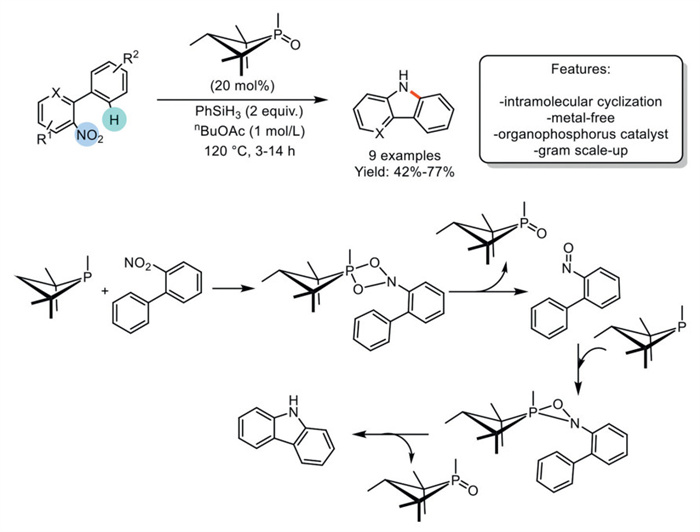
In 2018, Radosevich et al. developed a Cadogan cyclization catalyzed by biphilic organophosphorus compounds. They employed a small-ring phosphacycloalkane, 1,2,2,3,4,4-hexamethylphosphetane, to catalyze the intramolecular Csp2–N bond-forming heterocyclization of o-nitrobiaryl in the presence of a hydrosilane terminal reductant. This method achieved the synthesis of 9 variously functionalized carbazoles with yields ranging from moderate to good, even in gram-scale reactions. Experimental and computational modeling revealed that the turnover-limiting deoxygenation event involves a rate-determining (3 + 1) cheletropic addition between the organo-phosphetane catalyst and the 2-nitrophenyl substrate, forming a pentacoordinate spirobicyclic dioxazaphosphetane intermediate. This intermediate decomposes via (2 + 2) cycloreversion, producing phosphetane P-oxide and 2-nitrosobiphenyl. Further investigations suggest the involvement of an oxazaphosphirane (2 + 1) adduct between the phosphacycloalkane catalyst and 2-nitrosobiphenyl, which evolves through the loss of phosphetane P-oxide to yield the carbazole product via Csp2–H insertion in a nitrene-like manner. However, the need for a superstoichiometric amount of hydrosilane as the terminal reductant may affect the process mass intensity, thereby limiting its suitability for large-scale synthesis. (Scheme 20) [89].
Scheme 20
Scheme 20.
Cadogan cyclization catalyzed by biophilic organophosphorus.
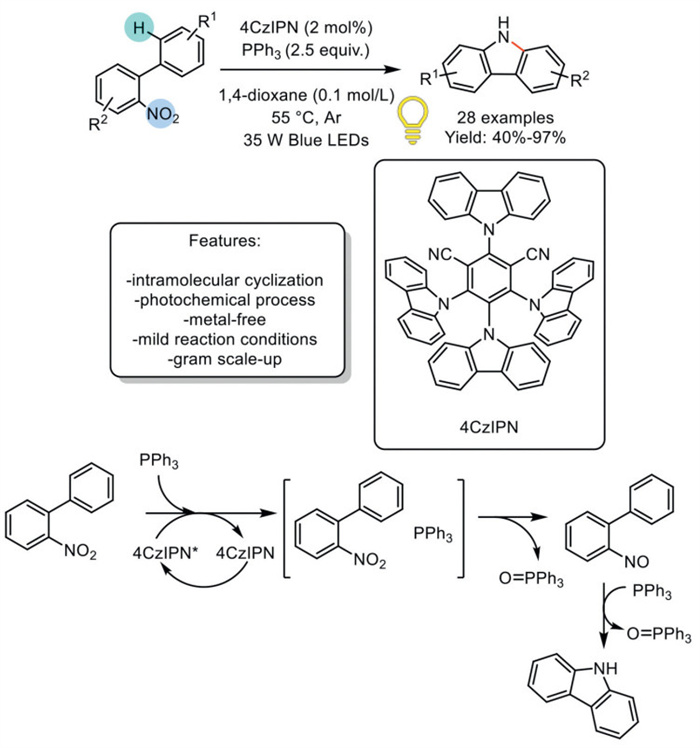
In 2021, Huang et al. reported a visible-light-driven photochemical Cadogan-type cyclization of o-nitrophenyl using PPh3 and the 4CzIPN photosensitizer under blue LEDs, enabling the formation of carbazole. The electronically contrasting properties of nitro biaryls and phosphines were proposed to facilitate the transient formation of photoactive electron donor–acceptor (EDA) complexes. The reaction conditions accommodated a broad range of functional groups, resulting in 28 examples with yields ranging from good to excellent. While the method demonstrates a broad substrate scope, substrates with electron-deficient groups typically show higher reactivity than those with electron-donating groups, which may necessitate longer reaction times. The process was highly effective on a gram scale; however, the study observed that some reactions did not achieve complete conversion on larger scales, even after modifying the reaction conditions. The authors also reported the synthesis of benzo[a]carbazoles, which were accessed in good to excellent yields. Density functional theory (DFT) calculations revealed that the electron-poor nitroarene and electron-rich PPh3 interact via coulombic attraction and π–π interaction, forming an atom transfer complex. Energy transfer from the excited 4CzIPN to the complex promotes it to its excited state, leading to oxygen transfer and producing nitroso biphenyl and P(O)Ph3. This explains why the photocatalysis system operates at mild temperatures, much lower than those required for traditional Cadogan cyclization (Scheme 21) [90].
Scheme 21
Scheme 21.
Blue LEDs driven photochemical Cadogan-type cyclization of o-nitrobiphenyl to carbazoles.
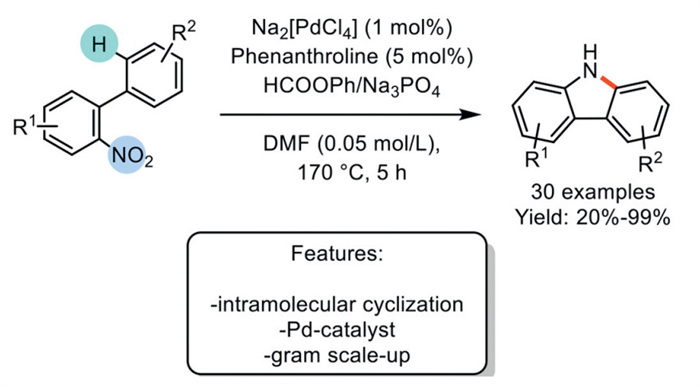
In 2022, Ragaini et al. developed a Pd/phenanthroline-catalyzed method for the reductive cyclization of o-nitrobiphenyls to carbazoles. This process utilizes phenyl formate as a reductant, which releases CO in situ, thus eliminating the need for high-pressure equipment. The species Na2[PdCl4] was used as the pre-catalyst in the presence of phenanthroline as a ligand. The selectivity of the reaction is highly influenced by the choice of base, which facilitates formate decomposition; Na3PO4 was identified as the preferred base. Although the optimized catalytic system demonstrates excellent stability and is tolerant to both moisture and air, enabling the reaction to proceed with low catalyst loadings, several critical factors affecting the system's stability have been identified. This protocol was effectively applied to synthesize 30 different substrates, producing products with yields ranging from good to excellent. Additionally, the overall procedure is easily scalable, and the products can be isolated without the need for chromatographic purification (Scheme 22) [91].
Scheme 22
Scheme 22.
Pd/phenanthroline-catalyzed reductive intramolecular cyclization of o-nitrobiphenyls to carbazoles.
Multi-component reactions offer significant advantages in terms of atom economy, as the formation of complex molecules is obtained in a single operation by combining three or more starting materials in one pot [92-96]. Among multi-component intermolecular synthetic routes to access carbazoles, arylation is a well-established reaction that allows the desired product to be obtained in a one-pot procedure. In multi-component intermolecular catalytic Csp2–H functionalization, both Csp2–Csp2 and Csp2–N bonds can be synthesized in a single step. This can be achieved through a Buchwald–Hartwig domino amination starting from 2-haloaniline and an aryl halide, or anilines and aryl dihalides [71,97,98]. Alternatively, a Suzuki-Miyaura tandem cross-coupling followed by an SNAr reaction of aniline-derived boronic esters with aryl dihalides can be employed [99].
This section highlights recent advancements in the development of novel, efficient catalytic Csp2–N bond-forming reactions, classifying the protocols as Csp2–H/Csp2–X (X = Cl, Br, I) coupling. Furthermore, to maximize the process's atom economy, intermolecular multi-component Csp2–H/Csp2–H coupling should be considered, although only a few examples have been reported.
3.1
Csp2–H/Csp2–X (X = Cl, Br, I) coupling
One-pot transition-metal catalysis, often utilizing Pd-based catalysts, is a powerful approach for synthesizing carbazoles through Csp2–N cross-coupling reactions. This section highlights recent advancements in the development of multi-component catalytic Csp2–N bond-forming reactions to access carbazoles.
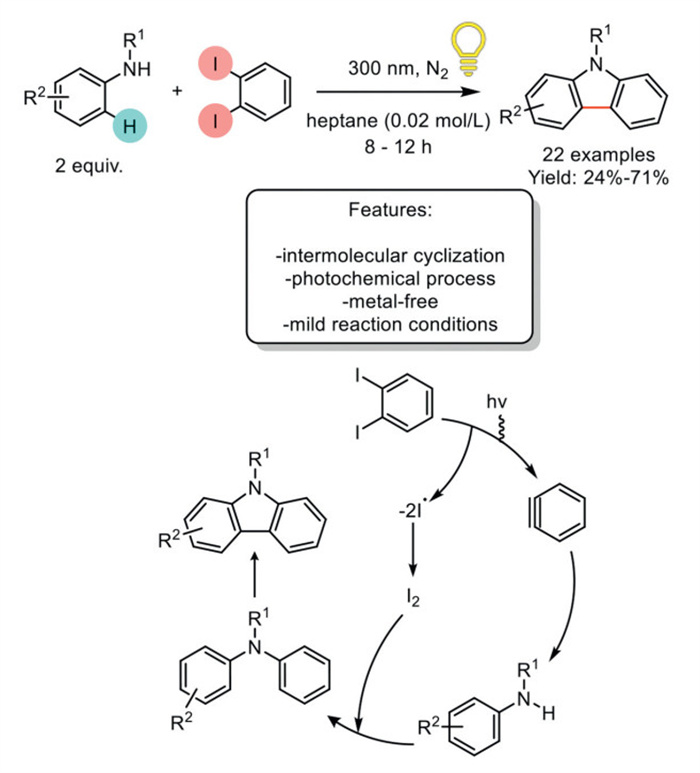
In 2018, Xia et al. introduced a photoinduced cross-coupling method for synthesizing carbazoles by reacting amines with 1,2-diiodobenzene under mild conditions. This approach successfully produced 22 examples with yields ranging from moderate to excellent, although the electron-withdrawing group resulted in lower yields. Halo- and trifluoromethyl-substituted substrates were tolerated, providing products with moderate yields. The scope was expanded to include tetrahydroquinoline, which also yielded moderate results. Substrates with meta-substituents produced regioisomeric mixtures, while ortho-substituted compounds yielded two distinct products. Steric effects were observed with bulkier groups. Mechanistic investigations revealed that the reaction proceeds via the formation of an excited benzyne intermediate, generated by the absorption of UV light by 1,2-diiodobenzene, leading to the elimination of an iodine molecule. The amine, acting as a nucleophile, interacts with the triple bond, followed by a proton transfer that results in the formation of a tertiary aminobenzene product. This intermediate undergoes oxidative cyclization, with the iodine molecule serving as an oxidant, to yield the final carbazole product (Scheme 23) [100].
Scheme 23
Scheme 23.
Photoinduced cross-coupling method for carbazole synthesis from amines with 1,2-diiodobenzene.
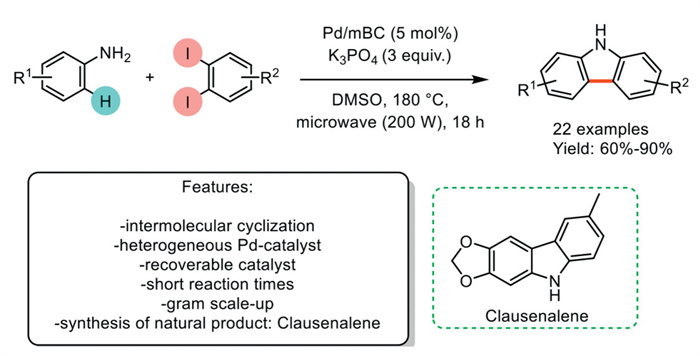
In 2021, D. C. Gerbino et al. developed an effective microwave-assisted method for synthesizing carbazoles through a Pd-catalyzed tandem reaction. This strategy involves a sequential Buchwald–Hartwig amination followed by direct arylation using aniline and 1,2-dihaloarenes. A novel, recoverable Pd nanocatalyst supported on magnetic biochar (mBC) was employed under ligand-free conditions, using K3PO4 as the base and microwave irradiation to promote the reaction. Compared to existing Pd-based methods, this protocol significantly reduces reaction times. It demonstrates exceptional compatibility with a wide range of functional groups, facilitating the synthesis of 22 different functionalized carbazoles with high yields and good regioselectivity. Demonstrating its practical utility, the authors present a straightforward and scalable synthesis of the bioactive carbazole alkaloid clausenalene (Scheme 24).
Scheme 24
Scheme 24.
Pd-catalyzed tandem Buchwald–Hartwig amination followed by direct arylation to carbazoles from aniline and 1,2-dihaloarenes.
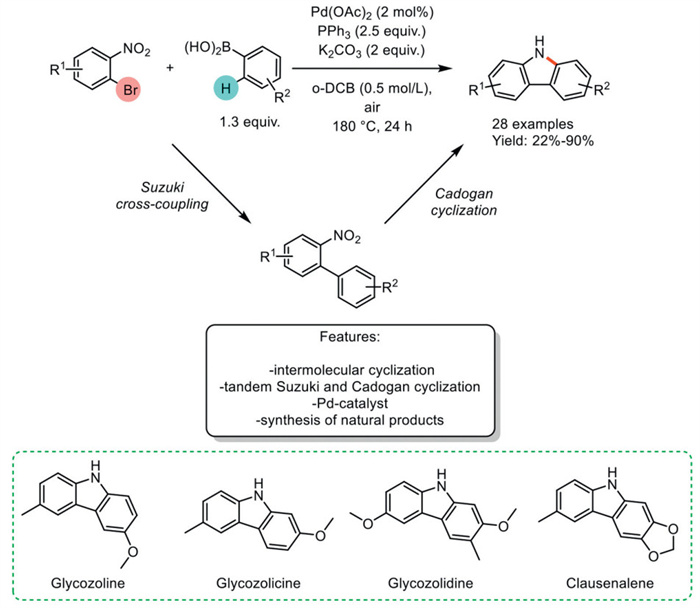
In 2016, Woo et al. [101] developed a highly efficient Pd-catalyzed one-pot tandem Suzuki reaction and reductive Cadogan cyclization of 2-dinitrobenzene with phenylboronic acid to synthesize carbazoles. Pd(OAc)2 in combination with triphenylphosphine in o-dichlorobenzene (o-DCB) at 150 ℃ was successfully employed in the synthesis of a diverse array of carbazoles, achieving moderate to excellent yields with both electron-neutral and electron-rich aryl boronic acids. Arylboronic acids with bulky groups at the para position were converted to carbazoles with excellent yields. Although electron-deficient aryl boronic acids generally yielded slightly lower results, 4-fluorophenyl boronic acid was an exception, providing good yields. The scope was further extended by using a wide range of substituted o-bromonitrobenzenes, which were converted to the corresponding carbazoles in good yields. Despite the excellent regioselectivity of the protocol for most starting materials, 3-tolylboronic acid and 4–methoxy-3-methylphenylboronic acid produced mixtures with low regioselectivity. This one-pot strategy was applied to the synthesis of four bioactive carbazole alkaloids, glycozoline, glycozolicine, glycozolidine, and clausenalene, with good yields and regioselectivity (Scheme 25) [102].
Scheme 25
Scheme 25.
Pd-catalyzed one-pot tandem Suzuki reaction and reductive Cadogan cyclization of 2-dinitrobenzene with phenylboronic acid to carbazoles.
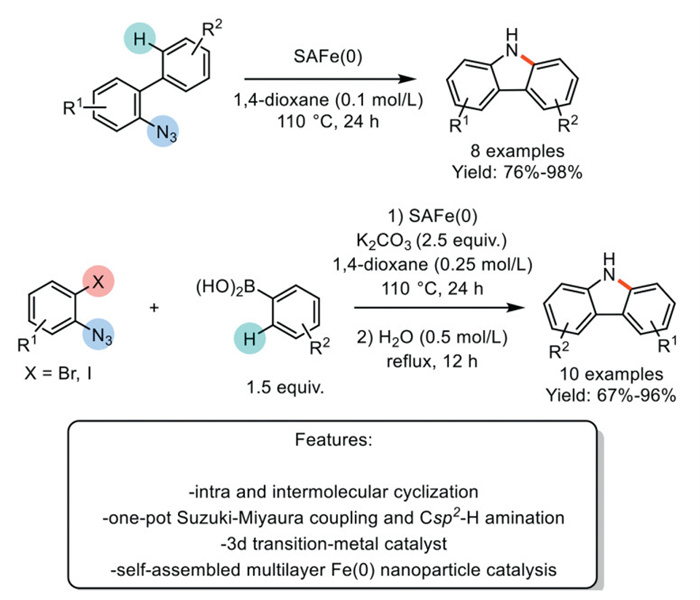
In 2020, Arisawa et al. developed a three-step method to synthesize catalytically active self-assembled multilayer iron(0) nanoparticles (SAFe(0)) for ligand-free Csp2−Csp2 and Csp2−N bond-forming reactions, such as Suzuki−Miyaura coupling and Csp2−H amination. Their investigation began with the SAFe-catalyzed intramolecular Csp2–H amination of 2-azide biphenyl to produce a carbazole derivative under ligand-free conditions. The optimized process was successfully applied to a variety of substrates, resulting in 10 examples with yields ranging from good to excellent. They then expanded the procedure to a one-pot, two-step Suzuki−Miyaura Csp2−H amination, catalyzed by SAFe(0), using 1-azide-2-iodobenzenes and phenylboronic acids in the presence of K2CO3 as the base. This approach achieved similar success with an additional set of substrates, producing 10 examples with yields ranging from good to excellent. Although this methodology is effective, it has been tested on only a limited number of substrates, all of which contain electron-donating groups (Scheme 26) [103].
To maximize the atom economy of carbazole synthesis, it is essential to avoid using halogenated starting materials and focus on direct Csp2–H/Csp2–H functionalization. However, the intermolecular version of this coupling to access carbazoles is not well documented. Only one reported instance involves a non-catalytic cross-dehydrogenative coupling (CDC) reaction.
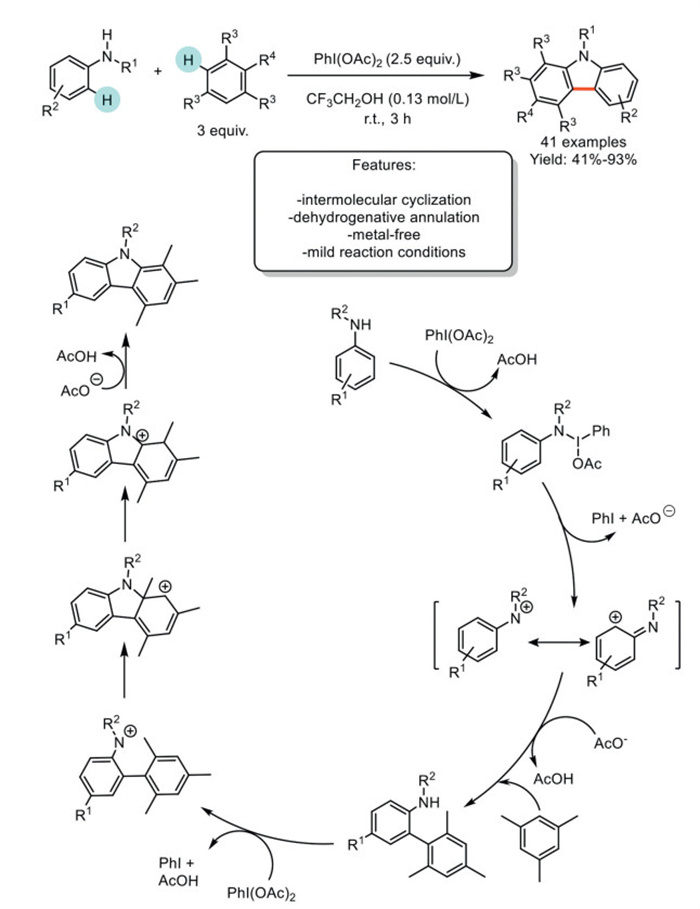
In 2017, Mal and Maiti reported an innovative intermolecular dehydrogenative annulation (IDA) method for synthesizing carbazoles via a one-pot sequential Csp2−Csp2/Csp2−N bond formation. The process involves the selective alkyl group migration of readily available 1,3,5-trialkylbenzenes and anilide substrates, utilizing the hypervalent iodine(Ⅲ) reagent PhI(OAc)2 (PIDA) as an oxidant under ambient conditions. The optimized protocol was successfully applied to the synthesis of 41 carbazoles, with products obtained in good to excellent yields. Mechanistically, PIDA reacts with anilides to generate a nitrenium ion or a more stabilized carbenium ion, which then reacts with 1,3,5-trialkylbenzenes to form a C-arylated intermediate. Regioselective arylation indicates that the reaction follows an ionic pathway. This intermediate is further oxidized by a second molecule of PhI(OAc)2, producing another ionic intermediate. Electrophilic aromatic substitution leads to a carbenium intermediate, which is further stabilized by the migration of an adjacent quaternary alkyl group. This generates the final cationic intermediate, which is deprotonated by the acetate ion to afford the carbazole product (Scheme 27) [104].
Scheme 27
Scheme 27.
Intermolecular dehydrogenative annulation of 1,3,5-trialkylbenzenes and anilide substrates using hypervalent iodine(Ⅲ) to synthesize carbazoles.
The hydrogen-borrowing strategy is a well-established methodology in transition-metal-catalyzed dehydrogenative aromatization [105-108]. This approach has recently emerged as an atom-economic process for accessing carbazoles. This section presents recent advancements in this methodology.
In 2022, Huang et al. introduced a visible-light-induced protocol for the hydrogen-borrowing reduction and dehydrogenative aromatization/cyclization of 2-cyclohexenyl nitroaromatic substrates to carbazoles. This process begins with the formation of active singlet species through efficient energy transfer from excited Ir[dF(CF3)ppy]2(dtbpy)PF6 to nitroarenes. The combination of PdCl2, PPh3, and Ir[dF(CF3)ppy]2(dtbpy)PF6 in catalytic amounts under blue LED irradiation promotes the feasibility of an intermolecular hydrogen-borrowing reduction/dehydrogenative aromatization transformation to carbazole. Although this method was applied to synthesize 17 different examples, the yields ranged from moderate to good. In some cases, the competitive formation of tetrahydrocarbazole byproducts led to reduced yields. Additionally, aza-carbazole was obtained in a modest 43% yield, which may limit the method's applicability for synthesizing polycyclic heteroaromatic frameworks. However, it was successfully used with reactants attached to drug molecules and complex natural product molecules. Under visible-light irradiation, the [IrⅢ] catalyst photosensitizes the reactant into its excited state via energy transfer, triggering a hydrogen atom transfer (HAT). This intermediate undergoes a SET followed by proton transfer and water elimination, resulting in the formation of nitrosobenzene. The nitrosobenzene then engages in a second sequential ET/HAT/SET process to form o-hydroxylaminebiphenyl. This intermediate eliminates water to produce a nitrene intermediate, which subsequently inserts into the structure to generate the desired carbazole (Scheme 28) [109].
Scheme 28
Scheme 28.
Visible-light-induced hydrogen-borrowing reduction and dehydrogenative aromatization/cyclization of 2-cyclohexenyl nitroaromatic substrates to carbazoles.
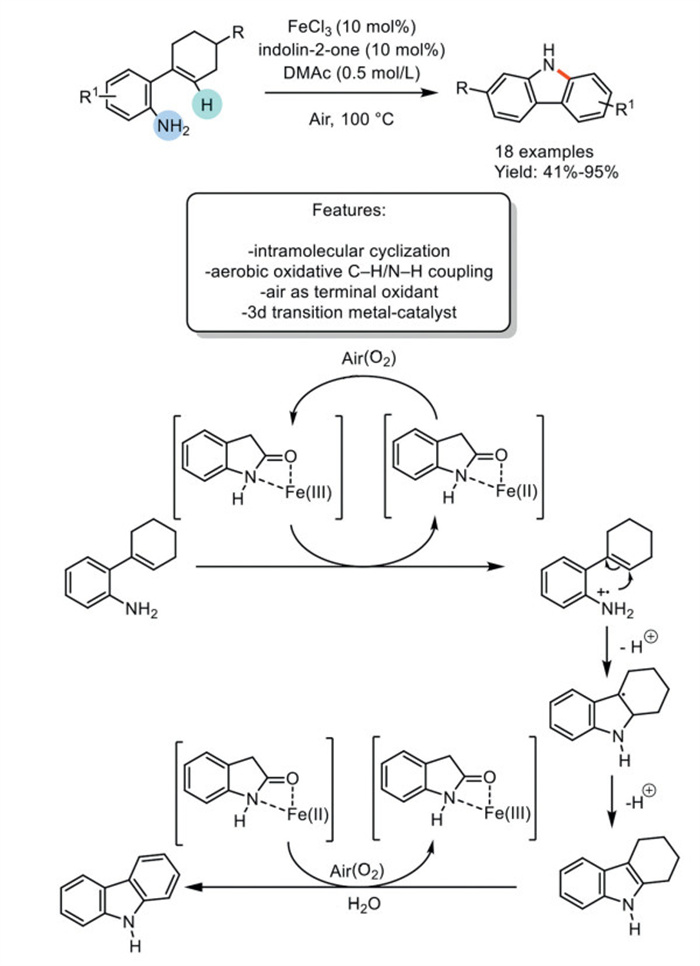
In 2023, Feng, Zhang et al. reported an iron-catalyzed cross-dehydrogenative coupling Csp2–H amination for synthesizing N–H carbazoles using air as the oxidant. In this study, Fe(Ⅲ) is continuously converted to Fe(Ⅱ) through SET, and Fe(Ⅱ) is re-oxidized to Fe(Ⅲ) in the presence of indolin-2-one and the green oxidant air. This protocol enables Csp2–N bond formation under environmentally friendly conditions with excellent functional group compatibility, resulting in 18 carbazoles with good to excellent yields. The authors proposed a plausible reaction mechanism where indolin-2-one, chelating with Fe(Ⅲ/Ⅱ), acts as both a radical donor and a hydrogen scavenger. The radical process is influenced by the presence of air (O2). Initially, the Fe(Ⅲ) complex undergoes a SET process, generating the Fe(Ⅱ) complex. Simultaneously, 2-cyclohexenyl aminoarene is oxidized to the corresponding radical cation by the Fe(Ⅲ) complex. The deprotonation and cyclization of the intermediate follow this. The intermediate then undergoes another SET and deprotonation, followed by aromatization to yield the desired product with the assistance of the Fe(Ⅱ) complex (Scheme 29) [110].
Scheme 29
Scheme 29.
Fe-catalyzed cross-dehydrogenative coupling Csp2–H amination to carbazoles with air as the oxidant.
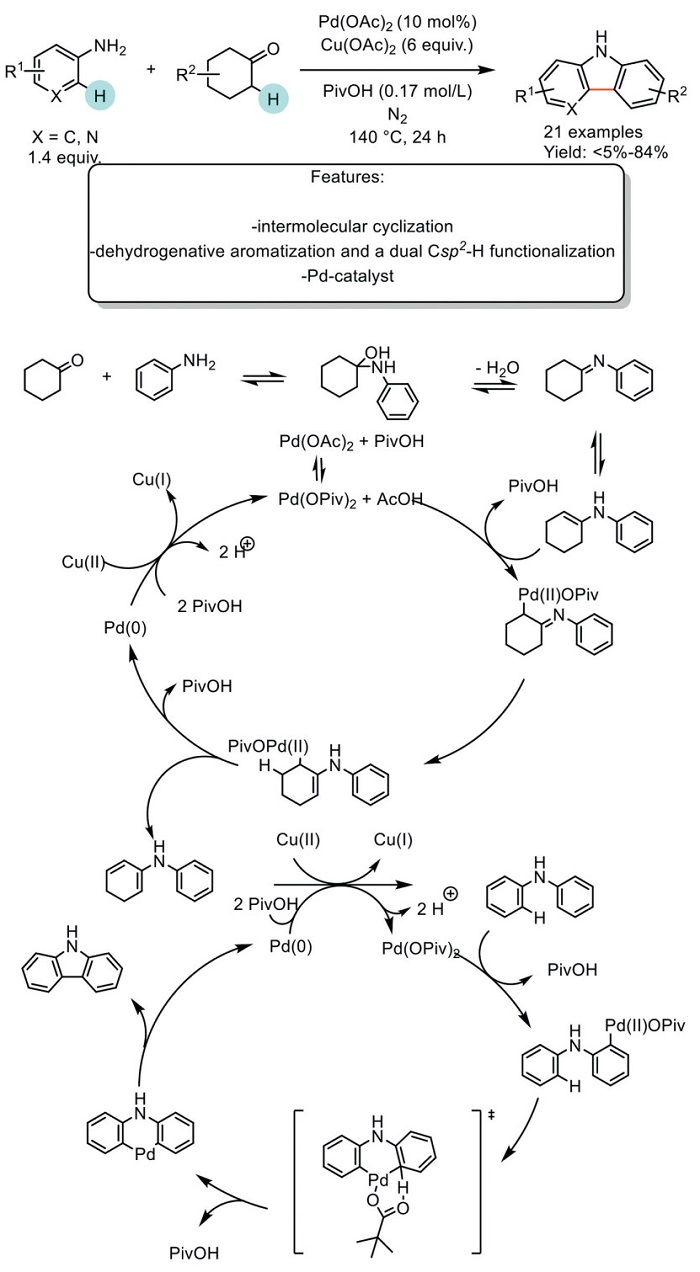
In 2016, Wang et al. developed a ligand-free Pd-catalyzed one-pot domino reaction via a dehydrogenative aromatization and a dual Csp2−H functionalization process for the synthesis of carbazoles from facile arylamines and cyclic ketones using Cu(OAc)2 as sacrificial oxidant. The reaction was performed in PivOH, which enables the formation of Pd(OPiv)2. The optimized reaction was applied to synthesize 21 products in good yield, except when a chloro substituent was present in the aromatic ring, which triggered a Buchwald−Hartwig amination side reaction. The proposed mechanism begins with the condensation of aniline and cyclohexanone, resulting in the formation of an enamine. This step is followed by palladation, producing the Pd(Ⅱ) species. Next, tautomerization and β-hydride elimination release the cyclic diene intermediate and a Pd(0) species. The Pd(0) species can be oxidized to Pd(Ⅱ) in the presence of Cu(Ⅱ). Subsequently, the intermediate goes through a second oxidation. The reaction is accelerated by the rapid electrophilic palladation of the diarylamine intermediate with Pd(OPiv)2. This is followed by further intramolecular Csp2−H cleavage, producing an intermediate that, through reductive elimination, yields the carbazole product and regenerates the Pd(0) species. The Pd(0) species is then oxidized back to Pd(Ⅱ) by Cu(Ⅱ), completing the catalytic cycle (Scheme 30) [111].
Scheme 30
Scheme 30.
Pd-catalyzed one-pot domino dehydrogenative aromatization and a dual Csp2−H functionalization.
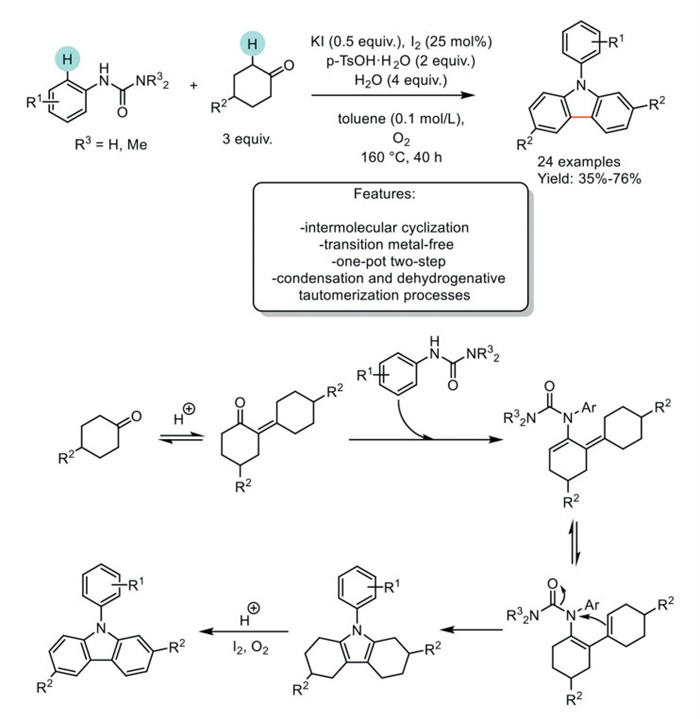
In 2016, Deng et al. developed an efficient transition metal-free strategy for synthesizing carbazoles from arylureas and cyclohexanones, utilizing a combination of potassium iodide (SSKI oral solution) and iodine (I2). This combination enhanced the yield, and through optimized reaction conditions, the authors investigated the applicability of the reaction conditions with various substituted cyclohexanones and phenylureas. The para-toluensulfonic acid (p-TsOH) provided the necessary acidic conditions, while the presence of a small amount of water proved beneficial for the reaction, which was carried out in toluene as the solvent. As a result, 24 carbazoles were synthesized, with yields ranging from modest to good. Despite the broad applicability of the method, using arylureas with electron-withdrawing groups results in noticeably lower reactivity. Additionally, moderate yields were observed when substituents were located at the meta-position of the nitrogen atom. The authors proposed a plausible reaction mechanism that begins with the condensation of two cyclohexanones under acidic conditions to form an unsaturated ketone intermediate. This intermediate then condenses with phenylureas to form an enamine complex. The enamine complex isomerizes to form a conjugated enamine intermediate, which then undergoes cyclization to produce a pyrrole compound. Ultimately, a dehydrogenative tautomerization of the pyrrole intermediate under oxidative conditions yields the desired carbazole product (Scheme 31) [112].
Scheme 31
Scheme 31.
Transition metal-free one-pot two-step process to carbazoles from arylureas and cyclohexanones.
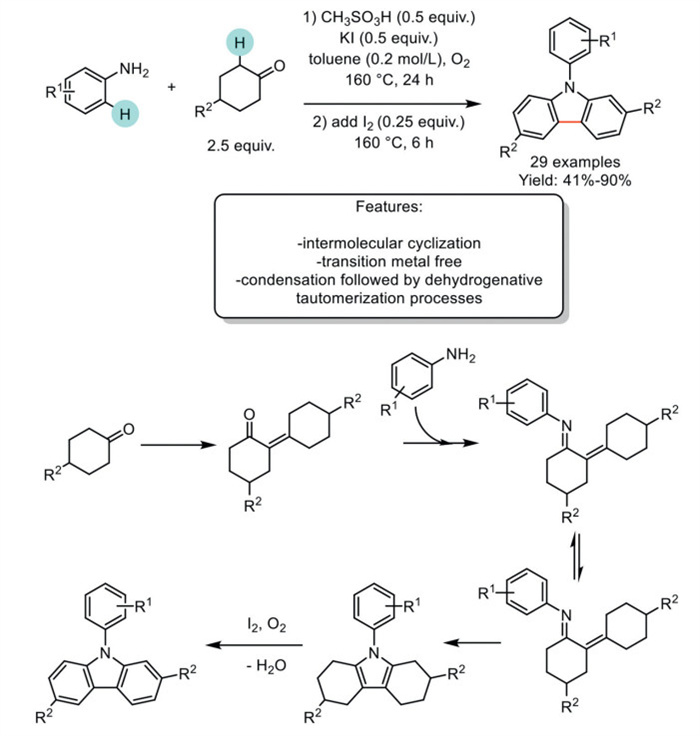
In 2017, Deng et al. developed a strategy for synthesizing carbazoles from anilines and cyclohexanones. In their previous work, the entire carbazole moiety, except for the nitrogen atom, was derived from nonaromatic cyclohexanones through a dehydrogenation-tautomerization sequence. However, only a portion of the arylurea was incorporated into the resulting products, reducing the reaction's atom economy. Additionally, arylureas are relatively inaccessible chemicals, with only a few being commercially available. This approach used readily available anilines and cyclohexanones as starting materials under transition-metal-free conditions, unveiling a novel route for the synthesis of substituted N-aryl carbazoles. The authors initially used KI/I2, but the dehydrogenation reaction was not completed under these conditions when nitrogen was used as the nitrogen source. In this work, the optimized reaction conditions involve a one-pot, two-step process in which KI demonstrated the highest efficiency. Acid proved to be crucial for this transformation, as no desired product was obtained when methanesulfonic acid was replaced with acetic acid. Iodine was added after 24 h for the dehydrogenation step. KI proved to be the most efficient iodide-containing additive for yielding the desired product. Acid additives play a crucial role in this transformation. Under the optimized reaction conditions, the scope was extended to a wide range of substituted anilines and cyclohexanones, resulting in 29 carbazoles with moderate to good yields. The mechanistic study revealed that the initial steps were the same as in the previous work, leading to an unsaturated ketone intermediate [112]. The subsequent condensation of the ketone with the aniline forms an imine intermediate, which can isomerize to an enamine intermediate. This enamine intermediate can then be converted into a pyrrole intermediate through a cyclization procedure, and the final dehydrogenative-tautomerization reaction under oxidative iodine conditions yields the final product (Scheme 32) [113].
Scheme 32
Scheme 32.
Transition metal-free one-pot two-step process to carbazoles from anilines and cyclohexanones.
This section reports the Csp2–H functionalization of simple unfunctionalized carbazoles. Leveraging the electron-rich nature of the C3 position, direct Csp2–H functionalization through electrophilic aromatic substitution (EAS) occurs predominantly at this position.
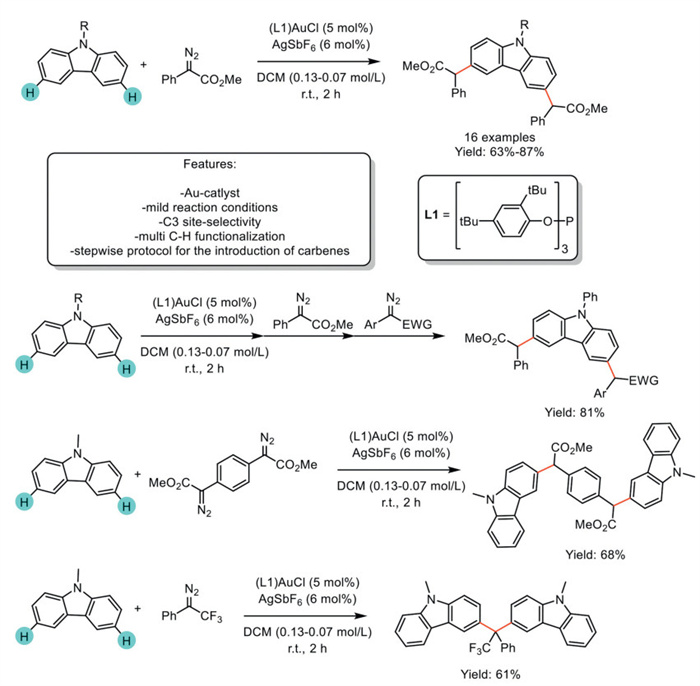
In 2024, Nguyen and Koenigs et al. reported a multi-C–H functionalization of aryldiazoacetates with carbazole via Au-catalyzed carbene transfer reactions. The Au-catalyst exhibits a distinct role, enabling the introduction of up to six carbene fragments onto (poly)carbazole frameworks, enabling the obtainment of 16 functionalized carbazoles and polycarbazoles under mild reaction temperatures and reduced reaction times. Moreover, the authors reported a stepwise protocol for the introduction of two different carbenes into carbazole moieties. The application of linchpin reagents bearing two carbene precursors allows the linkage of two carbazole fragments and opens new pathways toward polycarbazoles (Scheme 33) [114].
Scheme 33
Scheme 33.
C3 site-selective multi-Csp2–H functionalization of aryldiazoacetates with carbazole via Au-catalyzed carbene transfer reactions.
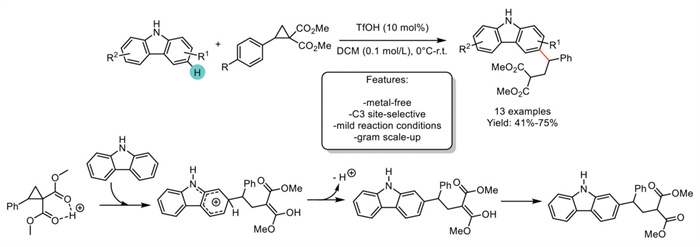
In 2021, Zhao et al. reported the ring-opening reactions of cyclopropane to functionalize simple, unfunctionalized NH-carbazoles. While the use of Lewis acid Sc(OTf)3 resulted in an N−H-functionalized reaction to furnish N-alkylated carbazoles, employing TfOH (triflic acid) as the Brønsted acid generated a Friedel-Crafts-type addition at the C3 position in the absence of any protection group. The strategy facilitates the synthesis of 13 Csp2–H functionalized carbazoles. However, 3,6-dibromo and 3,6-dibutyl instead of the Friedel-Crafts-type addition of the carbazole at the C3 position, the reaction site was switched to the relatively inert C1 position (Scheme 34) [115].
Scheme 34
Scheme 34.
Site-selective C3-functionalization of carbazoles via ring-opening reactions of cyclopropane.
Selective functionalization at other positions, particularly the C1 position, remains a significant challenge. This paper reports recent examples of C3 site-selectivity.
In 2023, Maiti, Ge et al. reported a site-selective C1 alkylation and acylation of carbazoles via a palladium/norbornene cascade process mediated by a unique six-membered palladacycle intermediacy. The reaction was carried out using Pd(OAc)2 as the catalyst, NBE as the ligand, and KOPiv as the base, with a small amount of water added to enhance the base's solubility in acetonitrile. Notably, high functional group compatibility was observed for this reaction, enabling the obtainment of 24 alkylated carbazoles and 6 acylated carbazoles. The authors observed that C2-substituted carbazoles provided the desired products with good yields and excellent C8 site-selectivity, exclusively due to the steric effect. The C3-substituted carbazoles showed different reactivities, while a C8 alkylated product was obtained using an electron-donating group-substituted carbazole substrate; carbazoles bearing an electron-withdrawing group favored the formation of C1 products. These results suggested that an EAS mode may not be applied to the Csp2–H bond cleavage step in the catalytic cycle. The authors reported a plausible reaction mechanism. First, ligand exchange on the Pd-catalyst with carbazole in the presence of a base generates the intermediate, which undergoes norbornene (NBE) insertion. Following this, site-selective Csp2–H palladation occurs to give a six-membered ring intermediate. The formation of the key 6-membered palladacycle intermediate, followed by the oxidative addition of this intermediate with an alkyl bromide, provides the palladium(Ⅳ) intermediate, which then proceeds through a reductive elimination to afford the palladium(Ⅱ) intermediate. Subsequently, it undergoes a norbornene extrusion followed by ligand exchange to yield the desired product (Scheme 35) [116].
Scheme 35
Scheme 35.
Site-selective C1 alkylation and acylation of carbazoles via a palladium/norbornene cascade process.
In 2023, Jia et al. reported the DFT theoretical calculations to investigate the site-selective C1 alkylation and acylation of carbazoles via a palladium/norbornene-mediated cascade process. The authors revealed the complete catalytic cycle of this alkylation reaction, as well as the insertion and extrusion mechanisms of the transient directing group NBE [117].
6.
Miscellaneous
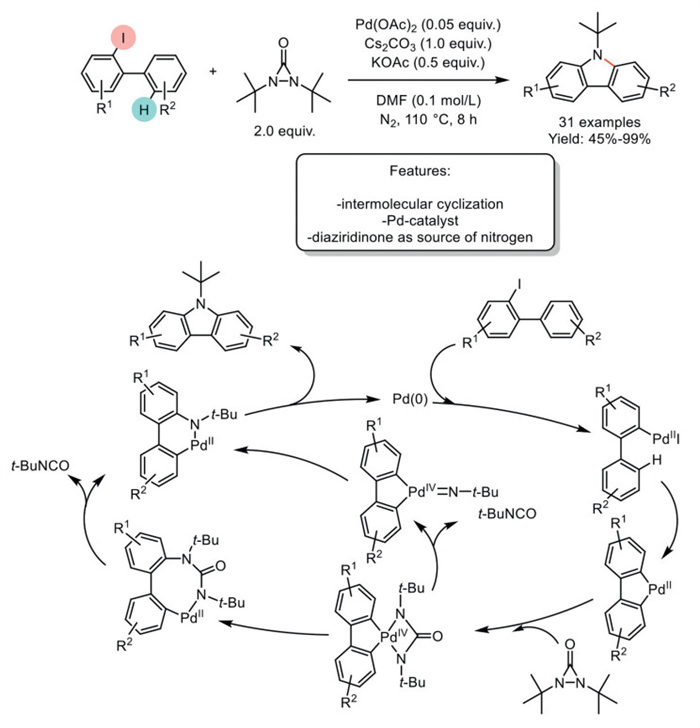
In 2018, Zhang et al. reported a Pd-catalyzed Csp2–H activation of 2-iodobiphenyls and diaziridinone via dual Csp2–N bond formation. The reaction was catalyzed by Pd(OAc)2 in the presence of Cs2CO3 and KOAc, suggesting that the bases play a crucial role in generating the active Pd(0) species. At the same time, KOAc may also facilitate C–H activation. This optimized protocol was used to synthesize 31 substrates, with yields ranging from good to excellent. The tert–butyl groups of the products could be readily removed using trifluoroacetic acid (TFA), yielding unprotected carbazoles. The limitations of this method include the use of diaziridinone as a nitrogen source and the formation of N-protected carbazoles, which necessitate an additional deprotection step. Both factors diminish the atom economy of the process. The authors proposed a catalytic cycle that begins with the oxidative addition of 2-iodobiphenyls to Pd(0), forming a Pd(Ⅱ) species. This species undergoes intramolecular Csp2–H activation to create a palladium(Ⅱ) cycle. Subsequent insertion into the N–N bond of diaziridinone via oxidative addition yields a palladium(Ⅳ) cycle. An eight-membered palladium(Ⅱ) cycle is then formed after reductive elimination. Finally, β-N elimination, followed by another reductive elimination, leads to the desired product and regenerates the Pd(0) catalyst. It should be noted that a Pd(Ⅳ)-nitrene pathway cannot be ruled out (Scheme 36) [118].
Scheme 36
Scheme 36.
Pd-catalyzed Csp2–H activation of 2-iodobiphenyls and diaziridinone via dual Csp2–N bond formation.
Bochet et al. reported a photochemical Csp2–H activation of N-arylbenzotriazoles leading to carbazoles. To demonstrate the effectiveness of their methodology, they synthesized Clausenawalline D, an anti-malarial carbazole alkaloid. Additionally, they applied this method to the synthesis of 11 carbazoles, achieving yields ranging from poor to good. The yields depend largely on the relative absorbance of the precursor and product, which compete during irradiation. Furthermore, the reaction demonstrated a strong solvent dependence, with acetonitrile proving to be the most effective solvent. (Scheme 37) [119].
Scheme 37
Scheme 37.
Photochemical Csp2–H activation of N-arylbenzotriazoles leading to carbazoles.
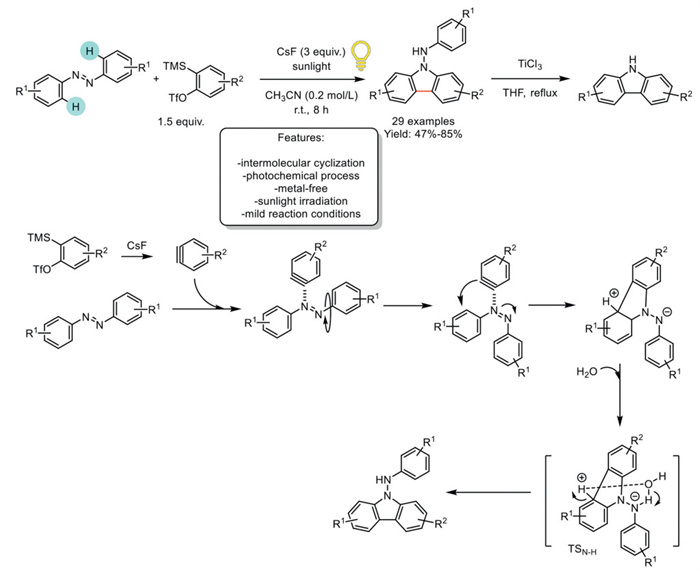
Li et al. developed a sunlight-mediated [3 + 2] cycloaddition of azobenzenes with arynes to synthesize carbazoles. Arynes, generated in situ from 2-(trimethylsilyl)aryl triflates, enable ortho-Csp2–H functionalisation of azobenzenes at room temperature. The cycloaddition products can be further converted into useful carbazoles via N−N bond cleavage. However, if N–H carbazoles are required, an additional step is necessary, involving reductive cleavage under TiCl3/THF reflux conditions. Under sunlight irradiation, CsF was identified as the optimal reagent for generating benzyne. Using the optimized reaction conditions, 29 variously functionalized carbazole scaffolds were obtained with yields ranging from good to excellent. The authors proposed a reaction mechanism in which treating 2-(trimethylsilyl)phenyl triflate with CsF generates benzyne species in situ. The interaction of azobenzene with benzyne forms an intermediate with an elongated N=N bond. This intermediate undergoes rapid rotation under sunlight irradiation, leading to a triplet-state intermediate that undergoes intramolecular cyclization to generate a five-membered intermediate. DFT calculations supported the proposed reaction mechanism (Scheme 38) [120].
Scheme 38
Scheme 38.
Sunlight-mediated [3 + 2] cycloaddition of azobenzenes with arynes to carbazoles.
In conclusion, this review presents the past decade of developments in Csp2–H functionalization for the synthesis of carbazoles. Our detailed analysis demonstrates that direct Csp2–H functionalization is an effective and cost-efficient strategy for synthesizing carbazoles from simple, readily available starting materials, offering excellent atom and step economy.
The privileged carbazole scaffold, widely present in natural alkaloids and APIs, continues to hold a central position in materials science and medicinal chemistry. Its structural versatility and biological relevance ensure that carbazole synthesis remains a compelling challenge for the scientific community. As such, the development of efficient and selective direct Csp2–H functionalization strategies remains a critical area of research.
Considering the future perspective, several promising directions are emerging in the field. The development of direct Csp2–H/Csp2–H and Csp2–H/N–H coupling methodologies offers a powerful strategy that enables the use of simple, readily available starting materials without the need for pre-functionalization. This approach enhances both atom economy and step economy, facilitating the efficient synthesis of natural products and bioactive compounds. Advances in sustainable catalysis, particularly the use of earth-abundant and less toxic 3d-transition metals, are gaining attention. The implementation of recyclable heterogeneous metal catalysts provides cost-effective and environmentally friendly alternatives to traditional precious metal systems, aligning synthetic methodologies with the principles of green chemistry.
Additionally, the integration of photoredox catalysis with transition metal catalysis unlocks new reaction pathways under mild and energy-efficient conditions. Electrosynthesis represents another compelling approach, employing electricity as a clean reagent to drive oxidative transformations. By avoiding the use of stoichiometric chemical oxidants, this technique reduces waste and enhances the overall sustainability of the process. Microwave-assisted synthesis has also proven to be a valuable alternative, particularly in the Csp2–H functionalization routes toward carbazole frameworks, offering improved reaction rates and energy efficiency. Although flow technology remains underutilized in the synthesis of these compounds, the development of continuous flow protocols holds great potential for simplifying scale-up processes.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Giulia Brufani: Writing – original draft. Edoardo Bazzica: Writing – original draft, Data curation. Yanlong Gu: Writing – review & editing, Writing – original draft. Francesco Mauriello: Writing – original draft, Methodology. Luigi Vaccaro: Writing – review & editing, Writing – original draft, Resources, Project administration.
Acknowledgments
This publication was prepared with support and funding by the European Union – NextGenerationEU under the Italian Ministry of University and Research (MUR) National Innovation Ecosystem (No. ECS00000041–VITALITY and also "Ecosistema TECH4YOU–(Spoke 3 – Goal 3.5). MUR is thanked for PRIN-PNRR 2022 project "P2022XKWH7 – CircularWaste. The University of Perugia is acknowledged for financial support to the university project "Fondo Ricerca di Ateneo, edizione 2022". The National PhD program in Catalysis coordinated by the University of Perugia is also thanked. Y. Gu is grateful for the financial supports of key research and development and technology transfer projects of Inner Mongolia Autonomous Region (No. 2025KJHZ0008) and major special projects of science and technology of Ordos (No. 2022EEDSKJZDZX003).
[1]
J. Molette, J. Routier, N. Abla, et al., ACS Med. Chem. Lett. 4 (2013) 1037–1041. doi: 10.1021/ml400015f
A. Banerjee, S. Kundu, A. Bhattacharyya, S. Sahu, M.S. Maji, Org. Chem. Front. 8 (2021) 2710–2771. doi: 10.1039/d1qo00092f
[21]
S.N. Georgiades, P.G. Nicolaou, Recent advances in carbazole syntheses, in: Eric F.V. Scriven, Christopher A. Ramsden (Eds.), Advances in Heterocyclic Chemistry, Advances in Heterocyclic Chemistry, 129, Academic Press, 2019, pp. 1–88.
J. Wu, Y. Xie, X. Chen, G.J. Deng, Adv. Synth. Catal. 358 (2016) 3206–3211. doi: 10.1002/adsc.201600673
[113]
J. Wu, X. Chen, Y. Xie, et al., J. Org. Chem. 82 (2017) 5743–5750. doi: 10.1021/acs.joc.7b00556
[114]
S. Jana, C. Empel, T.V. Nguyen, R.M. Koenigs, Chem. Eur. J. 27 (2021) 2628–2632. doi: 10.1002/chem.202004724
[115]
H. Zhao, P. Shen, D. Sun, H. Zhai, J. Org. Chem. 86 (2021) 9189–9199. doi: 10.1021/acs.joc.1c00494
[116]
M. Elsaid, R. Ge, C. Liu, D. Maiti, H. Ge, Angew. Chem. Int. Ed. 62 (2023) e202303110. doi: 10.1002/anie.202303110
[117]
F. Jia, Y. Yang, B. Zhang, Catal. Sci. Technol. 13 (2023) 6812–6822. doi: 10.1039/d3cy01137b
[118]
C. Shao, B. Zhou, Z. Wu, X. Ji, Y. Zhang, Adv. Synth. Catal. 360 (2018) 887–892. doi: 10.1002/adsc.201701039
[119]
I. Alimi, R. Remy, C.G. Bochet, Eur. J. Org. Chem. 2017 (2017) 3197–3210. doi: 10.1002/ejoc.201700300
[120]
W. Zhang, J. Bu, L. Wang, P. Li, H. Li, Org. Chem. Front. 8 (2021) 5045–5051. doi: 10.1039/d1qo00739d
Figure 1
Bioactive and synthetically relevant molecules incorporating the carbazole unit: Representative examples of natural products (A), pharmaceutically and biologically active compounds (B) and hole-transporting and light-emitting molecules (C).
Scheme 2
Consecutive Cu-catalyzed Chan-Lam N-arylation of o-iodoanilines and boronic acids followed by the intramolecular Pd-catalyzed aryl Csp2–H activation.
Scheme 19
Metal-free deoxygenation and reductive distillation method for nitroarenes with organosilicon-reducing reagents to synthesize carbazole structural moieties.
Scheme 27
Intermolecular dehydrogenative annulation of 1,3,5-trialkylbenzenes and anilide substrates using hypervalent iodine(Ⅲ) to synthesize carbazoles.
Scheme 28
Visible-light-induced hydrogen-borrowing reduction and dehydrogenative aromatization/cyclization of 2-cyclohexenyl nitroaromatic substrates to carbazoles.

 DownLoad:
DownLoad:







 下载:
下载:









































 下载:
下载:
