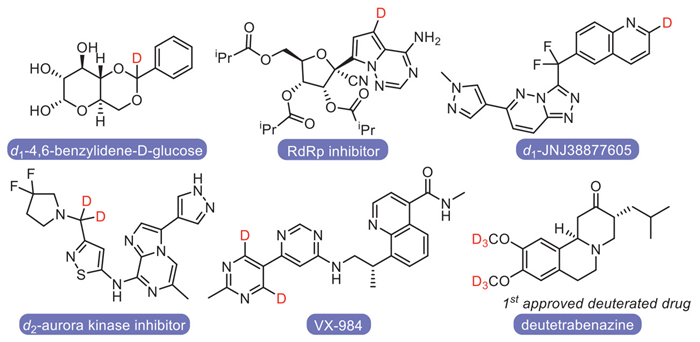
Figure 1.
Selected examples for deuterated drugs.
Divergent site-selective synthesis of deuterated pyrroles from radical initiated cyclizations of N-propargyl enamines
Baihui Zheng , Dandan Zhang , Baoping Ren , Yifei Li , Qun Liu , Ling Pan

The deuterium kinetic isotope effect facilitates the widespread applications of deuterated organic compounds (Fig. 1) in studying drug metabolisms, reaction mechanisms [1–3]. Precise label level of deuteration at functional groups bearing multi-H atoms also facilitated the detailed study for deuterium involved process. For example, mono-deuterated compounds had been applied in determination of long-range dipolar coupling [4] and in astrophysical detections [5]. Since the first successful development of deuterium-labeled nitrogen-containing drug, deutetrabenazine [2], the precise synthesis of deuterated drug analogues has attracted increasing interests. Except for the traditional synthesis starting from deuterium-containing building blocks such as D2 gas, hydrogen isotope exchange (HIE) has been proved to be the most popular method for deuterium-labelling [6–10], including the acid/base-mediated labelling or transition-metal catalyzed C–H functionalization. However, it’s still challenging to install deuterium in high selectivity for both reaction site and labelled number.
As shown in Scheme 1a, the site-selectivity could be achieved via simple single-site-HIE or directing group directed mono-deuteration [11–13] di-deuteration at two similar aromatic C–H bonds via transition-metal catalyzed coupling or at two adjacent unsaturated carbons of alkenes/alkynes via reductive addition [10,14–18]. Otherwise it’s difficult to achieve a high selectivity. It is also difficult to control the deuteration degree, installing one, two, and three deuterium atoms precisely at one site in high efficiency, especially the mono-label for functional groups bearing multi-H/D-exchange-sites like methyl or benzylic methylene. Besides the dehalogenative deuteration [19] and deuterium transfer (addition) to olefins [20] for partial deuteration of benzyl or methyl group, one of the few solutions was reported by Dong’s group in 2022 for selective deuterations at α-carbon of carbonyl group. The excellent one-pot Cu-catalyzed degree-controlled deacylative deuterations of alkyl radicals, generated from aromatization-driven C–C cleavage in the presence of N-methylpicolino-hydrazonamide (MPHA) at 90 ℃. The H/D exchange with D2O of both MPHA and the substrates bearing acidic ketone α-hydrogens facilitated the deuteration at different stages (Scheme 1a) [21]. The combination of HIE and radical processes may provide a promising solution for selective deuterations. Recently, elegant examples [21–34] for the radical involved selective deuterations have emerged [35–45] via deuterium atom transfer (DAT) [18,30,40,41]; deuterium atom abstraction (DAA) [29,31,32]; or deuterated water splitting (DWS) strategy, etc. (Scheme 1b) [12,33−34]. However, most of these radical transformations are limited to single-site deuterations, and there are almost no reports on the deuterations of C(sp2) and C(sp3) sites simultaneously. The divergent deuterations with precise site-selectively and controllable D-labeling are still challenging.
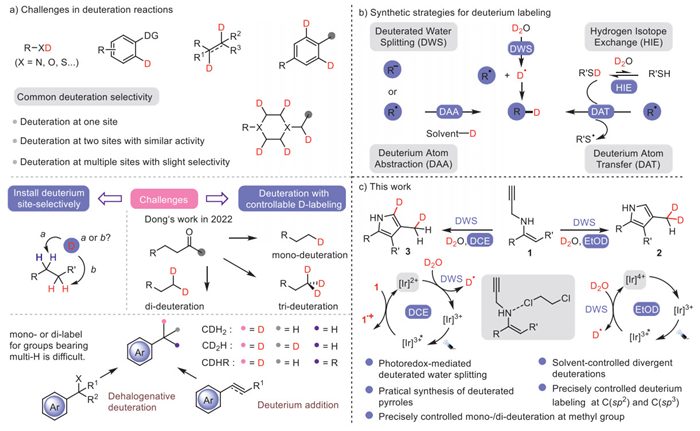
As the continuous interests in the construction of carbo-/heterocycles via visible-light induced tandem radical transformations [46–51], this work envisioned that deuterations at different stages of the tandem radical transformations may achieve site-specific deuterations, and different photoredox cycles may also achieve different deuterations, which may leding to controllable D-labeling. Deuterated water splitting was focused on the easily-available D2O deuterium source and its efficient radical transformations. Herein, photoredox DWS was investigated from the radical cyclizations of N-propargyl enamines, giving deuterated pyrrole derivatives with deuterations at the C(sp2) and C(sp3) carbon precisely (Scheme 1c). Different from the direct incorporation of the CD3 group [52–55], a precise mono- or di-deuteration to methyl group was achieved [19,20], which has been proved to be pretty useful but challenging in studying the therapeutic and metabolic profile of related drugs. Deuteration at the pyrrole motif could also be achieved by controlled deuterium labeling at specific sites in high efficiency. The halogen effect between solvents and substrates were proposed to initiate different catalytic cycles for these divergent deuterations from the same N-propargyl enamines.
Firstly, the feasibility of this radical-triggered cyclization/deuteration was investigated with N-propargyl enamine 1a as a model substrate (Table 1). To our delight, under 15 W blue LEDs irradiation with (Ir[dF(CF3)ppy]2(dtbpy))PF6 (PS1, 3 mol%) as a photosensitizer, deuterated pyrrole 2a was obtained in 83% isolated yield with an excellent deuteration ratio in a co-solvent under nitrogen atmosphere (entry 1). When other potential photosensitizers were screened, no better results were observed than that using PS1 (entries 2−4). Varying the loading of PS1 led to the decrease in both yields and deuteration efficiency (entries 5 and 6). Excessive deuterium oxide led to a poor solubility of 1a, resulting in a lower yield (entry 7). The reaction using 5 W blue LEDs irradiation could not give better result (entry 8). Moreover, other solvents were also screened. No desired 2a was observed when CD3COOD used as the co-solvent (entry 9). The reaction using acetonitrile gave a decreased yield (entry 10). It is interesting to find that deuterations at both the pyrrole moiety (Csp2) and 4-CH3 (Csp3) of 1a were achieved when CDCl3 or 1,2-dichloroethane (DCE) used as the co-solvents (entries 11 and 12). Further optimizations (entries 13−15) showed that deuterations at the C(sp2) and C(sp3) could be achieved in a nearly 1:1 ratio in as high as 84% yield in the presence of PS1 (2 mol%) and D2O (20 equiv.) in DCE (see Supporting information for details). It is also a good example for the mono-label at methyl group.
 DownLoad:
CSV
DownLoad:
CSV
 |
||
| Entry | Variations from the standard conditions | Yields of 2aa |
| 1 | None | 83% (1.75 D) |
| 2 | [Ir(dtbbpy)(ppy)2][PF6] instead of PS1 | Trace (\) |
| 3 | Ir(ppy)3 instead of PS1 | N.O.b (\) |
| 4 | Acr+-Mes ClO4– instead of PS1 | N.O.b (\) |
| 5 | PS1 (1 mol%) | 71% (1.65 D) |
| 6 | PS1 (5 mol%) | 68% (1.64 D) |
| 7 | 2 mL D2O, 3 mL EtOD instead of 1 mL D2O, 4 mL EtOD | 70% (1.72 D) |
| 8 | 5 W blue LEDs instead of 15 W | 63% (1.56 D) |
| 9 | CD3COOD instead of EtOD | N.O.b (\) |
| 10 | MeCN instead of EtOD | 44% (1.79 D) |
| 11c | CDCl3 instead of EtOD | 46% (1.98 D, 0.56 D) |
| 12c | DCE instead of EtOD | 64% (1.70 D, 0.22 D) |
| 13c | 10 W blue LEDs instead of 15 W, DCE instead of EtOD | 40% (1.25 D, 0.26 D) |
| 14c | On the basis of entry 13, D2O (20 equiv.), DCE (5 mL) | 51% (1.11 D, 0.89 D) |
| 15c | On the basis of entry 14, 0.1 mmol scale, PS1 (2 mol%) | 84% (1.00 D, 0.94 D) |
| a Isolated yields. b N.O. = Not observed. c Two-sites-deuterated product 3a was obtained. |
||
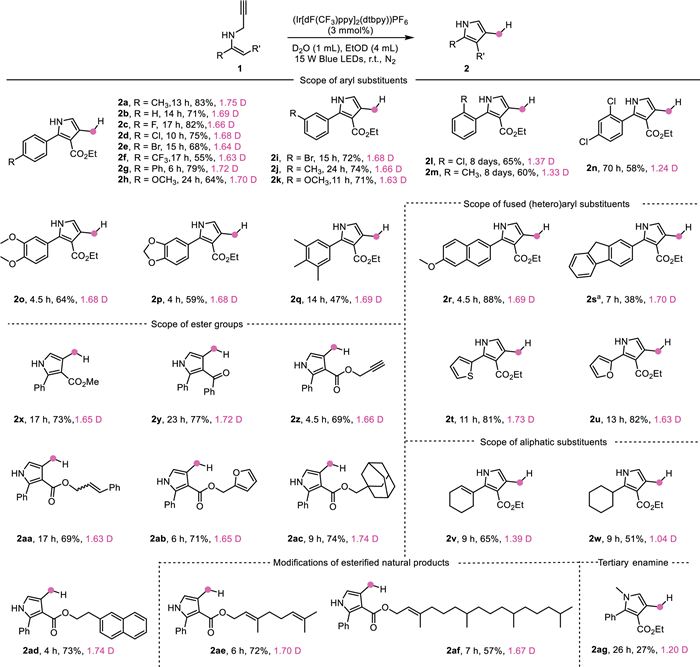
Encouraged by these exciting results, the scope of this highly selective DWS deuteration was studied. The results of photocatalytic deuterations at the 4-CH3 (Csp3) are summarized in Scheme 2. N-Propargyl enamines 1 bearing electron-withdrawing or electron-donating substituents at the ortho-, meta-, and para-positions of the phenyl group were all tolerated to this DWS deuteration, delivering the desired 2a–2m in moderate to excellent yields with high D incorporation (1.33–1.75 D) exclusively at 4-CH3 position. It was shown that ortho-substituted ones required a longer reaction time with a lower D incorporation. N-Propargyl enamines 1 bearing poly-substituents at the phenyl group were also tolerated, giving the corresponding 2n–2q smoothly. N-Propargyl enamines 1 bearing 2–methoxy-naphthyl, 9-fluorenyl, thienyl, furyl substituents were all suitable substrates (products 2r–2u). Due to the poor solubility of 1s in EtOD, DMF was used as the co-solvent instead, giving the desired 2s with a 1.70 D incorporation. It is glad to see that the reaction of N-propargyl enamines 1 bearing aliphatic substituents could be achieved successfully in moderate D incorporation (products 2v and 2w). This reaction also showed excellent tolerance to N-propargyl enamines 1 bearing various ester groups, affording the corresponding 2x–2ad in high yields with good D incorporation. Reactive alkyne and alkene units remained intact in products 2z and 2aa, facilitating further functionalizations. To testify the utility of this DWS deuteration, the modifications of esterified natural products were investigated, giving modified nerol and phytol derivatives 2ae and 2af in good yields with high levels of D incorporation. The reaction from tertiary enamine 1ag was also tested, similarly to the formation of 2l–2n, a longer reaction time was required with a lower D incorporation, giving the corresponding 2ag in 27% yield. It was deduced that the steric hindrance at the nitrogen atom was unfavorable to this transformation.
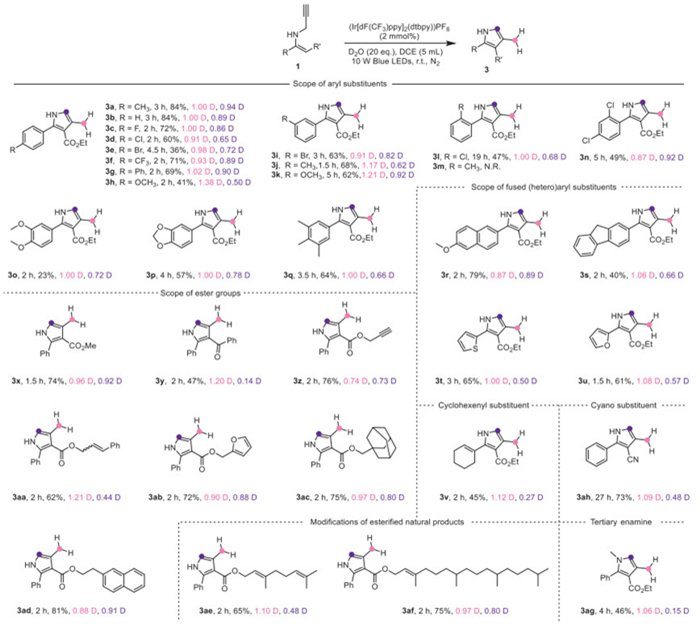
Accordingly, the precise installation of deuterium atoms at C(sp2) and C(sp3) simultaneously was further investigated as shown in Scheme 3 under the optimal reaction conditions (Table 1, entry 15). Generally, these reactions showed excellent tolerance to N-propargyl enamines 1, affording the desired two-sites-deuterated pyrroles 3 with approximately 1D per site in good yields with a shorter reaction time. Most of the substrates used in Scheme 2 could give the corresponding deuterated pyrroles 3, including the modifications of esterified natural products. It was noticed that the reaction of N-propargyl enamines 1 bearing 2-methyl or benzoyl substituents only gave the corresponding one-site-deuterated pyrroles 2m and 2y, while N-propargyl enamine 1 bearing cyano substituent only gave the two-sites-deuterated pyrrole 3ah. Although currently it’s difficult to explain, it indicates that different mechanisms may be involved for the formations of 2 and 3.
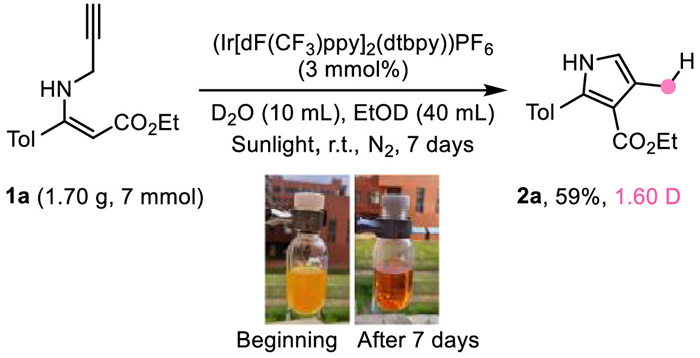
Furthermore, product 2a could be synthesized in 71% yield in a 0.2 mmol scale’s reaction under natural sunlight irradiation at nitrogen atmosphere (see Supporting information for details). The practicality of this reaction was further verified from the scale-up reaction under natural sunlight irradiation in 7 mmol scale. Product 2a could be obtained in 59% yield with similar D incorporation (Scheme 4).
To gain more insight into the key points for above different selectivity, an array of controlled experiments and spectroscopic analyses were investigated (Scheme 5, Fig. 2 and 3). Firstly, the formation of 2a and 3a were investigated with the light off/on over a period of time under the optimal reaction conditions respectively. Both reactions proceeded well under visible light irradiation, but no further transformations were observed without light irradiation (see Supporting information for details). It indicates that the continuous irradiation of visible light is essential for these catalytic reactions and the photo-induced pathways may be involved. Then, the radical trapping experiments with 2,2,6,6-tetramethyl-1-piperidyloxy (TEMPO) as the radical-inhibitor have been performed under the standard reaction conditions for the formation of products 2 and 3 respectively. Both reactions have been greatly inhibited, indicating the possibility of a radical process (Scheme 5a).
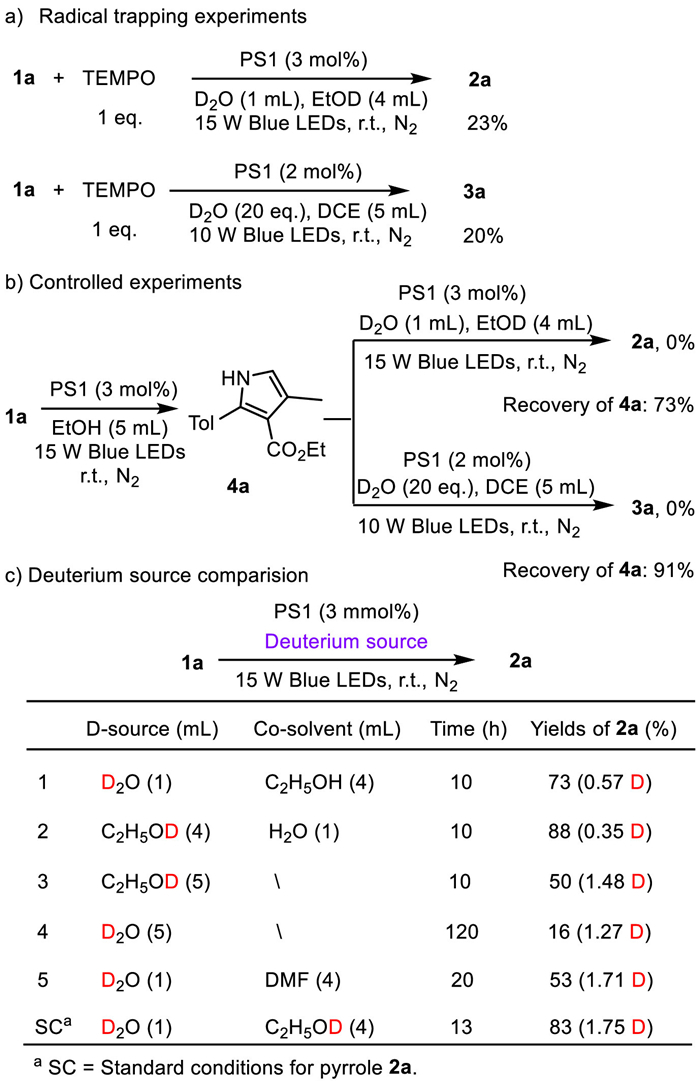
To further understand the mechanisms, the reaction of 1a was performed under the standard conditions for pyrrole 2a but using undeuterated ethanol as the sole solvent to get the corresponding undeuterated pyrrole 4a. Then, 4a was subjected to the standard conditions using EtOD or DCE as co-solvent with D2O respectively. It was found that no deuterated product was observed in both reactions (Scheme 5b), indicating these deuterations are not simple H/D exchange. By varying the deuterium sources and co-solvents (Scheme 5c and Table 1, entry 10), it was deduced that D2O is more efficient as the deuterium source while EtOD mainly provides a good solubility.
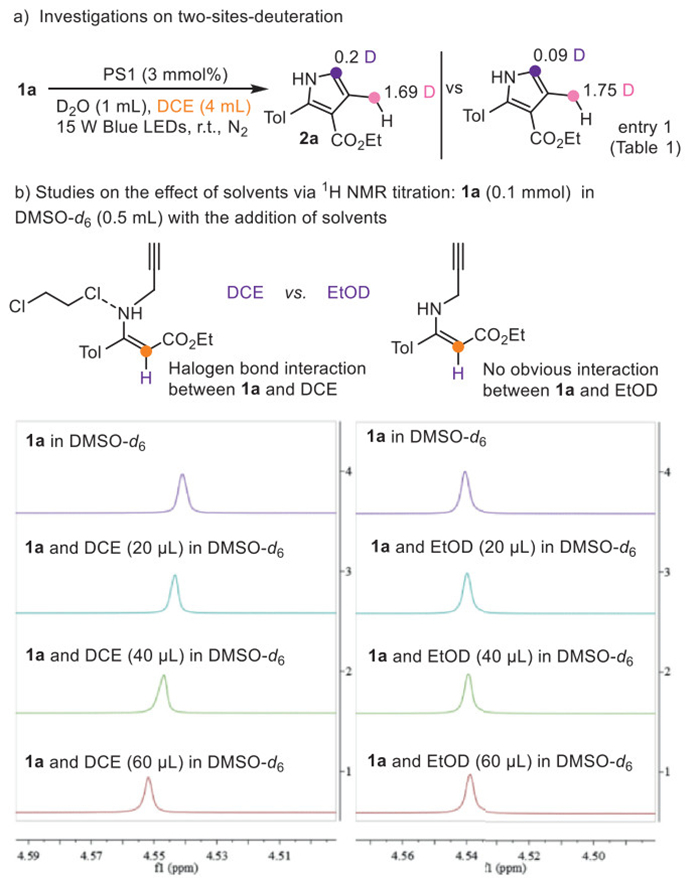
It was noticed that when DCE was used instead of EtOD as co-solvent under otherwise standard reaction conditions for 2a, deuteration at C(sp2) increased, indicating that chlorine containing solvents seem facilitating the two-sites-deuteration process (Fig. 2a). We speculate that halogen bond interaction [56–58] may be formed between DCE and 1a, facilitating the SET process between 1a and the photosensitizer. The 1H NMR titration experiment of the alkene hydrogen of 1a to higher chemical shift values was clearly observed with the increased addition of DCE, which also supported such an interaction between DCE and 1a (Fig. 2b). In contrast, no obvious changes in the 1H NMR chemical shift for the alkene hydrogen were observed when EtOD was added similarly.
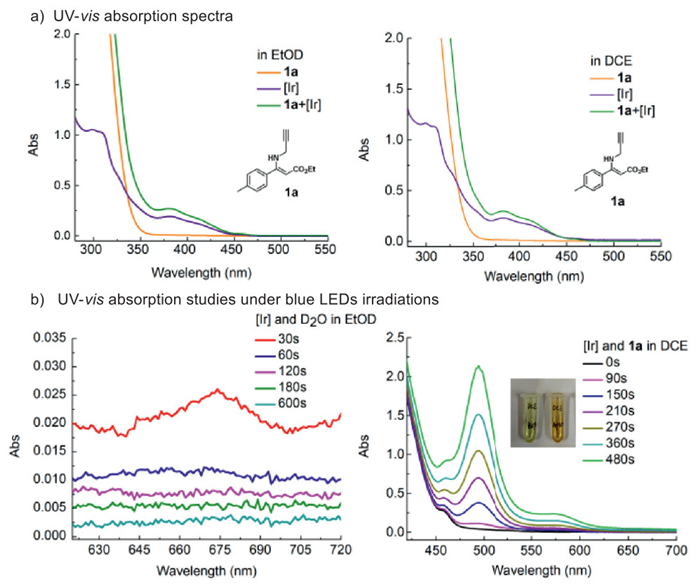
Moreover, the UV−vis absorption spectra confirmed that (Ir[dF(CF3)ppy]2(dtbpy))PF6 is the sole photosensitizer in both deuterations (Fig. 3a). The interaction of excited [Ir] photosensitizer with D2O and 1a were also investigated respectively in EtOD or DCE under blue LEDs irradiation (Fig. 3b). It was observed that an absorption at about 675 nm was observed for the irradiation of D2O and [Ir] photosensitizer in EtOD, which could be attributed to the formation of Ir(Ⅳ) (reference value between 600 nm and 700 nm) [59–61] during the SET process between Ir(Ⅲ) and D2O, while no obvious absorption was observed for 1a and [Ir] photosensiter in EtOD. On the contrary, a strong absorption at about 500 nm was observed for the irradiation of 1a and [Ir] photosensitizer in DCE with a significant color change of the solution, while no obvious absorption was observed for D2O and [Ir] photosensitizer in DCE. The Stern-Volmer emission quenching studies revealed that the excited [Ir] photosensitizer could be quenched by D2O in ethanol and by 1a in DCE (see Supporting information for details). It gave a strong support for our proposed solvent-controlled divergent deuteration pathways.
In light of all the experimental data and precedent literatures, two different plausible catalytic cycles were proposed for the formation of pyrroles 2 and 3 (Scheme 6, with the reactions of 1a as examples). For the formation of 2a, the [Ir] photosensitizer was excited to Ir(Ⅲ)* under blue LEDs irradiation and transformed to Ir(Ⅳ) via a SET process with D2O, realizing deuterated water splitting (DWS) [12,33,34]. The addition of generated deuterium radical to 1a initiated the radical cyclization, giving intermediate Ⅱ. Subsequent oxidative radical-polar crossover (RPC) process [62–64] of Ⅱ with Ir(Ⅳ) species [59−61] and the following deprotonation, achieving intermediate Ⅳ. Further addition of deuterium radical and the following SET/deprotonation process afforded one-site-deuterated pyrrole 2a. On the other hand, for the formation of 3a, the proposed halogen bond interaction [56–58] between DCE and N-propargyl enamine 1a (Fig. 2b) facilitates the SET process of excited photosensitizer Ir(Ⅲ)* with Ⅵ leading to the formation of intermediate Ⅶ, which was transformed to α-amino alkyl radical Ⅷ via deprotonation [65–69]. Then, intermediate Ⅸ, from the radical coupling of Ⅷ and deuterium radical [70], undergoes similar radical addition/cyclization/RPC/deprotonation process, with the final aromatization, afforded two-sites-deuterated pyrrole 3a. The high resolution mass spectra (HRMS) detections of the reaction intermediates (captured by TEMPO) also supported our proposed radical species (Schemes 6a and b). The capture of carbocation intermediates like intermediate Ⅲ was also testied by the addition of acetonitrile, and the corresponding derivative A-Ⅲ was captured by HRMS detections (Scheme 6c). It was proposed that the different solubility of deuterated reagent in solvent and halogen bond interaction between DCE and N-propargyl enamines may be essential for the selectivity. Thus, the solvent-controlled divergent deuterations from the same N-propargyl enamines have been achieved with precisely controlled deuterium labeling.

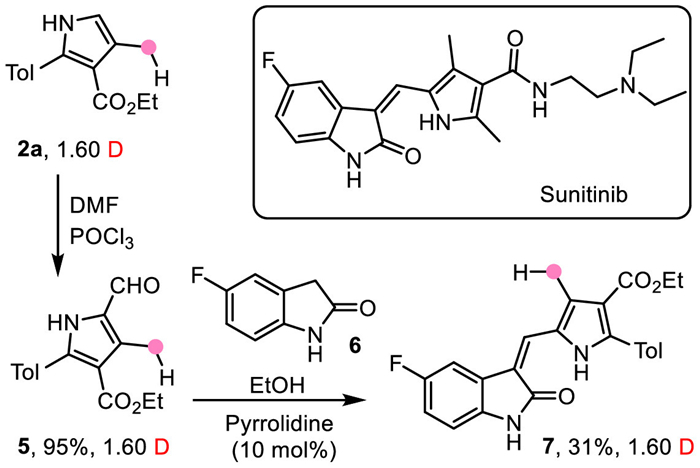
To further verify the practicality of this reaction, the synthesis of sunitinib, which is a novel oral multi-targeted anticancer drug [71], from one-site-deuterated pyrrole 2a was studied (Scheme 7). It is glad to see that sunitinib analogue 7 could be easily obtained via Vilsmeier-Haack reaction of 2a and the following condensation with indolinone 6 with deuterium labeling (1.60 D) remained [72], providing a potential method for the synthesis of deuterated drugs.
In conclusion, photoredox deuterated water splitting was achieved from the radical cyclizations of N-propargyl enamines, giving deuterated pyrroles with deuterations at the C(sp2) and C(sp3) precisely. The precise mono- or di-deuteration of methyl group have also been successfully achieved as a good example for the controllable deuteration label at multi-H/D-exchange-sites, which is especially useful but challenging in drug-studies. These solvent-controlled divergent deuterations have been achieved in high efficiency and selectivity, which may credit to the halogen effect between the solvents and substrates. The mechanistic difference between these different catalytic cycles have been discussed in detail. Furthermore, these deuterations showed broad tolerance to various N-propargyl enamines. The gram scale synthesis under natural sunlight irradiation and its application in the synthesis of drug analogues verified the practicality of these deuterations. Further applications for potential deuterated drugs are in progress.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Baihui Zheng: Methodology, Investigation. Dandan Zhang: Validation, Formal analysis. Baoping Ren: Validation, Formal analysis. Yifei Li: Formal analysis. Qun Liu: Formal analysis. Ling Pan: Writing – review & editing, Writing – original draft, Data curation, Conceptualization.
Financial support from Science and Technology Development Plan Project of Jilin Province, China (20220402046GH) is greatly acknowledged.
Supplementary material associated with this article can be found, in the online version, at doi:
J. Atzrodt, V. Derdau, W.J. Kerr, et al., Angew. Chem. Int. Ed. 57 (2018) 1758–1784. doi: 10.1002/anie.201704146
T. Pirali, M. Serafini, S. Cargnin, et al., J. Med. Chem. 62 (2019) 5276–5297. doi: 10.1021/acs.jmedchem.8b01808
S. Kopf, F. Bourriquen, W. Li, et al., Chem. Rev. 122 (2022) 6634–6718. doi: 10.1021/acs.chemrev.1c00795
C. Canlet, B.M. Fung, J. Phys. Chem. B 104 (2000) 6181–6185. doi: 10.1021/jp000509g
L.H. Coudert, L. Margulès, C. Vastel, et al., A&A 624 (2019) A70. doi: 10.1051/0004-6361/201834827
J. Atzrodt, V. Derdau, T. Fey, et al., Angew. Chem. Int. Ed. 46 (2007) 7744–7765. doi: 10.1002/anie.200700039
J. Atzrodt, V. Derdau, W.J. Kerr, et al., Angew. Chem. Int. Ed. 57 (2018) 3022–3047. doi: 10.1002/anie.201708903
A. Sattler, ACS Catal. 8 (2018) 2296–2312. doi: 10.1021/acscatal.7b04201
F. Bu, Y. Deng, J. Xu, et al., Nature 634 (2024) 592–599. doi: 10.1038/s41586-024-07989-7
G. Prakash, N. Paul, G.A. Oliver, et al., Chem. Soc. Rev. 51 (2022) 3123–3163. doi: 10.1039/d0cs01496f
M. Zhang, X.A. Yuan, C. Zhu, et al., Angew. Chem. Int. Ed. 58 (2019) 312–316. doi: 10.1002/anie.201811522
C. Liu, S. Han, M. Li, et al., Angew. Chem. Int. Ed. 59 (2020) 18527–18531. doi: 10.1002/anie.202009155
N. Li, Y. Ning, X. Wu, et al., Chem. Sci. 12 (2021) 5505–5510. doi: 10.1039/d1sc00528f
M. Valero, D. Bouzouita, A. Palazzolo, et al., Angew. Chem. Int. Ed. 59 (2020) 3517–3522. doi: 10.1002/anie.201914369
Y. Kuang, H. Cao, H. Tang, et al., Chem. Sci. 11 (2020) 8912–8918. doi: 10.1039/d0sc02661a
A. Kurimoto, R.S. Sherbo, Y. Cao, et al., Nat. Catal. 3 (2020) 719–726. doi: 10.1038/s41929-020-0488-z
X. Liu, R. Liu, J. Qiu, et al., Angew. Chem. Int. Ed. 59 (2020) 13962–13967. doi: 10.1002/anie.202005765
A. Suzuki, Y. Kamei, M. Yamashita, et al., Angew. Chem. Int. Ed. 135 (2023) e202214433. doi: 10.1002/ange.202214433
D. Wood, S. Lin, Angew. Chem. Int. Ed. 62 (2023) e202218858. doi: 10.1002/anie.202218858
Z.P. Vang, S.J. Hintzsche, J.R. Clarkv, Chem. Eur. J. 27 (2021) 9988–10000. doi: 10.1002/chem.202100635
X. Zhou, T. Yu, G. Dong, J. Am. Chem. Soc. 144 (2022) 9570–9575. doi: 10.1021/jacs.2c04382
Z. Zhang, C. Qiu, Y. Xu, et al., Nat. Commun. 11 (2020) 4722. doi: 10.1038/s41467-020-18458-w
M. Abdellaoui, T. Deis, M.A. Wiethoff, et al., Adv. Synth. Catal. 365 (2023) 884–891. doi: 10.1002/adsc.202201410
X. Dong, Z. Wang, H. Wang, et al., Chem. Sci. 11 (2020) 1026–1031. doi: 10.1039/c9sc05132e
J. Xu, J. Fan, Y. Lou, et al., Nat. Commun. 12 (2021) 3983. doi: 10.1038/s41467-021-24259-6
L. Wang, C. Liu, L. Li, et al., Chin. J. Chem. 40 (2022) 719–724. doi: 10.1002/cjoc.202100728
J. Dong, M. Yu, F. Yue, et al., Org. Lett. 24 (2022) 2064–2068. doi: 10.1021/acs.orglett.2c00722
Z. Sun, R. Ji, J. Wu, et al., Adv. Synth. Catal. 365 (2023) 476–481. doi: 10.1002/adsc.202201367
Y. Fan, W. Ou, M. Chen, et al., Org. Lett. 25 (2023) 432–437. doi: 10.1021/acs.orglett.2c04154
T. Constantin, M. Zanini, A. Regni, et al., Science 367 (2020) 1021–1026. doi: 10.1126/science.aba2419
T. Patra, S. Mukherjee, J. Ma, et al., Angew. Chem. Int. Ed. 58 (2019) 10514–10520. doi: 10.1002/anie.201904671
Z.Z. Zhou, J.H. Zhao, X.Y. Gou, et al., Org. Chem. Front. 6 (2019) 1649–1654. doi: 10.1039/c9qo00240e
B. Zhang, C. Qiu, S. Wang, et al., Sci. Bull. 66 (2021) 562–569.
X.L. Nan, Y. Wang, X.B. Li, et al., Chem. Commun. 57 (2021) 6768–6771. doi: 10.1039/d1cc02551a
R. Zhou, L. Ma, X. Yang, et al., Org. Chem. Front. 8 (2021) 426–444. doi: 10.1039/d0qo01299h
N. Li, Y. Li, X. Wu, et al., Chem. Soc. Rev. 51 (2022) 6291–6306. doi: 10.1039/d1cs00907a
W. Ou, C. Qiu, C. Su, Chin. J. Catal. 43 (2022) 956–970.
Y.Y. Loh, K. Nagao, A.J. Hoover, et al., Science 358 (2017) 1182–1187. doi: 10.1126/science.aap9674
R. Zhou, J. Li, H.W. Cheo, et al., Chem. Sci. 10 (2019) 7340–7344. doi: 10.1039/c9sc02818h
Y. Zhang, P. Ji, Y. Dong, et al., ACS Catal. 10 (2020) 2226–2230. doi: 10.1021/acscatal.9b05300
Y. Li, Z. Ye, Y.M. Lin, et al., Nat. Commun. 12 (2021) 2894.
K. Ban, K. Imai, S. Oyama, et al., Angew. Chem. Int. Ed. 62 (2023) e202311058. doi: 10.1002/anie.202311058
J. Zhang, M. Jiao, Z. Lu, et al., Angew. Chem. Int. Ed. 63 (2024) e202409862. doi: 10.1002/anie.202409862
M. Jiao J. Zhang, M. Wang, et al., Nat. Commun. 15 (2024) 5067. doi: 10.1038/s41467-024-48590-w
W. Xiao, Y. Tian, L. Du, et al., Org. Lett. 27 (2025) 1112–1117. doi: 10.1021/acs.orglett.4c04516
B. Zheng, X. Li, Y. Song, et al., Org. Lett. 23 (2021) 3453–3459. doi: 10.1021/acs.orglett.1c00915
Q. Xu, X. Zhou, S. Zhang, et al., Org. Lett. 23 (2021) 4870–4875. doi: 10.1021/acs.orglett.1c01596
B. Zheng, X. Li, S. Meng, et al., Org. Chem. Front. 10 (2023) 859–865. doi: 10.1039/d2qo01906j
B. Zheng, J. Zhi, N. Wang, et al., Org. Chem. Front. 11 (2024) 500–507. doi: 10.1039/d3qo01849k
Y. Song, B. Zheng, S. Yang, et al., Org. Lett. 25 (2023) 2372–2376. doi: 10.1021/acs.orglett.3c00890
L. Pan, B. Zheng, X. Yang, et al., Adv. Synth. Catal. 363 (2021) 234–243. doi: 10.1002/adsc.202001001
J. Steverlynck, R. Sitdikov, M. Rueping, Chem. Eur. J. 27 (2021) 11751–11772. doi: 10.1002/chem.202101179
R. Caporaso, S. Manna, S. Zinken, et al., Chem. Commun. 52 (2016) 12486–12489.
L. Hu, X. Liu, X. Liao, Angew. Chem. Int. Ed. 55 (2016) 9743–9747. doi: 10.1002/anie.201604406
K. Komeyama, Y. Yamahata, I. Osaka, Org. Lett. 20 (2018) 4375–4378. doi: 10.1021/acs.orglett.8b01863
H.F. Piedra, C. Valdés, M. Plaza, Chem. Sci. 14 (2023) 5545–5568. doi: 10.1039/d3sc01724a
R. Miao, D. Wang, J. Xiao, et al., Phys. Chem. Chem. Phys. 22 (2020) 10212–10218. doi: 10.1039/d0cp00946f
H.F. Piedra, M. Plaza, Chem. Sci. 14 (2023) 650–657. doi: 10.1039/d2sc05556b
K. Kwon, R.T. Simons, M. Nandakumar, et al., Chem. Rev. 122 (2022) 2353–2428. doi: 10.1021/acs.chemrev.1c00444
K. Zhang, D. Rombach, N.Y. Nçtel, et al., Angew. Chem. Int. Ed. 60 (2021) 22487–22495. doi: 10.1002/anie.202109235
R. Giri, I. Mosiagin, I. Franzoni, et al., Angew. Chem. Int. Ed. 61 (2022) e202209143.
S.K. Nanda, Adv. Synth. Catal. 365 (2023) 834–853.
B. Sahoo, J.L. Li, F. Glorius, Angew. Chem. Int. Ed. 54 (2015) 11577–11580. doi: 10.1002/anie.201503210
X.C. Xu, Y. Sang, M. Yang, et al., Org. Chem. Front. 11 (2024) 5502. doi: 10.1039/d4qo00652f
P. Wang, Q. Zhao, W. Xiao, et al., Green Synth. Catal. 1 (2020) 42–51.
J. Sun, G. Zheng, G. Zhang, et al., Org. Chem. Front. 12 (2025) 1671.
Y. Li, J. Bao, Y. Zhang, et al., Chem 8 (2022) 1147–1163. doi: 10.3390/electronics11071147
L. Wang, X. Huo, X. He, et al., Green Chem. 26 (2024) 8315–8322. doi: 10.1039/d3gc04109c
L. Niu, S. Jin, M. Zhu, et al., Org. Lett. 27 (2025) 3183–3187. doi: 10.1021/acs.orglett.5c00471
F. Parsaee, M.C. Senarathna, P.B. Kannangara, et al., Nat. Rev. Chem. 5 (2021) 486–499. doi: 10.1038/s41570-021-00284-3
C.N. Kondoh, M.D.Y. Miura, M.D.T. Yamanaka, N. Engl. J. Med. 379 (2018) 1877–1878.
G. Meng, C. Liu, S. Qin, et al., Res. Chem. Intermed. 41 (2015) 8941–8954. doi: 10.1007/s11164-015-1939-z

Scheme 2 Deuterations at the 4-CH3 (Csp3) of N-propargyl enamines 1. a DMF as the co-solvent.
Table 1. Effects of the reaction parameters.
|
|
||
| Entry | Variations from the standard conditions | Yields of 2aa |
| 1 | None | 83% (1.75 D) |
| 2 | [Ir(dtbbpy)(ppy)2][PF6] instead of PS1 | Trace (\) |
| 3 | Ir(ppy)3 instead of PS1 | N.O.b (\) |
| 4 | Acr+-Mes ClO4– instead of PS1 | N.O.b (\) |
| 5 | PS1 (1 mol%) | 71% (1.65 D) |
| 6 | PS1 (5 mol%) | 68% (1.64 D) |
| 7 | 2 mL D2O, 3 mL EtOD instead of 1 mL D2O, 4 mL EtOD | 70% (1.72 D) |
| 8 | 5 W blue LEDs instead of 15 W | 63% (1.56 D) |
| 9 | CD3COOD instead of EtOD | N.O.b (\) |
| 10 | MeCN instead of EtOD | 44% (1.79 D) |
| 11c | CDCl3 instead of EtOD | 46% (1.98 D, 0.56 D) |
| 12c | DCE instead of EtOD | 64% (1.70 D, 0.22 D) |
| 13c | 10 W blue LEDs instead of 15 W, DCE instead of EtOD | 40% (1.25 D, 0.26 D) |
| 14c | On the basis of entry 13, D2O (20 equiv.), DCE (5 mL) | 51% (1.11 D, 0.89 D) |
| 15c | On the basis of entry 14, 0.1 mmol scale, PS1 (2 mol%) | 84% (1.00 D, 0.94 D) |
| a Isolated yields. b N.O. = Not observed. c Two-sites-deuterated product 3a was obtained. |
||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们









 下载:
下载: