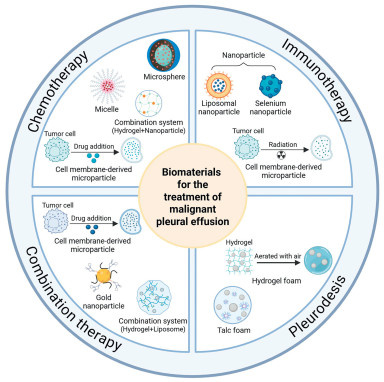
Figure 1.
Schematic diagram of biomaterials for the therapy of MPE, including those used in chemotherapy, immunotherapy, combination therapy, and pleurodesis.

Malignant pleural effusion (MPE) refers to pleural effusion caused by a primary malignant tumor of the pleura or the metastasis of a malignant tumor from other sites to the pleura. There are about 250,000 new cases in the U.S. and Europe according to statistics [1,2]. MPE is a complication of many cancers, the most common of which is being lung cancer, followed by breast cancer, lymphoma, gynecologic malignancies, and mesothelioma [3]. Non-small cell lung cancer (NSCLC) is the most common type of lung cancer, accounting for approximately 85% of all cases, and about 40% of patients with advanced NSCLC may develop MPE [4,5]. The median survival of patients with MPE is typically 3–12 months, depending on the stage and type of the underlying malignancy [6,7]. Patients with MPE caused by ovarian cancer have a longer survival time, whereas those with NSCLC-induced MPE have an average life expectancy of only 3.3 months [8,9]. MPE is caused by an imbalance between the production and absorption of pleural effusion. The invasion of tumor cells into thoracic lymph nodes and the pleura leads to fluid accumulation by impairing lymphatic outflow and increasing the permeability of both the pleura and tumor blood vessels [10]. The traditional clinical treatments for MPE are mainly palliative local thoracic treatment, such as thoracocentesis, indwelling pleural catheter, talc powder pleurodesis, hyperthermic intrathoracic chemotherapy, and intrathoracic antitumor drug therapy [11,12]. Traditional MPE treatments have poor efficacy, high recurrence rate and many adverse reactions. Therefore, the development of an efficient and selective treatment is urgently needed to solve the problem of MPE.
The mechanism of MPE is still unclear, but it is generally believed that tumor-induced inflammation, neovascularization, and high vascular permeability are the core of the pathogenesis of MPE [13]. Vascular endothelial growth factor (VEGF) is a part of the VEGF/platelet-derived growth factor gene family, which exhibits strong angiogenic, mitogenic and vascular permeability-enhancing effects, and is considered to be a key cytokine in the pathogenesis of MPE [14,15]. VEGF can activate vesicle vacuolar organelles, which form a pathway for fluid and solute to cross blood vessel walls; induce mitogenic and chemotactic effects on vascular endothelial cells to promote capillary sprouting; and stimulate endothelial differentiation and proliferation, contributing to tumor expansion and the progression of MPE [16,17]. VEGF is the first tumor expression factor to be identified as an important factor in the development of MPE, which is widely used in clinical treatment of the disease [18]. Commonly used antiangiogenic drugs in the clinic include bevacizumab (BEV) and apatinib (APA). BEV can inhibit the VEGF-receptor signaling pathway by binding to human VEGF (VEGF-A), thereby preventing new blood vessel formation in tumors and inducing tumor cell apoptosis [19,20]. In addition, APA can block VEGFR-2 phosphorylation by binding to intracellular adenosine triphosphate sites, thereby inhibiting the rapidly accelerated fibrosarcoma (Raf)/mitogen‑activated protein kinase (MEK)/extracellular signal‑regulated kinase (ERK) signaling pathway leading to endothelial cell proliferation and the p38 mitogen-activated protein kinase (p38 MAPK) signaling pathway stimulating endothelial cell migration [21,22]. As research on MPE has intensified, researchers have identified additional mediators that influence its development. C—C motif chemokine ligand 2 (CCL2), osteopontin, tumor necrosis factor (TNF), and angiopoietin 1 and 2 secreted by pleural tumors all promote the development of MPE by inducing new angiogenesis [23]. Interleukin-6 (IL-6) is the most typical cytokine associated with inflammation. Autocrine IL-6 activates the signal transducer and activator of transcription 3 (STAT3) pathway and upregulates VEGF expression, thereby contributing to the formation of MPE [24]. The host cell population may also play a role in the development of MPE: (1) monocytes and neutrophils are recruited into the pleural cavity by CCL2, promoting angiogenesis and vascular leakage; (2) eosinophils and myeloid suppressor cells are recruited by IL-5, which supports tumor cell survival in the pleural cavity and enhances vascular permeability; (3) mesothelial cells are stimulated by transforming growth factor β (TGF-β) to produce VEGF [25-27].
Chemotherapy is the main treatment for MPE, but unfortunately, traditional chemotherapy drugs are highly toxic, which limits the dose that can be administered to patients, reduces efficacy, and easily leads to drug resistance in tumor cells, thus resulting in metastasis [28]. Immunotherapy is an important approach in the clinical treatment of MPE. However, the MPE microenvironment contains a significant number of tumor-promoting myeloid immune cells and immunosuppressive cytokines, leading to an immunosuppressive tumor microenvironment (TME) and depletion of tumor-specific cytotoxic T lymphocyte (CTL) cells [29,30]. This ultimately diminishes the efficacy of antitumor responses. In recent years, intrapleural chemotherapy for MPE has no longer been used as a single treatment but has been integrated into other treatment strategies [31]. Combination therapy has become a major research focus both domestically and internationally for the treatment of secondary MPE, typically involving the combination of chemotherapeutic agents with immunotherapy and antiangiogenic drugs. Gandhi et al. used the programmed cell death ligand 1 (PD-L1) inhibitor pembrolizumab combined with platinum-based chemotherapy, and the experimental results revealed that both progression-free survival (PFS) and total survival were noticeably extended compared with chemotherapy alone [32]. Xiang et al. studied the therapeutic effects of APA and BEV combined with cisplatin on MPE [19]. Compared with control groups, the triple-drug combination group significantly prolonged the overall survival of tumor-bearing mice and markedly reduced VEGF levels in both serum and pleural effusion. However, combination therapy has yet to overcome the limitations of chemotherapy and immunotherapy, and its efficacy remains unsatisfactory. Therefore, developing novel therapeutic approaches that effectively mitigate the side effects of chemotherapy while enhancing the antitumor immune response in MPE patients is crucial.
Biomaterials include natural or synthetic materials, which are biodegradable and easy to accept by biological systems, with low toxicity and high cost effectiveness [33]. Compared with conventional clinical methods, various drug delivery systems (DDSs) developed by using these materials offer patients more precise and safer treatment options. DDSs based on biomaterials can overcome the limitations of traditional chemotherapy by increasing the solubility of poorly water-soluble therapeutic agents, reducing systemic toxicity, and improving drug efficacy [34,35]. Biomaterial-mediated immunotherapy, which is characterized by precise targeting, high stability, and low cytotoxicity, can effectively release immunotherapeutic drugs to remodel the TME and elicit rigorous immunogenicity [36,37]. In terms of combination therapy, biomaterials also show great potential. By encapsulating radioisotopes and anti-cytotoxic T-lymphocyte-associated protein-4 (CTLA-4) antibodies in sodium alginate (ALG) hydrogel, Chao et al. were able to reduce the in vivo toxicity of radioisotope therapy (RIT) and induce immune-memory effects, which in turn prevented tumor metastasis and recurrence [38]. Biomaterials, such as microcapsules, nanoparticles (NPs), microspheres, liposomes, and micelles, which have obvious advantages of high drug-loading capacity, controllable size and permeability of the delivery system, and the ability to transport large quantities of drugs to target sites via relatively small amounts of carrier molecules, are commonly employed in DDSs [39]. NPs optimize drug delivery by encapsulating therapeutic agents to enhance their aqueous solubility and bioavailability, prolong blood circulation for improved tumor accumulation via the enhanced permeation and retention effect, and mitigate systemic toxicity through controlled release kinetics and enzymatic protection of payloads, thereby positioning them as potent platforms for cancer therapy [40-43]. Liposomes exhibit high stability and drug-loading capacity, optimizing pharmacokinetics and tumor accumulation, thereby ensuring enhanced therapeutic efficacy and clinical translational potential [44]. In addition, hydrogels exhibit favorable biocompatibility and biodegradability coupled with a porous network structure that facilitates high drug-loading capacity [45]. Biomaterial-modified DDSs epitomize the convergence of nanotechnology, immunology, oncology, and biomedicine, offering a roadmap to bridge the gap between localized disease aggression and systemic therapeutic inadequacy. Their development not only addresses the unmet needs of MPE but also establishes a foundational strategy for intracavitary oncology, heralding a new era in the management of metastatic serosal malignancies. This review focuses on the latest applications and advancements of biomaterials in the treatment of MPE, emphasizing the advantages of DDSs based on these materials. We aim to increase awareness within the medical community regarding the potential of these delivery systems and to promote translational research in this field. Therefore, this paper discusses the application of biomaterials in the treatment of MPE from the perspectives of chemotherapy, immunotherapy, combination therapy, and pleurodesis, with a focus on their ability to overcome drug delivery challenges (Fig. 1).
DDSs based on biomaterials can address certain limitations of traditional chemotherapy and immunotherapeutic drugs. These systems increase drug stability and the accuracy of tumor cell targeting, reduce toxic side effects, and remodel TME, thereby increasing drug efficacy. Although APAs and BEVs have been approved for the clinical treatment of MPE, unfortunately, there is no precedent for the application of biomaterials in antiangiogenic drugs in current studies. This discrepancy underscores the importance of systematically evaluating how biomaterial-based DDSs could broaden therapeutic options for MPE beyond existing modalities.
Intrathoracic chemotherapy not only reduces pleural effusion but also enhances systemic antitumor effects. Currently, the approved intrathoracic antitumor therapies include chemotherapy (platinum-based drugs) and antiangiogenic treatments (recombinant human endostatin and BEV) [11]. Clinical guidelines recommend draining pleural effusion before initiating systemic chemotherapy. An experiment by Herrstedt et al. demonstrated that compared with patients without MPE, patients with small cell lung cancer (SCLC) and MPE have increased myelosuppression after treatment, possibly due to the accumulation of chemotherapy drugs in pleural effusion, which exacerbates toxicity [46]. In addition to conventional chemotherapy, hyperthermic intrathoracic chemotherapy (HITHOC) is also used in clinical practice. HITHOC directly exposes tumor cells to higher concentrations of chemotherapy drugs, reduces systemic side effects, and improves the survival rate of patients compared with those who do not receive it [47]. Unfortunately, the chemotherapy methods currently used in clinical practice still have significant limitations and fail to achieve the desired outcomes. To improve chemotherapy efficacy and address certain limitations, this review discusses the use of biomaterial-based chemotherapy drugs for the treatment of MPE, with a focus on microspheres, cell membrane-derived microparticles (MPs), micelles, biodegradable drug-eluting pellets, and some composite systems.
Doxorubicin (DOX) is a broad-spectrum antitumor drug with strong cytotoxic effects [48]. It interferes with DNA replication by inhibiting topoisomerase Ⅱ and generating free radicals, which induce damage to both DNA and the cell membrane, thereby triggering apoptosis in cancer cells [49]. Clinically, it is commonly used to treat breast cancer, lung cancer, ovarian cancer, and other malignancies. With its nontoxic nature and tunable degradation rate through molecular weight and polydispersity adjustments, poly(L-lactic acid) serves as an ideal biomaterial for DDSs. Ike et al. developed poly(L-lactic acid) microspheres (ADR-MS) containing DOX hydrochloride (ADR) as a sustained-release DDS for MPE [50]. The microspheres were prepared by mixing DOX and L-lactic acid via the oil-in-oil method. The average molecular weight was 3400 Da, the average particle size was 50 µmol/L, and the ADR content was 5%. The in vitro release experiment demonstrated that 50% of the ADR was released after 6.3 days and 100% was released after 20 days. Animal experiments on mice revealed that the ADR-MS treatment group presented an increased survival rate compared with the control groups [51]. Seven patients with MPE were treated clinically with ADR-MS, and ADR was detected in the drainage fluid more than two weeks after administration, indicating the sustained release of ADR-MS in the pleural cavity. Only one patient passed away after 4 months, whereas the remaining six patients survived for 21–31 months with no accumulation of fluid. The results showed that ADR-MS consistently released high local concentrations of ADR in the pleural cavity and was well tolerated. Minimal complications and no adverse systemic reactions were observed in patients. In summary, ADR-MS may improve the prognosis and overall well-being of patients with MPE.
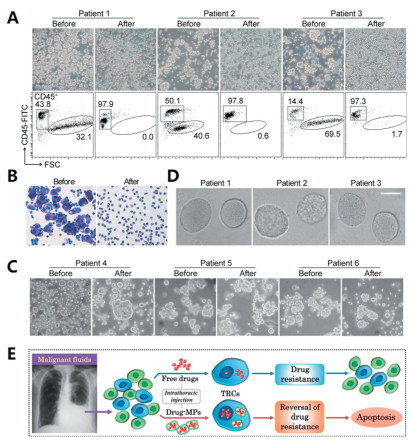
Tumor-repopulating cells (TRCs), a self-renewing subpopulation of tumorigenic cancer cells, play a pivotal role in cancer progression [52]. Microparticles derived from cellular membranes act as critical mediators of intercellular communication [53]. Tumor-cell-derived MPs (T-MPs) carry tumor-specific antigens identical to those expressed by their parental cells, inducing effective T-cell-dependent antitumor immune responses [54]. Given their capacity for uptake by both normal and cancer cells, MPs represent inherently biocompatible delivery vectors [55]. Ma et al. designed cisplatin-loaded T-MPs (CisMPs) to reverse the drug resistance of TRCs [56]. In this study, ultraviolet irradiated A549 cells were co-incubated with cisplatin to prepare CisMPs. Six lung cancer patients with MPE were recruited to assess therapeutic efficacy: three received intrapleural CisMPs, while other three received conventional cisplatin. After 7 days of treatment, 95% of tumor cells in MPE samples from the CisMPs group had disappeared (Fig. 2). Concurrently, marked improvements were observed in MPE volume, color, and turbidity, as well as the symptoms of the patients. In contrast, no therapeutic benefit was observed in the cisplatin group. These findings suggest that MP-mediated DDSs may have the potential to overcome tumor cell resistance, including cisplatin resistance, suggesting promising prospects for clinical application.
Cabazitaxel (CTX), a taxane chemotherapeutic agent, circumvents paclitaxel resistance through its reduced affinity for P-glycoprotein (P-gp) in neoplastic cells [57]. However, its therapeutic efficacy was reduced by inherent hydrophobicity [58]. To improve the aqueous solubility of CTX, Sun et al. introduced a so-called surfactant stripping method to increase the drug-to-surfactant ratio, subsequently employing mifepristone (MIF) as a co-loader to prevent taxane aggregation [59]. This methodology yielded a stable CTX-loaded micelle (MIF-sss-CTX). In this study, Lewis lung cancer (LLC) tumor and MPE mouse models were established. Both the Tween-80-based formulation and MIF-sss-CTX demonstrated significant reductions in pleural tumor burden and prolonged overall survival compared to phosphate-buffered saline (PBS) controls, while maintaining stable body weight. Although the accumulation of CTX in the two formulations was not evaluated, both delivery systems demonstrated comparable antitumor responses. The developed micellar system can improve the ability of water storage, thereby mitigating hydrophobic limitations. This advancement presents novel perspectives for the development of DDSs in MPE therapy.
Platinum-based chemotherapeutic agents play an important role in the process of cancer treatment. These compounds exert their cytotoxic effects through the formation of DNA adducts via crosslinking with bases, thereby impeding DNA repair mechanisms, inducing genomic damage, and triggering programmed cell death in malignant cells [60]. However, the clinical utility of cisplatin is frequently limited by its significant nephrotoxic and haematotoxic effects, often necessitating treatment discontinuation [61]. To mitigate the limitation and enhance its therapeutic efficacy, Chao et al. developed biodegradable drug-eluting pellets designed for sustained release of cisplatin [62]. In this study, biodegradable pellets with a diameter of 3 mm were constructed by mixing, compressing, and sintering poly(D, L-lactide-co-glycolide) (PLGA) copolymers with cisplatin. The in vitro release experiment in PBS demonstrated an initial burst release of 5% cisplatin within 24 h, followed by gradual elution of the remaining payload over 50 days. The in vivo drug release experiment employed New Zealand rabbits with bilateral 5 mm dorsal incisions. The control group received intrapleural cisplatin solution via the medial incision, while the experimental group underwent implantation of cisplatin-loaded pellets through the same incision. Both groups exhibited elevated pleural cisplatin concentrations during the initial 48-h period. However, while the solution group showed precipitous drug level decline thereafter, the pellet group maintained significantly elevated pleural concentrations for 18 subsequent days (P < 0.001). The control group experienced two fatalities with evidence of nephrotoxic accumulation, whereas no mortality occurred in the experimental group. These findings demonstrate that the pellets can achieve sustained intrapleural cisplatin delivery for at least two weeks with minimal systemic toxicity. This novel drug delivery method has the potential to serve as an effective adjunctive treatment for MPE in clinical practice.
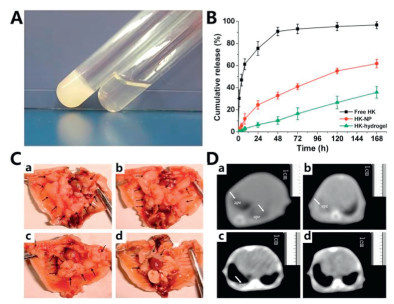
Honokiol (HK), a bioactive compound derived from magnolia bark, has been shown to inhibit the growth of transformed epithelial cells in vitro and exhibits antiangiogenic and antitumor activities [63]. However, its clinical translation is hampered by poor aqueous solubility. To overcome this limitation, Fang et al. designed a novel thermoresponsive hydrogel (HK hydrogel) using poly(ethylene glycol)-poly(ε-caprolactone)-poly(ethylene glycol) (PEG-PCL-PEG, PECE) loaded with HK-NPs [64]. HK-NPs were prepared via emulsion solvent evaporation technique using an F127 aqueous solution, while the PECE copolymer was produced by ring-opening copolymerization and coupling processes. The PECE aqueous solution exists as a free-flowing sol at room temperature but transitions into an in situ gel at approximately 37 ℃, forming a controlled DDS (Fig. 3A). In vitro release experiments revealed that significantly prolonged HK elution from the hydrogel formulation compared to control groups, maintaining therapeutic concentrations for seven days (Fig. 3B). To evaluate the therapeutic effects of HK hydrogels on MPE, the study employed LLC cell model in C57BL/6 mice with in situ gel administration. The experimental results demonstrated that, the HK hydrogel group exhibited a significant reduction in number of pleural tumor (Fig. 3C) and pleural effusion volume (Fig. 3D), compared with those of the HK-NP group, hydrogel group, and normal saline (NS) group. Specifically, compared with the NS group, the HK-NP group presented a 16% reduction in MPE volume, whereas the HK hydrogel group presented a 66.1% reduction in MPE volume and doubled the average survival time. Microvessel density (MVD) measurements among the four groups revealed that the HK hydrogel group had significantly reduced MVD. Immunohistochemical analysis of pleural tumors confirmed that HK hydrogel effectively inhibited angiogenesis (P < 0.05). Additionally, histological analysis of pleural tumors revealed an increased apoptosis rate in the HK hydrogel group. In summary, intrapleural injection of the HK hydrogel successfully induced tumor cell death, diminished pleural vascular permeability, and suppressed neovascularization in pleural tumors. These findings provide an ideal basis for future clinical trials of HK hydrogels.
MPE is an inflammatory condition within the pleural cavity. Despite the presence of immune-active substances in the TME, it is characterized predominantly by exhausted cells associated with large tumor masses and suspended tumor cells, lacking preexisting immune responses [65]. Functionally, MPE has been defined as "cold". By enhancing T cell activation, removing co-inhibitory signals, or providing co-stimulatory signals, "cold" tumors can be transformed into "hot" tumors [66,67]. These strategies for transforming "cold" tumors into "hot" ones have been explored through various therapeutic modalities, including the use of living cells and engineered immune cell therapies. For instance, numerous preclinical studies have demonstrated that living cell-mediated DDSs can be constructed by chemically conjugating or non-covalently binding therapeutic agents to cells with weak phagocytic capacity, thereby enhancing drug delivery efficacy [68]. Chimeric antigen receptor T-cell immunotherapy (CAR-T) cell therapy has also been demonstrated to possess substantial antitumor potential, wherein ex vivo modification enables the transfer of genetic constructs encoding antigen-specific recognition domains and T-cell activating signaling components into lymphocytes, thereby augmenting the immune system's tumoricidal efficacy [69]. To improve the efficacy of immunotherapy, this review discusses the role of biological material-based immunotherapy in MPE treatment, focusing on approaches involving NPs and cell membrane-derived microparticles.
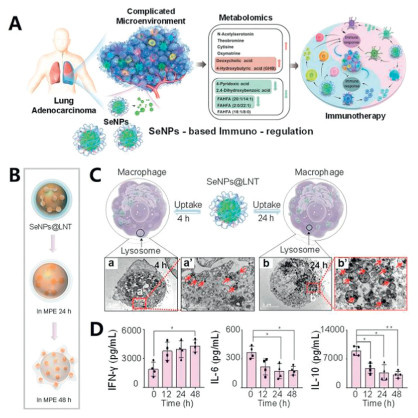
Elemental selenium NPs (SeNPs) demonstrate excellent biocompatibility, attenuated toxicity, potent free radical scavenging capacity, and a strong ability to traverse biological barriers [70]. Pretreatment of gamma delta T cells (γδT cells) with SeNPs can better inhibit tumor growth [71]. Lentinan (LNT), a polysaccharide isolated from shiitake mushrooms that is clinically approved in China as an adjunctive cancer therapy, possesses various biological functions, including antitumor, immunomodulatory, and anti-infective properties [72]. Based on these characteristics, Song et al. designed LNT-encapsulated SeNPs (SeNPs@LNT) to investigate innovative immunotherapeutic approaches for MPE (Fig. 4) [73]. The SeNPs@LNT composite was synthesized through Na2SeO3-LNT co-precipitation under magnetic stirring, followed by dialysis purification. The average diameter of SeNPs@LNT is approximately 160 nm. In vitro stability assessments revealed complete NP degradation within 48 h in MPE. Pleural effusions from MPE-LA patients treated with these NPs were collected to monitor immune cell interactions. The results indicated that macrophages treated with SeNPs@LNT presented reduced concentrations of IL-6, and the levels of inflammatory and immunosuppressive factor cytokines (IL-10) secreted by macrophages decreased 2.7-fold. These findings confirm SeNPs@LNT's dual mechanism of action: angiogenesis inhibition and inflammatory microenvironment modulation in MPE. Natural killer (NK) cells exhibit cytotoxic activity against various tumor cells, and their quantity and activity are associated with tumor patient prognosis [74]. Compared with that of the untreated samples, the percentage of NK cells induced by SeNPs@LNT increased twofold, and the proportion of γδT cells in MPE also tripled. These results indicate that LNT synergistically enhances the immune-stimulatory effects of SeNPs and triggers an antitumor immune response. Additionally, a higher risk of lung cancer has been proved to be linked with the increased catabolism of vitamin B6 [75]. After 24 h of co-incubation with SeNPs@LNT, the levels of a key component in the vitamin B6 metabolic pathway were significantly decreased. While this study offers a novel strategy for MPE treatment, the therapeutic effects of SeNPs@LNT in MPE patients have not been directly validated. Further clinical studies involving larger patient cohorts are needed to confirm the hypothesis regarding the regulation of immune cells by SeNPs in MPE.
Building on the aforementioned study, Liu et al. utilized a similar method to prepare LNT SeNPs (LET-SeNPs), investigating their immunomodulatory mechanisms on NK cells in MPE [76]. NK cells are innate immune lymphocytes, having the capacity to kill tumor cells and produce cytokines to regulate adaptive immune responses [77]. Previous studies have shown that LET-SeNPs can increase the secretion of interferon-gamma (IFN-γ) and IL-15, thereby promoting NK cell proliferation and functional activation [73]. The results demonstrated that LET-SeNPs significantly enhanced the quantity and activity of NK cells via the TrxR1-IL18RAP-pSTAT3 signaling pathway. MPE mouse models were established via A549-GFP cells, and the antitumor effects of LET-SeNPs were evaluated through tail vein injection. Compared with the control groups, the SeNPs+NK92 cell group presented a marked decrease in the number of pulmonary metastatic nodules and tracheal tumor growth area. Compared with that in the NK group, tumor proliferation was markedly suppressed, neural cell adhesion molecule (CD56) expression was significantly greater, the pleural effusion volume was reduced, and the color of the effusion was restored. In conclusion, prestimulation of NK cells with LET-SeNPs harnessed the innate immune system, resulting in robust antitumor effects. The development of this system provides a solid foundation for the potential clinical application of immunotherapy in MPE.
The stimulator of the interferon genes (STING) pathway plays a critical role in antitumor immunity [78]. Cyclic dinucleotides (CDNs) activate STING through binding, inducing the production of IFNs and thereby augmenting anti-tumor immune responses [79]. Liu et al. developed CDN-loaded lipid NPs (LNP-CDNs) to assess the effect of intrapleural administration on MPE [80]. The LNP-CDNs exhibited an average diameter of approximately 120 nm and a surface charge of −15 mV. In this study, the mouse MPE model was established using LLC cells. Chemical signaling analysis revealed that LNP-CDNs were predominantly internalized by macrophages and dendritic cells, facilitating targeted delivery to MPE and solid tumors. Subsequent analyses revealed that LNP-CDN treatment significantly increased the number of CD8+ T cells in pleural tumors, tumor-draining lymph nodes (TDLNs), and the spleen, with more than a tenfold increase in CD8+ T cells within the MPE. These findings demonstrated that LNP-CDNs effectively enhanced cross-priming of tumor-specific CD8+ T cells and expanded the population of multifunctional effector CD8+ T cells and stem-like memory CD8+ T cells in MPE. Additionally, LNP-CDN enhanced NK receptor activation, driving NK cells from an immature state to a mature state, while upregulating the gene expression of effector molecules and activating transcription factors. At the protein level, LNP-CDNs also heightened PD-L1 expression on tumor cells. To mitigate the overexpression of PD-L1, anti-PD-L1 therapy was combined with LNP-CDN, resulting in a synergistic immunotherapeutic approach. This combination therapy further diminished MPE volume and pleural tumor burden while significantly prolonging the survival of the mice. Moreover, the immune effects induced by LNP-CDNs were also observed in clinical MPE samples, strongly supporting its potential in clinical MPE immunotherapy.
Radiotherapy (RT) is employed in approximately 60% of cancer patients, functioning through targeted delivery of radiation beams to tumor sites to induce DNA damage and subsequent tumor cell death [81]. During RT, non-irradiated cells may receive signals from irradiated counterparts, resulting in oxidative stress and DNA damage, a phenomenon termed the radiation-induced bystander effect (RIBE) [82]. To exploit this mechanism, Wan et al. sought to remodel the TME for the treatment of tumors ineligible for RT [83]. Their study demonstrated that microparticles released by irradiated tumor cells (RT-MPs) mediated RIBE by inducing ferroptosis in tumor cells. This process enhanced macrophage phagocytosis and activated the Jak-STAT and MAPK pathways, thereby reprogramming macrophages from a tumor-promoting phenotype to a tumor-suppressing phenotype. This study used 6 MV X-rays to irradiate tumor cells and isolated exosomes via gradient centrifugation to prepare microparticles (RT-MPs). An MPE mouse model was established, and RT-MPs were administered intrapleurally. Results demonstrated that RT-MPs delayed tumor progression and enhanced survival rates compared to controls, alongside a 3.2-fold increase in PD-L1 expression on macrophages. Subsequent combination therapy using RT-MPs and anti-programmed cell death protein 1 (PD-1) monoclonal antibodies significantly prolonged survival, achieving complete remission in 1 of 15 mice without recurrence. Administration of six RT-MP injections elevated treatment success rates to 38.5%, surpassing outcomes from anti-PD-1 or RT-MP monotherapy. Furthermore, in a cisplatin-resistant MPE murine model, the combination therapy cured approximately 20% of mice, whereas cisplatin monotherapy showed no efficacy. These findings indicate that RT-MPs mimic RIBE to exert distinct anti-tumor effects, with anti-PD-1 combination strategies exhibiting promising therapeutic potential for MPE. For clinical translation, RT-MPs derived from autologous primary tumor cells could offer a non-toxic, personalized cancer treatment paradigm.
Immunotherapy as a monotherapy exhibits suboptimal clinical outcomes due to low response rates in immune activation, inadequate tumor infiltration of immune cells, and the intricate immunosuppressive nature of the TME [84]. Contemporary studies have demonstrated that multimodal therapeutic strategies integrating immunotherapy with complementary treatment approaches enhance the effectiveness of cancer treatment. This review discusses the therapeutic effects on MPE from two perspectives: chemotherapy, immunotherapy combinations and gene, immunotherapy combinations modified with biomaterials.
Chemotherapeutic agents exert direct cytotoxic effects on tumor cells, whereas immunotherapeutic interventions host immune responses to achieve targeted oncological destruction, with these therapeutic modalities demonstrating temporally complementary efficacy. The rapid-onset but transient therapeutic impact of chemotherapeutic agents contrasts with the delayed yet sustained antitumor response elicited by immunotherapeutic approaches, playing a good synergistic role [85].
Tumor cell-derived MPs (TMPs) demonstrate the capacity to induce effective T-cell-dependent antitumor immunity [54]. The antineoplastic agent methotrexate (MTX) exerts its therapeutic effects via folate antagonism, inhibiting nitric oxide-mediated pro-angiogenic signaling pathways critical for tumor neovascularisation [86,87]. Guo et al. designed a bifunctional particle (TMPs-MTX) through ultraviolet radiation b (UVB) irradiation-induced microparticle biogenesis from tumor cells, subsequently utilizing TMPs as targeted carries for MTX delivery [88]. Characterisation revealed TMPs-MTX exhibited polydisperse sizing (30‒930 nm range) with a mean diameter of 264 nm. To evaluate therapeutic potential in MPE, two mouse models were utilized: the LLC model and the MC38 cell-induced pleural tumor foci model, with systemic administration via intravenous injection. The experimental results demonstrated that TMPs-MTX treatment considerably lowered the burden of pleural tumors, the number of pleural tumor foci, and pleural effusion volume. Compared with the control group and the free MTX group, TMPs-MTX treatment significantly improved the survival rate and median survival time in both mouse models. After TMPs-MTX treatment, the CD4/CD8 ratios of both models were significantly lower (P = 0.003 in LLC model), while the proportions of immune cells such as neutrophils and macrophages in the blood increased. Based on the observed benefits and minimal toxicity of TMPs-MTX, a human clinical trial was conducted to evaluate intrapleural administration of autologous TMPs-MTX (ATMP-MTX) in MPE patients (Fig. 5). After ATMP-MTX treatment, patients achieved an objective clinical response rate of 90.91%. The volume of pleural effusion decreased, with notable improvements in its color and vascular permeability, and a reduction in the tumor cell count. Immunological analysis of the TME revealed that changes in the immune cell infiltrates and cytokine/chemokine levels were both observed (TNF-α, IFN-γ, and IL-17 all increase). These data suggest that ATMPs-MTX can influence the secretion of chemokines, thereby promoting the activation of antitumor effector cells in the TME of MPE. ATMPs-MTX as a novel therapeutic paradigm that combines chemotherapy with microenvironmental immunomodulation provides a promising therapeutic platform for the treatment of MPE.
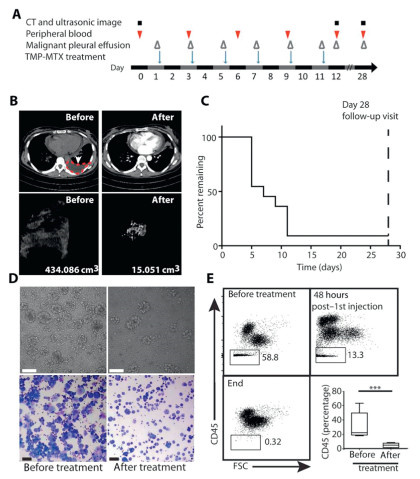
Neutrophils, the most abundant innate immune cells, play a crucial role in tumor progression by significantly influencing the TME through cytokine and chemokine production, thereby modulating inflammatory cell recruitment and activation [89]. Building upon Guo's research, Xu et al. adopted an analogous methodology to prepare MTX-encapsulated tumor cell-derived microparticles (MTX-MPs), investigating their mechanism in recruiting neutrophils to the pleural cavity for the treatment of MPE [90]. The study enrolled NSCLC MPE patients with prior systemic chemotherapy. Participants were divided into two groups, receiving intrapleural injections of either MTX-MPs or saline on day five, followed by alternate-day administrations. Subsequent MPE analysis revealed marked improvements in effusion clarity and color within the MTX-MPs group, with effective elimination of CD45− tumor cells and predominant CD45+ immune cell infiltration. In contrast, the saline group showed no changes in the proportions of CD45− and CD45+ cells. After 4 weeks' treatment, the volume of MPE in the MTX-MPs group was significantly reduced. Neutrophils, including CD11b+ and CD15b+ cells, were detected within MTX-MP-treated MPE, and their abundance was correlated with the reduction in MPE volume. Further investigations revealed MTX-MPs can induce macrophages to release C-X-C motif chemokine ligand1 (CXCL1) and CXCL2, which attract neutrophils to the pleural cavity. These neutrophils exerted antitumor effects through reactive oxygen species (ROS) and neutrophil extracellular trap (NET)-mediated tumor cell cytotoxicity. Additionally, the released NETs sealed damaged endothelial cells, thereby mitigating plasma extravasation. This study further underscores the therapeutic potential of MPs in MPE treatment.
To assess the safety and efficacy of TMPs-MTX, Dong et al. conducted a controlled trial administering intrathoracic injections of TMPs-MTX alongside TMPs-encapsulated saline combined with pemetrexed and cisplatin chemotherapy for the treatment of MPE [91]. Participants were equally allocated to TMPs-MTX or TMPs-saline groups, receiving daily treatments for six consecutive days commencing five days post-chemotherapy. Compared with that in the TMPs-saline group, the objective response rate of MPE in the TMPs-MTX group was considerably greater (P = 0.0237). Unfortunately, there was no discernible improvement in overall survival, PFS, or the objective response rate (ORR) of patients with target lesions. Study limitations included restricted sample size and unexamined immunological parameters. Nevertheless, the ORR of MPE indicated a positive safety profile and therapeutic potential in MPE.
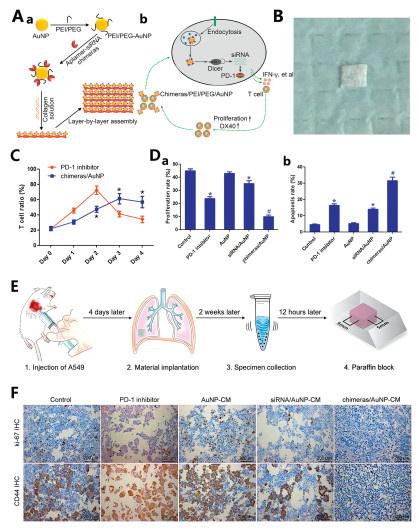
PD-1 serves as a T-cell inhibitory receptor, with its ligand PD-L1 playing a key role in tumor "immune escape". Antibodies targeting the PD-1/PD-L1 immune checkpoint can activate T cells, leading to dynamic and durable tumor regression [92]. The immune costimulatory molecule OX40 (CD134) is a critical marker for T-cell activation, and its ligand, OX40L, activates T cells by stimulating OX40 and initiating the activation of signaling pathways [93]. In a previous study, Chen et al. developed an aptamer-siRNA chimera and demonstrated its ability to specifically target endothelial progenitor cells while delivering siRNA [94]. The construct exhibited high stability and potent gene silencing efficacy. Gold NPs (AuNPs) can function not only as immunomodulators but also as DDSs, enhancing the efficacy and safety of cancer immunotherapy [95]. To achieve a more sustained PD-1 inhibition for MPE treatment, Chen et al. developed an aptamer-siRNA chimera/polyethyleneimine (PEI)/PEG/AuNP/collagen membrane complex [96]. The synthesis involved sequential addition of PEI/PEG solution to HAuCl4 under agitation and centrifugation, followed by the addition of an OX40 aptamer-PD-1 siRNA chimera to produce aptamer-siRNA chimera/PEI/PEG/AuNP. These NPs were then dissolved in a collagen solution to form a chimera/PEI/PEG/AuNP/collagen membrane (Fig. 6). The size of aptamer-siRNA chimera/PEI/PEG/AuNPs ranged from 150‒300 nm and their zeta potential was approximately 10.3 mV. MPE samples from lung cancer patients were collected and treated with either a PD-1 inhibitor or the chimera/PEI/PEG/AuNP formulation. After 48 h, the percentage of T cells peaked in the PD-1 inhibitor group but then began to decrease. At 72‒96 h, the T-cell proportion in the chimera/AuNP group was significantly greater than in the PD-1 inhibitor group. Post-96-h lymphocyte co-cultured with lung adenocarcinoma cells demonstrated 93.51% enhanced apoptosis induction and 58.46% proliferation reduction in the chimera/AuNP group versus PD-1 inhibitor controls. Murine MPE models receiving in situ nanocomposite membrane implantation exhibited 71.31% reduction in pleural tumor cell counts compared to PD-1 inhibitor group, with absent erythrocyte contamination or necrotic debris. Immunohistochemical profiling showed marked downregulation of proliferation marker Ki-67 and metastatic marker CD44 in the chimera/AuNP group. These findings collectively demonstrate superior therapeutic durability and reduced adverse effects of the nanocomposite approach over conventional PD-1 inhibition, proposing an innovative immunotherapeutic strategy for MPE management.
The STAT3 exhibits constitutive activation in NSCLC. Tumor cell-intrinsic and extrinsic STAT3 signaling in NSCLC promotes angiogenesis, cell survival, cancer cell stemness, drug resistance, and evasion of anti-tumor immunity [97,98]. Knockdown of small interfering RNA (siRNA) enables specific suppression of STAT3 expression, effectively disrupting its signal cascade [99]. To enhance siRNA targeting precision and in vivo stability, Fu et al. pioneered the development of a localized hydrogel system MSL@LID@SOG for postoperative NSCLC studies [100]. This study first prepared endoplasmic reticulum membrane (EM)-modified liposomes (MSLs) encapsulating siSTAT3 via the film dispersion method. The MSLs were then co-incubated with oxidized SOG and lidocaine hydrochloride (LID) to prepare the localized hydrogel MSL@LID@SOG. Experimental validation demonstrated the hydrogel's capacity for controlled LID release, facilitating both postoperative pain alleviation and NK activation. A clinical in situ NSCLC mouse model (LLC-Luc) was established in C57BL/6 mice, with intrapleural administration. Ultrasound monitoring revealed superior MPE volume reduction in the MSL@LID@SOG group compared to controls. Cytological analysis of MPE specimens showed diminished tumor cell counts alongside reduced M2-like macrophage infiltration, a cell population associated with tumor cell survival. These findings collectively indicate the hydrogel's capacity to suppress local tumor recurrence, mitigate MPE accumulation, provide analgesic effects, and enhance patient quality of life, thereby establishing a clinically translatable drug delivery platform for MPE therapeutics.
Pleurodesis constitutes a therapeutic intervention for MPE where chemical agents are applied to the pleura to create an adhesion between the parietal and visceral pleurae, sealing the pleural cavity to inhibit subsequent fluid reaccumulation [101,102]. Fibrin adhesion and fibrosis formation appear to be essential processes for the permanent adhesion between the visceral and parietal pleurae. Most sclerosing agents induce non-specific organizing fibrinous pleuritis, ultimately resulting in fibrotic pleural changes [103]. A meta-analysis of database literature indicates that talc and talc slurry are effective methods for achieving pleurodesis, with lower failure rates compared to many other commonly used interventions [104]. Nevertheless, clinical outcomes following talc administration exhibit considerable variability, with treatment failures not infrequently encountered—a phenomenon potentially attributable to suboptimal distribution of talc particles within the pleural cavity. Biomaterials have demonstrated promising potential in addressing the poor dispersibility of talc powder during pleurodesis procedures. This review systematically investigates functional modifications of talc through biomaterial-based approaches, aiming to provide novel therapeutic directions for MPE (Fig. 7).
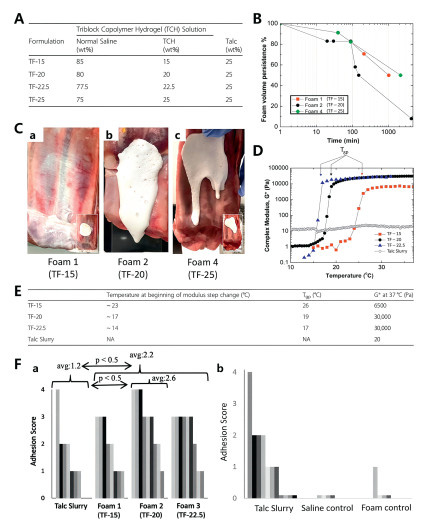
Baxter et al. developed a liquid foam formulation comprising 25% by weight talc combined with poly(ethylene oxide)-poly(propylene oxide)-poly(ethylene oxide) tri-block copolymer (TCH) at various concentrations [105]. When cold foam is delivered into the pleural cavity and comes into contact with the warm tissue, the liquid phase undergoes gelation, adhering to the tissue and extending the contact time between talc powder and the tissue. This enhances the formation of adhesions in the pleural space. The study utilized small amplitude oscillatory shear rheology (SAOS) to assess three formulations, talc powder, saline, and reverse triblock copolymer hydrogels (TF-15, TF-20, and TF-22.5), from the perspectives of rheology, foam stability, in vitro coating, and bioadhesive characteristics. Therapeutic effectiveness was subsequently validated through a New Zealand rabbit pleural fixation model. The sol-gel transition temperature (Tgp) was determined rheologically as 17 ℃ for TF-22.5, 26 ℃ for TF-15, and 19 ℃ for TF-20. Maintenance of foam temperature below Tgp during delivery is critical to prevent catheter occlusion, given that the liquid-state formulation must traverse delivery apparatus prior to encountering the 37 ℃ pleural environment. The temporal progression to Tgp following intrapleural administration governs coverage characteristics (Fig. 8): TF-15 had a prolonged gelation time, with strong fluidity prior to gelation, leading to uneven foam coverage. TF-22.5 exhibited the fastest gelation but failed to fully cover the tissue. TF-20 demonstrated an effective balance, forming a coating and gel within the pleural cavity, leading to more uniform talc deposition and increased pleural adhesion. TF-20 shows promising clinical potential, with minimal side effects observed, in addition to a transient decrease in heart rate in the rabbits. On the basis of these findings, subsequent investigations employing larger animal models with pleural physiology more closely approximating human anatomy should be expected to become the focus of future clinical research.
Beck et al. formulated a reverse thermosensitive hydrogel delivery system by constructing a tri-block copolymer with alternating polyethylene oxide and polypropylene oxide segments [106]. The hydrogel was air-agitated and cooled to produce a foam consisting of a thin liquid, which transitions rapidly into a viscous gel at physiological temperatures. An immunocompetent MPE model was established using C57BL/6 mice, with subjects allocated to four experimental groups: talc slurry (TS, 2 mg/g), talc foam (TF, 2 mg/g), saline control, and foam-only control (F). Post-interventional CT imaging at 10 days revealed the TF group demonstrated a statistically significant 30%‒40% reduction in right lung volume loss compared to controls (P < 0.05), whilst the TS group showed no statistically significant disparity (P > 0.05). The TF group significantly prevented the loss of air volume while the TS group did not exhibit such an effect. This discrepancy could be explained by the pleural cavity's increased foam dispersion. Foam-mediated delivery mechanisms facilitate superior spatial distribution of sclerosing agents and therapeutic compounds. This experimental framework establishes a methodological foundation for translational clinical investigations into talc foam applications and provides novel investigative pathways for MPE studies.
Building upon previous research, Lima et al. explored the therapeutic effects of non-hydrogel talc foams alongside physicochemical determinants affecting talc pleurodesis outcomes [107]. Three novel non-hydrogel foam formulations, ST-45, P-80, and L-23, were formulated and benchmarked against poloxamer 407 hydrogel foam (P-407) and traditional talc slurry. Experimental data indicated L-23 and P-80 achieved 76% and 40% greater mean adhesion scores respectively than talc slurry, whereas P-407 and ST-45 demonstrated doubled adhesion capacity relative to the slurry control. The P-80 formulation proved suboptimal, exhibiting premature foam collapse prior to intrapleural administration in New Zealand rabbits, resulting in substantial volume reduction and no therapeutic superiority over standard slurry. Conversely, P-407, ST-45, and L-23 displayed marked efficacy improvements compared to slurry controls. Foam stability was quantified through temporal volume retention analysis, with structural degradation as critical destabilization factor. According to the study, P-80 quickly deflated in less than 10 min, but L-23 and ST-45 retained their initial volume even after 16 h. Intrapleural administration of P-407 hydrogel foam revealed delayed gelation permitting unrestricted diffusion and collapse. Owing to limited heat transfer, P-407 gelled only in thin layers, and the foam continued to diffuse within the body. This retarded stabilization mechanism accounts for P-407′s superior efficacy compared to ST-45 and L-23. These results indicate that the contact time between foam and tissue is a crucial factor influencing the effectiveness of pleurodesis, which is related to the viscosity of non-hydrogel foams and the gelation process of hydrogel foams. This investigation clarifies the formulation-performance correlation in foam-mediated pleurodesis, establishing a conceptual framework for developing therapeutic foam delivery systems.
Liposomes serve as effective drug delivery and targeting vehicles, offering advantages including enhanced drug stability, reduced toxicity, and improved dosage administration [108,109]. To explore their therapeutic potential for MPE, Marazioti et al. used in vivo imaging systems to monitor liposomes which were labeled with fluorescence dye 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide (DiR), monitoring the retention of different types of liposomes within the pleural cavity [110]. Using the thin-film hydration method, the researchers prepared both multilamellar vesicles (MLVs) and small unilamellar vesicles (SUVs). The results showed that PEG-coated SUVs exhibited superior retention properties, leading to their selection for MPE murine models. Comparative analysis revealed significantly enhanced retention at the injection site for both SUVs and PEG-coated SUVs compared to MLVs and unmodified SUVs, attributable to particle size and surface coating effects on pleural cavity interactions. The improved structural integrity conferred by hydrophilic polymer coatings, particularly PEG, accounts for these enhanced retention characteristics [111]. Investigations comparing MPE-afflicted and healthy murine models showed no significant intergroup differences in pleural retention profiles, indicating that effusion presence does not substantially influence DiR-labelled liposome behavior. Tumor-associated DiR signals in MPE mice demonstrated comparable intensity to hepatic signals, suggesting significant tumor-targeting potential following intrapleural administration. The demonstrated prolonged retention capacity of intrapleurally administered liposomal formulations presents promising therapeutic prospects for MPE management.
Curcumin, a polyphenolic compound extracted from turmeric, exhibits pharmacological properties including anti-inflammatory, antioxidant, and antitumor activity [112]. It modulates key mediators like VEGF-A, IL-6 and TNF-α, which are implicated in MPE fluid accumulation [113]. Therefore, curcumin is considered to have potential therapeutic benefits for the treatment of MPE. To address its inherent limitations in aqueous solubility and stability, researchers have developed a liposomal curcumin formulation validated as safe for intravenous use in Phase Ⅰ trials [114]. Hocking et al. conducted comparative studies in Fischer rats, contrasting intrapleural administration of liposomal curcumin with intravenous delivery [115]. Their findings demonstrated an absence of pleural or pulmonary toxicity, with the intrapleural route permitting higher concentrations without hematological alterations, thereby confirming its viability for localized treatment. On this basis, Hocking et al. established a Phase Ⅰ clinical trial protocol to validate the safety and feasibility of liposomal curcumin for the treatment of MPE via pleural administration [116]. The study employed a 3 + 3 dose-escalation design to determine predefined dose levels, with dose cohorts of 100, 200, and 300 mg/m2. Participants underwent tunneled indwelling pleural catheter (TIPC) placement seven days prior to inpatient drug administration, with toxicity assessments and serious adverse event (SAE) monitoring determining dose escalation eligibility. Longitudinal data collection occurred at weeks 0, 4, 8, 12, 20, and 24. While comprehensive clinical investigations into liposomal curcumin's efficacy against MPE remain lacking, necessitating further preclinical and clinical evaluation, these foundational studies propose novel therapeutic avenues worthy of exploration.
Biomaterial-based DDSs are capable of achieving precise and efficient drug delivery, playing a critical role in treatments such as chemotherapy, immunotherapy, combined chemotherapy‒immunotherapy, combined gene‒immunotherapy, and pleurodesis. These systems can control MPE volume, reduce toxic side effects, and enhance antitumor efficacy, demonstrating significant potential in the treatment of MPE. Antiangiogenic drugs are important therapeutic agents for MPE, but there is no precedent for biomaterial-modified antiangiogenic drugs or associated combined therapy in the treatment of MPE so far. The development of effective DDSs for the antiangiogenic therapy of MPE may play an important role in preclinical drug research. In addition, most of the biomaterial-based DDSs for the therapy of MPE are under development. Although certain biomaterial-based DDSs, such as DOX liposomes (TLC-D99), vincristine sulfate liposome injection (VSLI), and albumin-bound paclitaxel NPs, have achieved clinical approval, the majority of DDSs still face significant challenges in translational progression from fundamental research to clinical application due to unresolved issues in biocompatibility, therapeutic performance, and predictive preclinical validation [117]. Therefore, future investigations should prioritize safety-centric approaches by implementing safe-by-design principles, utilizing advanced strategies such as predictive toxicology, high-throughput screening, and multi-omics methodologies to systematically evaluate the biocompatibility, toxicokinetics, and biodistribution of diverse biomaterials. Concurrently, structural optimization of DDSs must be implemented through the development of novel ligands capable of recognizing disease-specific biomarkers, thereby enhancing drug delivery efficiency and enabling stimuli-responsive precision drug release. Conventional animal models are limited by interspecies differences that compromise the accuracy of physiological and pathological response predictions. These limitations can be addressed by developing bioengineered human disease models, such as patient-derived organoids, functionalized tissue constructs, and organ-on-a-chip systems, which more faithfully replicate human-specific disease mechanisms and improve alignment between preclinical studies and clinical outcomes.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Yiyao Wan: Writing – original draft, Investigation, Conceptualization. Wen Chen: Writing – review & editing, Investigation, Conceptualization. Yan Yu: Investigation. Meng Pan: Investigation. Kun Shi: Writing – review & editing, Supervision, Funding acquisition, Conceptualization. Zhiyong Qian: Writing – review & editing, Supervision, Funding acquisition, Conceptualization.
We gratefully acknowledge financial support from the Noncommunicable Chronic Diseases-National Science and Technology Major Project (Nos. 2024ZD0522800, 2024ZD0522803); the National Natural Science Foundation of China (Nos. U21A20417, 31930067, 31800797); the Natural Science Foundation of Sichuan Province (No. 2024NSFSC0046); the Sichuan Science and Technology Program (No. 2022YFS0333); the 1·3·5 Project for Disciplines of Excellence, West China Hospital, Sichuan University (No. ZYGD24003). The schematic illustrations in the figures were created entirely or partially via BioRender (biorender.com).
K. Skok, G. Hladnik, A. Grm, A. Crnjac, Medicina 55 (2019) 490. doi: 10.3390/medicina55080490
A.G. Villanueva, Management of malignant pleural effusions. In: A. Ernst, F. Herth (eds), Principles and Practice of Interventional Pulmonology. Springer, New York, 2013, pp. 665–674.
E. Penz, K.N. Watt, C.A. Hergott, et al., Cancer Manag. Res. 9 (2017) 229–241. doi: 10.2147/CMAR.S95663
Z. Chen, C.M. Fillmore, P.S. Hammerman, et al., Nat. Rev. Cancer 14 (2014) 535–546. doi: 10.1038/nrc3775
J.M. Porcel, A. Gasol, S. Bielsa, et al., Respirology 20 (2015) 654–659. doi: 10.1111/resp.12496
M.E. Roberts, E. Neville, R.G. Berrisford, et al., Thorax 65 (2010) ii32-ii40.
A.C. Bibby, P. Dorn, I. Psallidas, et al., Eur. Respir. J. 52 (2018) 1800349. doi: 10.1183/13993003.00349-2018
Y. Chen, N.W. Mathy, H. Lu, Mol. Med. Rep. 17 (2018) 8019–8030.
M.M. Zamboni, C.T. da Silva, Jr., R. Baretta, et al., BMC Pulm. Med. 15 (2015) 29. doi: 10.1186/s12890-015-0025-z
R. Asciak, N.M. Rahman, Clin. Chest Med. 39 (2018) 181–193. doi: 10.1016/j.ccm.2017.11.004
Chinese Thoracic Society, Chinese Medical Association, Chin. J. Tuberculosis Respirat. Dis. 46 (2023) 1189–1203.
F. Gonnelli, W. Hassan, M. Bonifazi, et al., Respir. Res. 25 (2024) 47. doi: 10.1186/s12931-024-02684-7
G.T. Stathopoulos, I. Kalomenidis, Curr. Opin. Pulm. Med. 15 (2009) 343–352. doi: 10.1097/MCP.0b013e32832af07c
F. Economidou, G. Margaritopoulos, K.M. Antoniou, N.M. Siafakas, Exp. Ther. Med. 1 (2010) 3–7. doi: 10.3892/etm_00000001
S. Wen, K. Zhang, Y. Li, et al., Chin. Chem. Lett. 31 (2020) 3153–3157. doi: 10.1016/j.cclet.2020.03.077
D.O. Bates, Cardiovasc. Res. 87 (2010) 262–271. doi: 10.1093/cvr/cvq105
D. Marquez-Medina, S. Popat, Clin. Transl. Oncol. 18 (2016) 760–768. doi: 10.1007/s12094-015-1464-y
Y. Sun, Y. Hu, C. Wan, et al., Biomater. Sci. 9 (2021) 6381–6390. doi: 10.1039/d1bm00971k
Z. Xiang, X. Deng, W. He, et al., Ann. Med. 54 (2022) 1357–1371. doi: 10.1080/07853890.2022.2071977
C.G. Willett, Y. Boucher, E. di Tomaso, et al., Nat. Med. 10 (2004) 145–147. doi: 10.1038/nm988
D. Zhao, H. Hou, X. Zhang, OncoTargets Ther. 11 (2018) 4137–4147. doi: 10.2147/ott.s172305
Q. Du, J. Zou, Z. Huang, et al., Chin. Chem. Lett. 34 (2023) 107763. doi: 10.1016/j.cclet.2022.107763
G.T. Stathopoulos, I. Kalomenidis, Am. J. Respir. Crit. Care Med. 186 (2012) 487–492. doi: 10.1164/rccm.201203-0465PP
H.H. Yeh, W.W. Lai, H.H. Chen, et al., Oncogene 25 (2006) 4300–4309. doi: 10.1038/sj.onc.1209464
A. Marazioti, C.A. Kairi, M. Spella, et al., PLoS One 8 (2013) e71207. doi: 10.1371/journal.pone.0071207
G.T. Stathopoulos, T.P. Sherrill, S.P. Karabela, et al., Am. J. Respir. Crit. Care Med. 182 (2010) 1273–1281. doi: 10.1164/rccm.201001-0001OC
Y.C. Gary Lee, D. Melkerneker, P.J. Thompson, et al., Am. J. Respir. Crit. Care Med. 165 (2002) 88–94. doi: 10.1164/ajrccm.165.1.2104006
E. Fernandes, J.A. Ferreira, P. Andreia, et al., J. Control. Release 209 (2015) 288–307. doi: 10.1016/j.jconrel.2015.05.003
Y. Fan, A. Chen, J. Zhu, et al., Cancer Lett. 588 (2024) 216777. doi: 10.1016/j.canlet.2024.216777
S. Taefehshokr, A. Parhizkar, S. Hayati, et al., Pathol. Res. Pract. 229 (2022) 153723. doi: 10.1016/j.prp.2021.153723
B. Shen, M. Tan, Z. Wang, et al., J. Oncol. 2022 (2022) 1476038.
L. Gandhi, D. Rodríguez-Abreu, S. Gadgeel, et al., N. Engl. J. Med. 378 (2018) 2078–2092. doi: 10.1056/nejmoa1801005
R. Joshi, A. Gupta, C.D. Kaur, Application of biomaterials in cancer research. In: Malviya, R., Sundram, S. (eds), Targeted Cancer Therapy in Biomedical Engineering, Springer Nature Singapore, Singapore, 2023, pp. 245–289.
K. Krukiewicz, J.K. Zak, Mater. Sci. Eng. C Mater. Biol. Appl. 62 (2016) 927–942. doi: 10.1016/j.msec.2016.01.063
W. Chen, K. Shi, Y. Yu, et al., Chin. Chem. Lett. 35 (2024) 109159. doi: 10.1016/j.cclet.2023.109159
Y. Li, J. Cui, C. Li, et al., Chin. Chem. Lett. 34 (2023) 108180. doi: 10.1016/j.cclet.2023.108180
D. Katiyar, Mater. Today Proc (2023), doi: 10.1016/j.matpr.2023.05.460.
Y. Chao, L. Xu, C. Liang, et al., Nat. Biomed. Eng. 2 (2018) 611–621. doi: 10.1038/s41551-018-0262-6
.P. Torchilin, Eur. J. Pharm. Sci. 11 (2000) S81–S91. doi: 10.1016/S0928-0987(00)00166-4
Y.B. Meng, J. Wu, Chin. J. Polym. Sci. 40 (2022) 1016–1027. doi: 10.1007/s10118-022-2701-9
J. Xie, Y. Lu, B. Yu, et al., Chin. Chem. Lett. 31 (2020) 1173–1177. doi: 10.1016/j.cclet.2019.10.030
J. Huang, X. You, P. Xin, et al., Chin. Chem. Lett. 32 (2021) 1737–1742. doi: 10.1016/j.cclet.2020.12.006
X. You, L. Wang, J. Zhang, et al., Chin. Chem. Lett. 34 (2023) 107720. doi: 10.1016/j.cclet.2022.07.063
X. Wu, X. Chen, X. Wang, et al., Chin. Chem. Lett. 35 (2024) 108756. doi: 10.1016/j.cclet.2023.108756
Y. Wang, C. Zhang, S. Han, et al., Chin. Chem. Lett. 35 (2024) 109578. doi: 10.1016/j.cclet.2024.109578
J. Herrstedt, P. Clementsen, O.P. Hansen, Eur. J. Cancer 28a (1992) 1070–1073.
H. Zhou, W. Wu, X. Tang, et al., Medicine 96 (2017) e5532. doi: 10.1097/MD.0000000000005532
Y. Zhang, T. Li, Y. Hu, et al., Chin. Chem. Lett. 33 (2022) 2507–2511. doi: 10.1016/j.cclet.2021.11.076
S. Rivankar, J. Cancer Res. Ther. 10 (2014) 853–858. doi: 10.4103/0973-1482.139267
O. Ike, Y. Shimizu, S. Hitomi, et al., Chest 99 (1991) 911–915. doi: 10.1378/chest.99.4.911
O. Ike, Y. Shimizu, Y. Ikada, et al., Biomaterials 12 (1991) 757–762. doi: 10.1016/0142-9612(91)90026-7
Y. Tan, A. Tajik, J. Chen, et al., Nat. Commun. 5 (2014) 4619. doi: 10.1038/ncomms5619
Z.H. Wu, C.L. Ji, H. Li, et al., Eur. Rev. Med. Pharmacol. Sci. 17 (2013) 2420–2427.
H. Zhang, K. Tang, Y. Zhang, et al., Cancer Immunol. Res. 3 (2015) 196–205. doi: 10.1158/2326-6066.CIR-14-0177
K. Tang, Y. Zhang, H. Zhang, et al., Nat. Commun. 3 (2012) 1282. doi: 10.1038/ncomms2282
J. Ma, Y. Zhang, K. Tang, et al., Cell Res. 26 (2016) 713–727. doi: 10.1038/cr.2016.53
M. Pourmadadi, A. Ghaemi, M. Shaghaghi, et al., J. Drug Deliv. Sci. Technol. 82 (2023) 104338. doi: 10.1016/j.jddst.2023.104338
T. Liu, H. Zou, J. Mu, et al., Chin. Chem. Lett. 32 (2021) 1751–1754. doi: 10.1016/j.cclet.2020.12.008
B. Sun, H. Jing, M.T. Mabrouk, et al., Pharm. Dev. Technol. 25 (2020) 1281–1288. doi: 10.1080/10837450.2020.1818780
Z.H. Siddik, Oncogene 22 (2003) 7265–7279. doi: 10.1038/sj.onc.1206933
K. Hotta, K. Matsuo, H. Ueoka, et al., J. Clin. Oncol. 22 (2004) 3852–3859. doi: 10.1200/JCO.2004.02.109
Y.K. Chao, Y.W. Wen, K.S. Liu, et al., Int. J. Pharm. 484 (2015) 38–43. doi: 10.1016/j.ijpharm.2015.02.048
X. Bai, F. Cerimele, M. Ushio-Fukai, et al., J. Biol. Chem. 278 (2003) 35501–35507. doi: 10.1074/jbc.M302967200
F. Fang, C. Gong, Z. Qian, et al., ACS Nano 3 (2009) 4080–4088. doi: 10.1021/nn900785b
P. Murthy, C.N. Ekeke, K.L. Russell, et al., Oncoimmunology 8 (2019) e1554969. doi: 10.1080/2162402x.2018.1554969
T.L. Whiteside, S. Demaria, M.E. Rodriguez-Ruiz, et al., Clin. Cancer Res. 22 (2016) 1845–1855. doi: 10.1158/1078-0432.CCR-16-0049
T.F. Gajewski, L. Corrales, J. Williams, et al., Adv. Exp. Med. Biol. 1036 (2017) 19–31. doi: 10.1007/978-3-319-67577-0_2
X. Li, J. He, W. He, Acta Pharm. Sin. B 14 (2024) 5515–5517. doi: 10.1016/j.apsb.2024.08.011
Y. Xie, X. Li, J. Wu, et al., Chin. Chem. Lett. 34 (2023) 108202. doi: 10.1016/j.cclet.2023.108202
A. Khurana, S. Tekula, M.A. Saifi, et al., Biomed. Pharmacother. 111 (2019) 802–812. doi: 10.1016/j.biopha.2018.12.146
Y. Huang, Y. Fu, M. Li, et al., Angew. Chem. Int. Ed. 59 (2020) 4406–4414. doi: 10.1002/anie.201910615
Y. Zhang, M. Zhang, Y. Jiang, et al., J. Cancer Res. Clin. Oncol. 144 (2018) 2177–2186. doi: 10.1007/s00432-018-2718-1
Z. Song, W. Luo, H. Zheng, et al., Adv. Healthc. Mater. 10 (2021) e2100149. doi: 10.1002/adhm.202100149
L. Chiossone, P.Y. Dumas, M. Vienne, E. Vivier, Nat. Rev. Immunol. 18 (2018) 671–688. doi: 10.1038/s41577-018-0061-z
H. Zuo, P.M. Ueland, Ø. Midttun, et al., Ann. Oncol. 30 (2019) 478–485. doi: 10.1093/annonc/mdz002
S. Liu, N. Li, H. Lai, et al., Adv. Funct. Mater. 34 (2024) 2401264. doi: 10.1002/adfm.202401264
D. Meng, H. Pan, W. He, et al., Adv. Funct. Mater. 32 (2022) 2202603. doi: 10.1002/adfm.202202603
C. Li, Y. Zhang, Y. Wan, et al., Chin. Chem. Lett. 32 (2021) 1615–1625. doi: 10.1016/j.cclet.2021.01.001
L. Sun, J. Wu, F. Du, et al., Science 339 (2013) 786–791. doi: 10.1126/science.1232458
Y. Liu, L. Wang, Q. Song, et al., Nat. Nanotechnol. 17 (2022) 206–216. doi: 10.1038/s41565-021-01032-w
F.G. Herrera, J. Bourhis, G. Coukos, CA Cancer J. Clin. 67 (2017) 65–85.
C. Mothersill, C.B. Seymour, Nat. Rev. Cancer 4 (2004) 158–164. doi: 10.1038/nrc1277
C. Wan, Y. Sun, Y. Tian, et al., Sci. Adv. 6 (2020) eaay9789. doi: 10.1126/sciadv.aay9789
J.L. Liang, G.F. Luo, W.H. Chen, X.Z. Zhang, Adv. Mater. 33 (2021) e2007630. doi: 10.1002/adma.202007630
W. Mu, Q. Chu, Y. Liu, N. Zhang, Nanomicro Lett. 12 (2020) 142.
M.S. Feng, P. Guo, L.X. Jiang, et al., Chin. Chem. Lett. 20 (2009) 178–180. doi: 10.1016/j.cclet.2008.10.027
C.S. Wu, C.H. Wang, J.L. Zhang, et al., Chin. Chem. Lett. 25 (2014) 447–450. doi: 10.1186/1556-276X-9-447
M. Guo, F. Wu, G. Hu, et al., Sci. Transl. Med. 11 (2019) eaat5690. doi: 10.1126/scitranslmed.aat5690
A.D. Gregory, A.M. Houghton, Cancer Res. 71 (2011) 2411–2416.
P. Xu, K. Tang, J. Ma, et al., Cancer Immunol. Res. 8 (2020) 1193–1205. doi: 10.1158/2326-6066.cir-19-0789
X. Dong, Y. Huang, T. Yi, et al., Front. Immunol. 13 (2022) 1002938. doi: 10.3389/fimmu.2022.1002938
J. Sunshine, J.M. Taube, Curr. Opin. Pharmacol. 23 (2015) 32–38. doi: 10.1016/j.coph.2015.05.011
L.H. Yan, X.L. Liu, S.S. Mo, et al., Am. J. Transl. Res. 13 (2021) 923–934.
W. Chen, W. Zeng, J. Sun, et al., ACS Nano 9 (2015) 6069–6076. doi: 10.1021/acsnano.5b01203
J.S. He, S.J. Liu, Y.R. Zhang, et al., Front. Pharmacol. 12 (2021) 687399. doi: 10.3389/fphar.2021.687399
W. Chen, F. Guo, Z. Ren, et al., Front. Bioeng. Biotechnol. 10 (2022) 973892. doi: 10.3389/fbioe.2022.973892
Z. Kang, S. Li, Y. Li, et al., Chin. Chem. Lett. 36 (2024) 110447.
S. Parakh, M. Ernst, A.R. Poh, Cancers (Basel) 13 (2021) 6228. doi: 10.3390/cancers13246228
E. Albesiano, M. Davis, A.P. See, et al., Cancer Res. 70 (2010) 6467–6476. doi: 10.1158/0008-5472.CAN-09-4058
X. Fu, Y. Shi, Z. Gu, et al., Asian J. Pharm. Sci. 19 (2024) 100925.
F. Rodriguez-Panadero, A. Montes-Worboys, Respiration 83 (2012) 91–98. doi: 10.1159/000335419
M. Mierzejewski, P. Korczynski, R. Krenke, J.P. Janssen, Respir. Res. 20 (2019) 247. doi: 10.1186/s12931-019-1204-x
V.B. Antony, N. Nasreen, K.A. Mohammed, et al., Chest 126 (2004) 1522–1528. doi: 10.1378/chest.126.5.1522
A. Dipper, H.E. Jones, R. Bhatnagar, et al., Cochrane Database Syst. Rev. 4 (2020) Cd010529.
J. Baxter, T.A. Lima, R. Huneke, et al., J. Cardiothorac. Surg. 15 (2020) 58. doi: 10.1186/s13019-020-01098-y
T.N. Beck, A.Y. Deneka, L. Chai, et al., BMC Cancer 19 (2019) 614. doi: 10.1186/s12885-019-5777-z
T.A. Lima, R.A. Coler, G.W. Laub, et al., Drug Deliv. 28 (2021) 733–740. doi: 10.1080/10717544.2021.1895910
S. Antimisiaris, S. Mourtas, K. Papadia, Int. J. Pharm. 525 (2017) 293–312. doi: 10.1016/j.ijpharm.2017.01.056
K. Thapa Magar, G.F. Boafo, X. Li, et al., Chin. Chem. Lett. 33 (2022) 587–596. doi: 10.1016/j.cclet.2021.08.020
A. Marazioti, K. Papadia, A. Giannou, et al., Int. J. Nanomedicine 14 (2019) 3773–3784. doi: 10.2147/ijn.s202568
A.H. Matloob, S. Mourtas, P. Klepetsanis, S.G. Antimisiaris, Int. J. Pharm. 476 (2014) 108–115. doi: 10.1016/j.ijpharm.2014.09.041
J. Lal, S.K. Gupta, D. Thavaselvam, D.D. Agarwal, Chin. Chem. Lett. 27 (2016) 1067–1072. doi: 10.1016/j.cclet.2016.03.032
M.K. Shanmugam, G. Rane, M.M. Kanchi, et al., Molecules 20 (2015) 2728–2769. doi: 10.3390/molecules20022728
R. Greil, S. Greil-Ressler, L. Weiss, et al., Cancer Chemother. Pharmacol. 82 (2018) 695–706. doi: 10.1007/s00280-018-3654-0
A. Hocking, S. Tommasi, P. Sordillo, S. Klebe, Int. J. Nanomedicine 15 (2020) 943–952. doi: 10.2147/ijn.s237536
A.J. Hocking, A.L. Farrall, S. Newhouse, et al., BMJ Open 11 (2021) e047075. doi: 10.1136/bmjopen-2020-047075
X. Li, Y. Lai, G. Wan, et al., Chin. J. Nat. Med. 22 (2024) 1100–1116.

Figure 1 Schematic diagram of biomaterials for the therapy of MPE, including those used in chemotherapy, immunotherapy, combination therapy, and pleurodesis.

Figure 2 (A) Comparison of MPE fluids after one-week treatment with CisMPs (magnification: 200×). (B) Cytologic analysis of pleural effusion in the CisMPs group (magnification: 200×). (C) Observation of MPE in the cisplatin group (magnification: 200×). (D) Formation of spheroids from primary tumor cells. Scale bar: 50 µm. (E) Differences between drug-MPs and free drugs. Copied with permission [56]. Copyright 2016, Springer Nature.

Figure 3 (A) Images of HK hydrogels at various temperatures. (B) Results of the in vitro release experiment. (C) Malignant pleural neoplasms in mice following intrapleural injection. (a) NS group; (b) hydrogel group; (c) HK group; (d) HK−hydrogel group. (D) Computed tomography (CT) imaging of MPE in mice after intrapleural administration. (a) NS group; (b) hydrogel group; (c) HK group; (d) HK−hydrogel group. Copied with permission [64]. Copyright 2009, American Chemical Society.

Figure 4 (A) Strategic design of nanotherapeutics. (B) Morphological and structural changes in SeNPs@LNT in MPE. (C) SeNPs@LNT uptake by macrophages. (a) TEM examination for 4 h. (a') Uptake and accumulation of SeNPs@LNT. (b) TEM examination for 24 h. (b') Uptake and accumulation of SeNPs@LNT. (D) The levels of IFN-γ, IL-6 and IL-10 in MPE. Copied with permission [73]. Copyright 2021, John Wiley and Sons.

Figure 5 (A) ATMP-MTX treatment and biomarker assessment schedule. (B) Comparison of pre- and post-treatment chest CT images in a patient. (C) Kaplan-Meier curve of pleurodesis success in all 11 patients. (D) Comparison of pre- and post-treatment light microscope examination of MPE cells from a patient. Scale bar: 100 µm. (E) Comparison of proportions of CD45− cells in MPE samples before and after ATMP-MTX treatment. Copied with permission [88]. Copyright 2019, The American Association for the Advancement of Science.

Figure 6 (A) Construction of aptamer-siRNA chimeras/PEI/PEG/AuNP/collagen membrane. (a) Schematic diagram. (b) Activation of T cells. (B) General picture of chimera/AuNP membrane. (C) PD-1 inhibitor group and the chimeras/AuNP group activate T cells. (D) Lymphocytes were co-cultured with lung adenocarcinoma cells after treatment for 96 h. (a) Proliferation rate. (b) Apoptosis rate. (E) Flow chart of animal experiments. (F) Expression of Ki-67 and CD44 in immunohistochemistry. Reproduced with permission [96]. Copyright 2022, Frontiers.

Figure 7 Schematic illustration of biomaterial modified talc for pleurodesis for the therapy of MPE. Mesothelial cells exposed to biomaterial-modified talc initiate an inflammatory cascade by secreting mediators, recruiting and proliferating fibroblasts, inducing nonspecific organizing fibrinous pleuritis, ultimately leading to pleural fibrosis.

Figure 8 (A) Composition of the formulations TF-15, TF-20, TF-22.5, and TF-25. (B) In vitro experiment of foam persistence. (C) Ex vivo vertical flow test on porcine chest walls. (a–c) Test results of TF-15, TF-20, and TF-22.5. (D) Temperature-dependent material properties of the formulations. (E) Summary of the important parameters. (F) The degree of adhesions formed after 28 days in a rabbit model. (a, b) Analysis of adhesion scores. Copied with permission [105]. Copyright 2020, Springer Nature.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
 下载:
下载: