Structure-activity relationship studies of 3,4-disubstitued indole-2-ketone derivatives identify a potent glutaminyl cyclase inhibitor with antitumor activity against breast cancer
Citation:
Hui Lei, Yingying Jiang, Jiayu Liu, Weifeng Zhang, Fanbo Meng, Jun Mou, Wenyi Liu, Pengcheng Lei, Rui Xiong, Zan Xu, Hang Zhang, Yanjun Wang, Guo-Bo Li, Lingling Yang, Hua-Li Wang. Structure-activity relationship studies of 3,4-disubstitued indole-2-ketone derivatives identify a potent glutaminyl cyclase inhibitor with antitumor activity against breast cancer[J]. Chinese Chemical Letters,
2026, 37(6): 111511.
doi:
10.1016/j.cclet.2025.111511
Structure-activity relationship studies of 3,4-disubstitued indole-2-ketone derivatives identify a potent glutaminyl cyclase inhibitor with antitumor activity against breast cancer
English
Structure-activity relationship studies of 3,4-disubstitued indole-2-ketone derivatives identify a potent glutaminyl cyclase inhibitor with antitumor activity against breast cancer
College of Food and Bioengineering, Xihua University, Chengdu 610039, China
b.
Department of Clinical Oncology, The University of Hong Kong-Shenzhen Hospital, Shenzhen 518053, China
c.
Key Laboratory of Drug Targeting and Drug Delivery System of Ministry of Education, West China School of Pharmacy, Sichuan University, Chengdu 610041, China
Received Date:
15 March 2025 Accepted Date:
25 June 2025 Revised Date:
24 June 2025 Available Online:
15 June 2026
Abstract:
Glutaminyl cyclase (QC), a key enzyme catalyzing the pyroglutamate modification of bioactive peptides, has emerged as a promising pharmacological target in cancer and other human diseases. Inhibition of QC activity represents a potential therapeutic strategy for diseases associated with QC dysregulation, suggesting that QC inhibitors might be as promising therapeutic approach for these diseases. Building upon our previous lead compound 1 (half maximal inhibitory concentration (IC50): Golgi resident QC (gQC), 3.22 µmol/L; secretory QC (sQC), 4.04 µmol/L), we performed systematic structural optimization to develop novel 3,4-disubstituted indole-2-ketone derivatives. Structure-activity relationship (SAR) studies identified highly active gQC and sQC inhibitors (compounds 24, 28, and 29) with significantly enhanced QC inhibitory activity. Among them, 28 exhibited superior inhibitory activity against gQC (IC50 = 0.095 µmol/L) compared to sQC (IC50 = 1.33 µmol/L), and could most significantly improve the thermal stability of gQC protein. Using compound 17 with similar structure but 130 times lower gQC inhibitory activity than 28 as a negative control, the in vitro anti-tumor activity study found that 28 can bind to gQC in MDA-MB-231 breast cancer cells and significantly inhibit the growth and migration, by affecting cell proliferation and inducing G0/G1 phase cell cycle arrest. In nude mice bearing MDA-MB-231 cell xenograft, intratumor injection of 28 (25 mg/kg) produced 40.6% inhibition rates. This work not only provides structural basis and new leads for drug discovery targeting QC, but also presents compelling evidence supporting QC, especially gQC as a potential target for breast cancer therapy.
Glutaminyl cyclase (QC), alternatively termed glutaminyl-peptide cyclotransferases (QPCT), consists of two isoenzymes, secretory QC (sQC/QPCT) and Golgi resident QC (gQC/QPCTL) [1–3]. Both types of QC catalyze irreversible post-translational chemical reactions in proteins or peptides by releasing ammonia or a water molecule to convert N-terminal glutamine or glutamic acid residues into N-terminal pyroglutamate (pE) [4–6]. This pE modification confers enhanced proteolytic resistance against aminopeptidase degradation while enhancing protein stability, hydrophobicity, and auto-aggregation propensity [7,8]. Notably, the pE moiety may also potentiate ligand-receptor binding interactions [9]. Mounting evidence implicates abnormal overexpression of QC in the pathogenesis of multiple disorders, including cancers [10–14], neurodegenerative diseases [3,15–17], periodontitis and related disorders [18,19] and inflammatory diseases [1,15,20–22].
Of particular oncological relevance, gQC has been shown to be associated with the CD47-signal regulatory protein α (SIRPα) immune checkpoint axis. Its overexpression enables tumor immune evasion by reinforcing "don't eat me" signals to phagocytes [10,23], while inhibiting gQC can enhance the macrophage-mediated phagocytosis of tumor cells [12]. Additionally, a series of studies have demonstrated that gQC plays a significant role in promoting neoplastic proliferation and migration [24], with genetic silencing attenuating growth in thyroid carcinoma [25] and reducing migratory capacity in lymphoma models [26]. Elevated gQC expression correlates with diminished survival across multiple cancer types [15]. In summary, these studies collectively indicate that gQC is an attractive target, and inhibiting QC activity presents a potential therapeutic strategy for treating these pE-related diseases, including cancers.
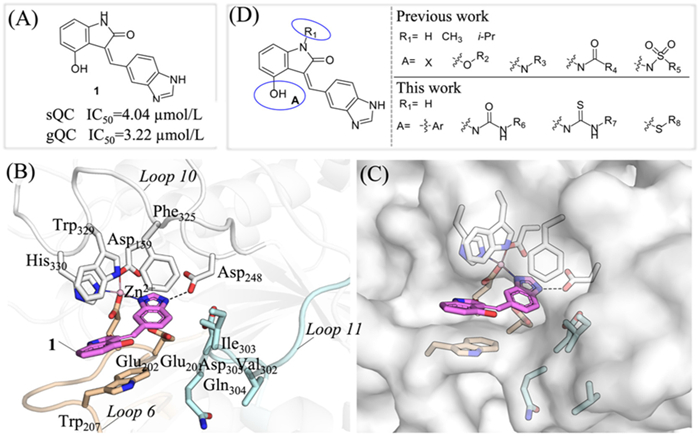
Although QC has been proven to be an effective target for the treatment of various diseases, and a number of inhibitors targeting gQC and sQC have been reported [27–33]. However, so far, only the benzimidazole inhibitor PQ912 is currently undergoing clinical trials for the treatment of Alzheimer's disease (AD) [33–36], and inhibitors application in cancer therapy has not been thoroughly explored. Thus, there is still a need to develop new gQC and sQC inhibitors to provide more candidate drugs for innovative drug development targeting gQC and sQC. In our previous study, guided by the X-ray crystal structure of sQC in complex with PQ912, a series of new benzimidazole compounds were designed and synthesized using an in-house Frag-SD tool, ultimately leading to the discovery of a novel gQC and sQC inhibitor, (Z)-3-((1H-benzo[d]imidazol-5-yl)methylene)-4-hydroxyindolin-2-one (1, Fig. 1A) [34]. And the X-ray crystal structure of sQC in complex with 1 (PDB: pdb:8XGT) confirms that the benzimidazole of 1 form a coordination bond with the active site zinc ion and exhibits hydrogen-bonding interactions with the carboxyl group of Asp248 (Figs. 1B and C). This structure also reveals the importance of different substituents at the 4-position of indolone for activity (Fig. 1D). Based on the structure-activity relationship (SAR) of 4-halogenation, 4-ether, 4-secondamine, 4-amide, and 4-sulfonamide substitution in the early stage, hereon we will continue to explore the influence of 4-aryl, 4-urea, 4-thiourea, and 4-thioether substitution compounds on the activity of gQC and sQC, in order to discover highly active and specific gQC and sQC or selective gQC inhibitors for further anti-tumor activity research (Fig. 1D) [37].
Figure 1
Figure 1.
Design Strategies for novel QC inhibitors. (A) Structure and inhibitory activity of lead compound 1 against gQC and sQC. (B, C) View of a crystal structure of sQC: 1 complex (PDB: 8XGT). (D) Research idea for structural optimization.
To ultimately discover the ideal gQC and sQC inhibitors against cancer, a series of 3,4-disubstituted indole-2-ketone derivatives were synthesized via the synthetic routes outlined in Schemes S1–S3 (Supporting information) [34,38–41]. In our previous study, we found that the substitutions at the 4-position of indole-2-ketone have significant impact on the inhibitory activity of compounds against gQC and sQC. In this investigation, we retained 1H-benzo[d]imidazol as the key pharmacophore group at 2-position and investigated the activity and SAR of 4-aryl-substituted, 4-ureido-substituted and 4-thioether-substituted compounds against gQC and sQC. As we previously reported, the inhibitory activity of compounds against pyroglutamyl aminopeptidase-1 (PGP-1) was used to exclude the false positive results caused by inhibiting PGP-1 [34]. The inhibitory activity of 4-aryl-substituted compounds 2–9 against sQC, gQC and PGP-1 are shown in Table S1 (Supporting information). Compared to lead compound 1, compounds 2 (3-furan) and 3 (benzene ring) exhibited enhanced inhibitory activity against both gQC and sQC, while compound 4 containing 3-pyridine showed no inhibitory activity against gQC either sQC. Given the stronger plasticity of the structure of phenyl, we synthesized the target compounds 5–9 with different substituents at benzene ring. Unfortunately, except for compound 8 which maintained comparable activity to compound 3, the inhibitory activity and ligand efficiency (LE) to gQC and sQC of other compounds were significantly reduced. Based on these results, further modifications of the 4-aryl-substituted compounds were not pursued, and the inhibitory activity and SAR of 4-ureido-substituted and 4-thioether-substituted compounds were explored instead.
The inhibitory activity of 4-ureido-substituted compounds 10–32 against gQC, sQC and PGP-1 are shown in Table S2 (Supporting information). Compared with 1 bearing 4–hydroxy-substituted, compounds 10–14 with 4-alkyl ureido-substituted had improved biochemical properties. Among them, compound 14 bearing 3,3-difluorocyclobutyl showed about 6-fold better activity to gQC and sQC (IC50: 0.58 and 0.77 µmol/L). Compounds obtained by replacing the alkyl group with phenyl (15) or monosubstituted phenyl (16–22) have less than ideal activity to both gQC and sQC, with only compounds 19 and 22 exhibiting nanomolar inhibitory activity against gQC (IC50: 0.44 and 0.30 µmol/L). Based on this result, we speculate that it may be due to the larger binding pocket of gQC compared to sQC, which can better accommodate larger substituents. Therefore, we further designed and synthesized the disubstituted phenyl compounds 23–29 with the aim of enhancing their activity towards gQC. As shown in Table S2, except for compound 29 (IC50: 0.30 µmol/L), the other disubstituted compounds 22–28 still exhibit weaker inhibitory activity against sQC, however multiple compounds displayed good inhibitory activity against gQC, especially compounds 24 and 28 (IC50: 0.082 and 0.095 µmol/L). Additionally, these two compounds have demonstrated moderate ligand efficiency with LE values of 0.32 and 0.29, respectively, and did not exhibit inhibitory activity against PGP-1. Furthermore, by replacing urea moiety with thiourea, three corresponding compounds 30 (vs.15), 31 (vs.18), and 32 (vs.19). Unfortunately, these compounds exhibited unsatisfactory activity against both gQC and sQC.
Finally, we investigated the inhibitory activity and SAR of 4-thioether-substituted derivatives 33–40 against gQC and sQC. As shown in Table S3 (Supporting information), although none of these compounds exhibited inhibitory activity against PGP-1, their inhibitory activity against gQC and sQC are not as strong as compounds 24, 28, or 29. Among of these, compounds 37 (0.97 and 0.97 µmol/L, respectively) and 38 (0.38 and 0.49 µmol/L, respectively) exhibit relatively good dual inhibitory activity against gQC and sQC. In summary, through the study of the 4-aryl-substituted, 4-ureido-substituted and 4-thioether-substituted derivatives, two compounds, 24 and 28 with high inhibitory activity against gQC, as well as one compound 29 with nanomolar inhibitory activity against both gQC and sQC. Given this, further research will be conducted using compounds 24, 28, and 29.
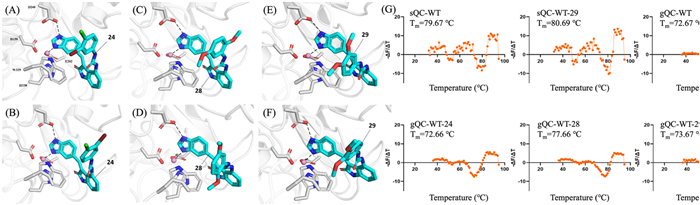
Molecular docking was performed to elucidate the potential binding modes of chosen compounds 24, 28 and 29 with gQC and sQC, respectively. As shown in Figs. 2A–F, all three compounds are positioned to occupy the catalytic active sites of gQC and sQC. The 1H-benzo[d]imidazol of compounds 24, 28 and 29 makes a coordination bond with the active site zinc ion and forms hydrogen-bonding interactions with the carboxyl group of D269 (gQC) or D248 (sQC) of the catalytic triad. Additionally, the indolin-2-one of compounds 24, 28 and 29 engage in π-π stacking interactions with W350 (gQC) or W329 (sQC). The substituent on the benzene ring of the urea functional group may affect the activity of the target compounds against gQC or sQC by regulating this π-π stacking interaction. The thermal shift assay is commonly employed to measure protein thermal stability upon the binding of ligands, which can be used to reflect the binding potencies of ligands. In this study, we tested the thermal stability of the binding of compound 29 with sQC, and compounds 24, 28 and 29 with gQC respectively. As shown in the melting curves and melting temperature (Tm) in Fig. 2G, the complexes of gQC: 29 or sQC: 29 had the melting temperature shifts (ΔTm) of 1.00 and 1.02 ℃ compared with the QC-WT proteins, respectively, indicating that 29 can bind and moderately stabilize gQC and sQC proteins. Furthermore, compound 28 substantially enhancing the thermal stability of gQC, reflected in a ΔTm value of 4.99 ℃ (72.67 vs. 77.66 ℃), yet 24 seems to have no effects on the thermal stability of gQC (72.67 vs. 72.66 ℃). These suggest that compound 28 may have strong binding with gQC to stabilize the gQC protein, by contrast, the binding of 24 to gQC appears to be weaker.
Figure 2
Figure 2.
Compounds 24, 28 and 29 occupy the catalytic active sites of gQC and sQC, and alter their thermal stability. Predicted interaction mode of compound 24 with sQC (A) and gQC (B). Predicted interaction mode of compound 28 with sQC (C) and gQC (D). Predicted interaction mode of compound 29 with sQC (E) and gQC (F). (G) The melting curve (first-derivative of dissociation) of gQC and sQC in the presence or absence of compounds 24, 28 or 29 (100 µmol/L).
Considering that the preferred compound exhibits better inhibitory activity against gQC than sQC (selective index (SI) > 10), we investigated gQC expression across malignancies. Primary analysis of pan-cancer mRNA expression profiles (see Table S4 in Supporting information for cancer type abbreviations) revealed significant gQC upregulation in tumor tissues relative to normal tissues, particularly in esophageal carcinoma (ESCA), liver hepatocellular carcinoma (LIHC), rectum adenocarcinoma (READ), and breast invasive carcinoma (BRCA, Fig. S1A in Supporting information). Integration of TCGA and GTEx datasets further validated elevated gQC transcript levels in BRCA and other tumors (Figs. S1B and S2 in Supporting information). Notably, across all molecular subtypes of BRCA, including luminal, HER2-enriched, and basal-like, gQC mRNA expression was consistently elevated, a finding corroborated by independent database analyses (Figs. S1A–C in Supporting information). Additionally, progressive upregulation of gQC mRNA was observed during sequential stages of lymph node metastasis (Fig. S1D in Supporting information), strongly implicating gQC in BRCA progression and metastatic dissemination.
To evaluate gQC expression at the protein level, we analyzed data from the National Cancer Institute's Clinical Proteomic Tumor Analysis Consortium (CPTAC) and immunohistochemical (IHC) evidence from the Human Protein Atlas (HPA) database. As shown in Fig. S3 (Supporting information), quantitative proteomic analysis of CPTAC samples revealed significantly elevated gQC expression in breast primary tumors, with consistent overexpression patterns observed across major molecular subtypes (Figs. S3A and B). In contrast, no statistically significant difference was detected in sQC protein levels between tumor and normal tissues (Figs. S3C and D), suggesting gQC represents the predominant isoform in breast carcinogenesis. Immunohistochemically stained tissue sample images demonstrated that gQC was nearly undetectable in normal breast tissues but overexpressed in tumor tissues, with a quantity of 75%–25% (Fig. S3E), consistent with the analysis result of the mRNA expression level (Fig. S1). The concordant elevation of both mRNA and protein levels specifically implicates gQC, rather than sQC, as the clinically relevant isoform driving BRCA pathogenesis. These findings position gQC as a potential molecular contributor to BRCA tumorigenesis and progression.
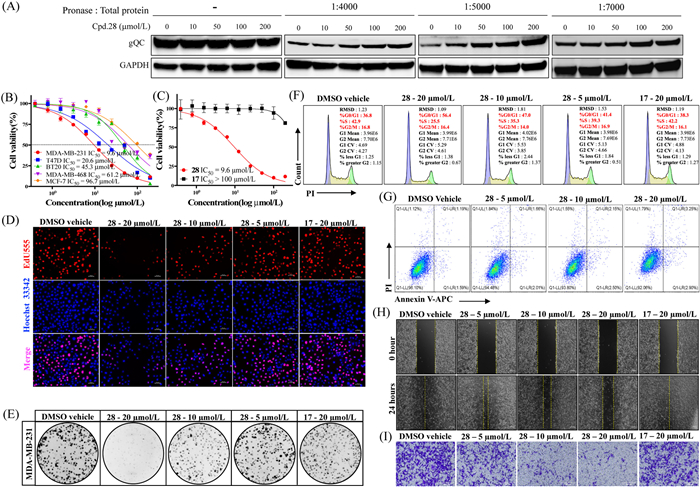
The binding of a drug to its target protein can induce conformational changes which increase proteolytic stability of the target protein by decreasing its protease sensitivity [42]. To evaluate whether compound 28 directly interacts with and stabilizes gQC in tumor cells, we performed a drug affinity responsive target stability (DARTS) assay. As demonstrated in Fig. 3A, preincubation of 28 with MDA-MB-231 cell lysates conferred pronounced protection against gQC proteolysis, even at pronase-to-protein ratios of 1:7000, 1:5000, and 1:4000. Furthermore, 28 stabilized gQC in a dose-dependent manner, evidenced by increased gQC band intensity. Notably, 100 and 200 µmol/L 28 significantly attenuated gQC degradation compared to the vehicle control (Fig. 3A).
Figure 3
Figure 3.
Compound 28 inhibits MDA-MB-231 cell proliferation through gQC-targeted mechanisms. (A) Target engagement of compound 28 with gQC demonstrated by DARTS assay. (B) The anti-viability activities (IC50, µmol/L) of compound 28 against various BRCA cell lines following a 72-h treatment period. (C) The profile of cell ability after 72-h treatment with compound 28 or 17. (D) The fluorescence microscopic appearance of EdU and Hoechst 33342 on MDA-MB-231 cells after treatment with compound 28 or 17. Scale bar: 50 µm. (E) Long-term colony formation assay on MDA-MB-231 cells. (F) Flow cytometry histograms of cell cycle distribution in MDA-MB-231 cells treated with 28 or 17 for 24 h. (G) Apoptosis rate quantified by Annexin V/PI dual staining after 24-h treatment with 28 (representative cytograms shown). (H) Representative images of wound healing assays at 0 and 24-h post-treatment with 28 (scale bar: 200 µm). (I) Effect of 28 on cell invasion tested by transwell assay 28 (scale bar: 200 µm). GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Given that gQC inhibition represents a therapeutic strategy for pE-associated pathologies including cancer [24–26], and considering the robust gQC inhibitory activity, thermal stabilization capacity, and cellular target engagement of compound 28, we investigated its anti-tumor effects in BRCA models. Cell counting kit-8 (CCK-8) viability assays across five BRCA cell lines (MDA-MB-231, MDA-MB-468, MCF-7, BT20 and T47D) revealed subtype-dependent sensitivity, with MDA-MB-231 cells exhibiting maximal responsiveness to 28 (IC50 = 9.6 μmol/L) (Fig. 3B). This differential efficacy aligns with proposed heterogeneity in gQC functional roles across BRCA subtypes. To confirm that the inhibitory effect of 28 on MDA-MB-231 cells is mediated by the inhibition of gQC, we further selected compound 17, which has a similar structure but 130 times lower gQC inhibitory activity to 28 (Table S2), as a negative control for cell activity testing. Consistent with its minimal enzymatic inhibition (Table S2), 17 showed negligible anti-viability effects in MDA-MB-231 cells (IC50 > 100 µmol/L, Fig. 3C). To distinguish target-specific activity from nonspecific cytotoxicity, compound 28 was evaluated in three normal cell lines (MCF-10A, NP460, Het-1A), demonstrating the IC50 values of compound 28 in all tested models were consistently greater than 100 µmol/L (Fig. S4 in Supporting information). Subsequent EdU incorporation assays revealed concentration-dependent suppression of proliferating MDA-MB-231 cells (5–20 µmol/L, 0.5–2 × IC50), with 28 significantly reducing red fluorescent nuclei counts compared to DMSO and compound 17 controls (Fig. 3D and Fig. S5A in Supporting information). Colony formation assays further corroborated these findings, 10 and 20 µmol/L 28 reduced MDA-MB-231 clonogenicity, whereas 17 showed no significant effect (Fig. 3E). Collectively, these data demonstrate that 28 selectively inhibits MDA-MB-231 cell proliferation through gQC-targeted mechanisms. To elucidate the inhibitory mechanism of compound 28, cell cycle progression in MDA-MB-231 cells was analyzed via flow cytometry. Treatment with 5 µmol/L 28 exhibited no significant alteration in cell cycle distribution compared to the DMSO vehicle control. However, dose-dependent G0/G1 phase accumulation was observed at higher concentrations, with 10 and 20 µmol/L 28 increasing the proportion of G0/G1-phase cells by 8.5% (P<0.001) and 17.7% (P<0.0001), respectively (Fig. 3F and Fig. S5B in Supporting information). In contrast, 20 µmol/L compound 17 showed no discernible effect on cell cycle dynamics (Fig. 3F and Fig. S5B), confirming the specificity of 28 in inducing G0/G1 arrest. Flow cytometry analysis using Annexin V/propidium iodide (PI) staining revealed no statistically significant differences in apoptotic rates between 28-treated groups (5–20 µmol/L) and controls (Fig. 3G), suggesting apoptosis-independent growth inhibition. These findings demonstrate that compound 28 selectively impedes MDA-MB-231 cell proliferation through dose-dependent induction of G0/G1 phase cell cycle arrest, without eliciting significant apoptotic effects under the tested conditions. Cell migration and invasion are recognized as a crucial factor in tumor metastasis, and gQC overexpression promotes tumor cell migration [24]. To assess the anti-migratory effects of compound 28, wound healing assays demonstrated dose-dependent inhibition of MDA-MB-231 cell migration following 24-h treatment with 28 (5, 10, and 20 µmol/L; Fig. 3H and Fig. S5C in Supporting information). Parallel transwell invasion assays corroborated these findings, showing significant reduction in invasive cell numbers across tested concentrations. In contrast, the structurally related but inactive analog 17 (20 µmol/L) exhibited negligible effects on migration and invasion (Figs. 3H and I, Figs. S5C and D in Supporting information), suggesting a mechanism involving specific gQC inhibition. These results underscore the therapeutic potential of targeted gQC inhibition in breast cancer metastasis and warrant further investigation into downstream molecular pathways modulated by this inhibitor class.
Considering the favorable antitumor activity of compound 28 observed in vitro, this study established a subcutaneous xenograft model using human breast cancer MDA-MB-231 cells in BALB/c nude mice to evaluate its in vivo antitumor efficacy. All experimental protocols were approved by the Institutional Animal Care and Use Committee of the University of Hong Kong-Shen Zhen hospital (protocol No. TOP-IACUC-2025–0068). When tumor volumes reached approximately 100 mm3, tumor-bearing mice were randomly assigned to three groups (n = 6) and administered the following treatments via intraperitoneal injection, including solvent control group, low-dose treatment group (28, 25 mg/kg) and high-dose treatment group (28, 75 mg/kg). All therapeutic regimens were administered three times weekly for a total duration of 21 days. As shown in Fig. S6 (Supporting information), no statistically significant tumor growth inhibition was observed in the compound 28-treated group compared to the control group (P > 0.05).
To investigate potential underlying reasons for this outcome, we employed SwissADME [43] to predict the physicochemical properties, drug-likeness, and pharmacokinetic characteristics of compound 28. The results revealed that while compound 28 exhibits favorable drug-like properties (good compliance with Lipinski's rule of five) and gastrointestinal absorption potential (topological polar surface area (TPSA) = 117.37 Å2), its poor water solubility (LogS = −8.64, experimental results demonstrate that the solubility in UPW at 25 ℃ is<10 μg/mL) may substantially compromise systemic bioavailability and tissue distribution ultimately affecting therapeutic efficacy following systemic administration (Table S5 and Fig. S7 in Supporting information). These predictive findings provide crucial guidance for subsequent structural optimization of compound 28 and the development of its evidence-based dosing regimen in biomedical applications.
Simultaneously, we characterized the pharmacokinetic (PK) profile of compound 28 in Sprague-Dawley rats after a single 10 mg/kg intraperitoneal injection, collecting plasma via the retro-orbital venous plexus. The plasma concentration-time profile of compound 28 is presented in Fig. S8 (Supporting information), with corresponding PK parameters summarized in Table S6 (Supporting information). The test results indicate that compound 28, characterized by low water solubility and high lipophilicity, may exhibit secondary absorption in vivo due to mechanisms such as tissue redistribution and enterohepatic recirculation. The data reveal an inter-peak interval exceeding 20 h, with the compound failing to clear completely within 72 h post-injection, leading to abnormally prolonged systemic exposure. However, this exposure profile demonstrates significant inter-individual variability and a pronounced food effect, rendering its absorption, distribution, metabolism and excretion (ADME) properties complex and unpredictable. These PK characteristics preliminarily suggest that developing compound 28via intravenous injection or other systemic administration routes may pose major challenges.
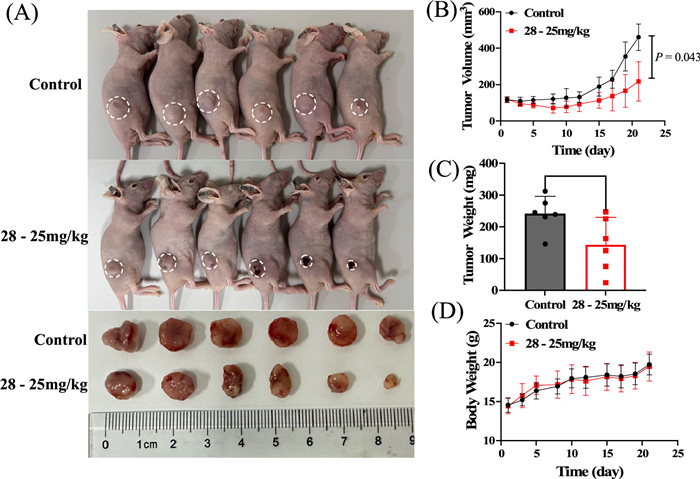
To validate the intrinsic antitumor bioactivity of compound 28, we established a localized administration evaluation system in tumor-bearing mice. Intratumoral administration (25 mg/kg, three times weekly, three weeks) demonstrated significant antitumor efficacy, achieving 40.6% tumor inhibition rates compared to controls (P<0.05, Figs. 4A–C). Notably, no significant body weight alterations occurred in treated groups compared to controls throughout the study period (Fig. 4D and Fig. S6D), suggesting favorable systemic tolerability of 28 at therapeutic doses. Our findings underscore the superiority of localized intratumoral delivery in maximizing antitumor efficacy while minimizing systemic exposure [44–46]. These data advocate for further exploration of intratumoral delivery strategies for compound 28, particularly in combination with immunomodulatory agents to amplify localized immune activation, as suggested by prior studies on intratumoral immunotherapies [47]. Collectively, these findings confirm that the core pharmacophore of compound 28 possesses definitive bioactivity, while emphasizing the necessity to optimize its physicochemical properties (particularly solubility parameters) to enhance systemic exposure.
Figure 4
Figure 4.In vivo antitumor efficacy of compound 28 in MDA-MB-231 xenograft models. Mice bearing MDA-MB-231 tumors (n = 6 per group) were treated with compound 28via intratumoral injection at doses of 25 mg/kg. (A) Representative images of excised tumors post-treatment. (B) Tumor volume dynamics, (C) terminal tumor weight, and (D) body weight changes (toxicity assessment) are shown. Data expressed as mean ± SD. Statistical comparisons: unpaired t-test (terminal comparisons) or two-way ANOVA (longitudinal tumor volume). P<0.05.
Emerging evidence highlights QC, particularly gQC, as a therapeutic target for neurodegenerative (e.g., AD, Huntington's disease (HD), Parkinson's disease), inflammatory, and malignancies [10,15,20]. QC inhibitor development has thus become a key focus in targeted therapy research. Current QC inhibitors demonstrate translational promise: PQ912 has entered phase Ⅱ trials for AD [35], and SEN177 effectively reduces HD-associated phenotypes by suppressing huntingtin (HTT) aggregation [17]. Both compounds inhibit glutaminyl cyclase (gQC), enhancing tumor cell phagocytosis and synergizing with immunotherapies [10,12]. In this study, we identified compound 28 as a novel QC inhibitor with distinct pharmacological advantages. Compound 28 demonstrates moderate selectivity (SI > 10) toward gQC, suggesting reduced off-target risks-a critical feature for long-term therapeutic applications. More notably, QC inhibitor 28 exhibited robust non-immuno-dependent antitumor activity in preclinical models, challenging the prevailing paradigm that QC-mediated anticancer effects primarily rely on immune modulation. This unexpected property implies two groundbreaking possibilities: (1) Compound 28 may engage a unique mechanism-of-action diverging from established inhibitors, potentially involving unexplored signaling nodes downstream of QC; and (2) QC itself might catalyze previously unrecognized substrates that directly contribute to tumor progression, independent of immune pathways. These dual implications expand oncobiological functions of QC and enable precision therapies targeting QC-linked vulnerabilities in immune-refractory cancers. Future studies should prioritize deciphering cryptic substrates of QC and delineating their pathophysiological relevance, which may uncover novel QC-driven oncogenic mechanisms. Furthermore, the non-canonical antitumor mechanism of QC inhibitor 28 warrants systematic exploration of its combination potential with established therapeutic regimens to address drug resistance. Collectedly, this study not only establishes a strategic molecular probe for unraveling the multifaceted roles of QC in disease pathogenesis but also illuminates promising therapeutic avenues targeting QC-associated malignancies.
In this study, we designed and synthesized a series of 3,4-disubstitued indole-2-ketone derivatives based on previous research, and the SAR studies led to the discovery of highly active gQC and sQC inhibitors taking 24, 28, and 29 as represent. Thermal shift assays confirmed that the inhibitor 28, which exhibits better inhibitory activity against gQC (IC50 = 0.095 µmol/L) than sQC (IC50 = 1.33 µmol/L), can significantly improve the thermal stability of gQC protein. Preliminary mechanistic evaluation using DARTS assays in MDA-MB-231 cells further confirmed that 28 protects gQC from proteolytic degradation in a concentration-dependent manner, establishing its target specificity. For functional characterization, 28 was selected as the target compound, while structurally analogous compound 17 (exhibiting 130-fold lower gQC inhibitory potency) served as a negative control. In vitro evaluations revealed that 28 effectively suppressed proliferation of MDA-MB-231 cells induction of G0/G1 phase arrest, concurrently inhibiting clonogenicity, migration, and invasion. In stark contrast, 17 exhibited negligible activity across all assays, underscoring the critical role of gQC inhibition in mediating these effects. In vivo studies using intratumoral administration of 28 confirmed significant antitumor efficacy without observable systemic toxicity. These findings substantiate gQC as a therapeutic target and position 28 as a privileged chemotype for further optimization.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Hui Lei: Visualization, Investigation, Data curation. Yingying Jiang: Investigation, Data curation. Jiayu Liu: Validation, Investigation. Weifeng Zhang: Validation, Investigation. Fanbo Meng: Validation, Investigation. Jun Mou: Investigation. Wenyi Liu: Validation, Investigation. Pengcheng Lei: Validation, Investigation. Rui Xiong: Validation, Investigation. Zan Xu: Validation. Hang Zhang: Validation. Yanjun Wang: Investigation. Guo-Bo Li: Visualization, Methodology, Data curation. Lingling Yang: Writing – review & editing, Writing – original draft, Validation, Methodology, Data curation. Hua-Li Wang: Writing – review & editing, Writing – original draft, Methodology, Investigation, Data curation.
Acknowledgments
This work was supported by Shenzhen Science and Technology Program (No. RCBS20221008093232045), Natural Science Foundation General Project of Sichuan Province (No. 2023NSFSC0606), and National Natural Science Foundation of China (No. 82202857).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.111511.
[1]
R. Barreira da Silva, R.M. Leitao, X. Pechuan-Jorge, et al., Nat. Immunol. 23 (2022) 568–580. doi: 10.1038/s41590-022-01153-x
K.P. Fabian, G. Santiago-Sanchez, M.R. Padget, et al., J. Immunother. Cancer 12 (2024) e009712. doi: 10.1136/jitc-2024-009712
Figure 1
Design Strategies for novel QC inhibitors. (A) Structure and inhibitory activity of lead compound 1 against gQC and sQC. (B, C) View of a crystal structure of sQC: 1 complex (PDB: 8XGT). (D) Research idea for structural optimization.
Figure 2
Compounds 24, 28 and 29 occupy the catalytic active sites of gQC and sQC, and alter their thermal stability. Predicted interaction mode of compound 24 with sQC (A) and gQC (B). Predicted interaction mode of compound 28 with sQC (C) and gQC (D). Predicted interaction mode of compound 29 with sQC (E) and gQC (F). (G) The melting curve (first-derivative of dissociation) of gQC and sQC in the presence or absence of compounds 24, 28 or 29 (100 µmol/L).
Figure 3
Compound 28 inhibits MDA-MB-231 cell proliferation through gQC-targeted mechanisms. (A) Target engagement of compound 28 with gQC demonstrated by DARTS assay. (B) The anti-viability activities (IC50, µmol/L) of compound 28 against various BRCA cell lines following a 72-h treatment period. (C) The profile of cell ability after 72-h treatment with compound 28 or 17. (D) The fluorescence microscopic appearance of EdU and Hoechst 33342 on MDA-MB-231 cells after treatment with compound 28 or 17. Scale bar: 50 µm. (E) Long-term colony formation assay on MDA-MB-231 cells. (F) Flow cytometry histograms of cell cycle distribution in MDA-MB-231 cells treated with 28 or 17 for 24 h. (G) Apoptosis rate quantified by Annexin V/PI dual staining after 24-h treatment with 28 (representative cytograms shown). (H) Representative images of wound healing assays at 0 and 24-h post-treatment with 28 (scale bar: 200 µm). (I) Effect of 28 on cell invasion tested by transwell assay 28 (scale bar: 200 µm). GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Figure 4In vivo antitumor efficacy of compound 28 in MDA-MB-231 xenograft models. Mice bearing MDA-MB-231 tumors (n = 6 per group) were treated with compound 28via intratumoral injection at doses of 25 mg/kg. (A) Representative images of excised tumors post-treatment. (B) Tumor volume dynamics, (C) terminal tumor weight, and (D) body weight changes (toxicity assessment) are shown. Data expressed as mean ± SD. Statistical comparisons: unpaired t-test (terminal comparisons) or two-way ANOVA (longitudinal tumor volume). P<0.05.

 DownLoad:
DownLoad:

 下载:
下载:





 下载:
下载:
