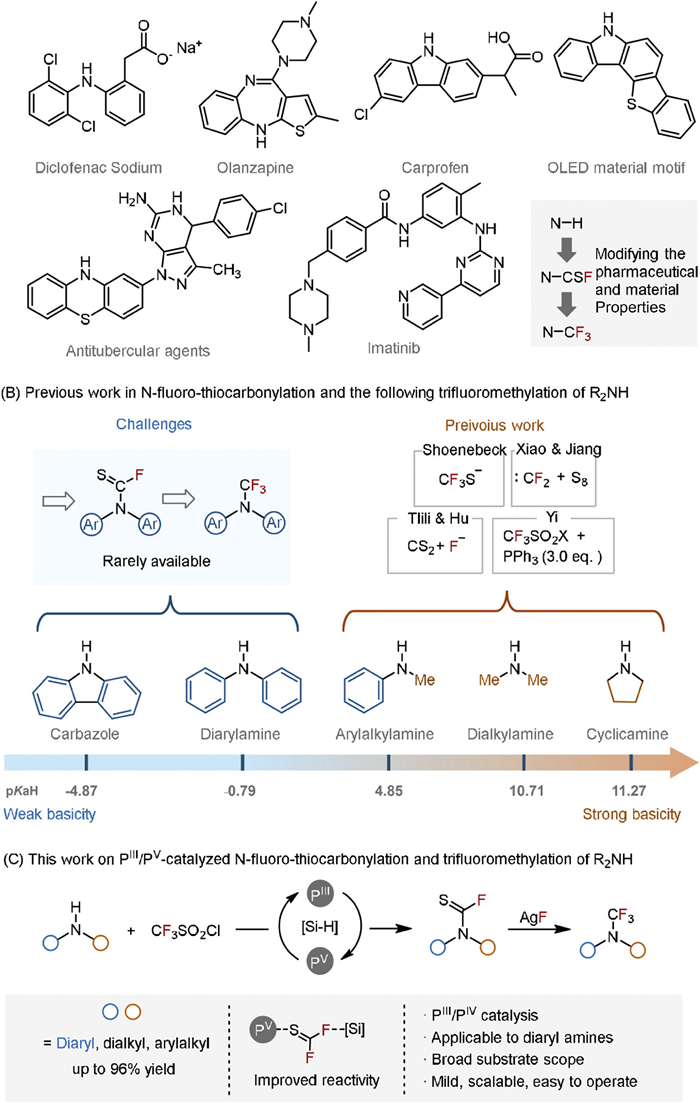
Figure 1.
The background and our study on N-fluoro-thiocarbonylation and the following N-trifluoromethylation of diaryl amines.
PⅢ/PⅤ-catalytic platform enabling N-fluoro-thiocarbonylation of diaryl amines for modular synthesis of tertiary N-trifluoromethylamines
Dongke Zhang , Li-Ao Ding , Qiuyu Xiang , Zhuojun Li , Qian Wu

Diaryl amine structures are key components in pharmaceuticals, biologically active compounds, and material science (Fig. 1A) [1–11]. Notably, due to their common occurrence in kinase inhibitors like imatinib (anti-cancer drug), diaryl amines are often referred to as "kinase-privileged fragments" [12–14]. The incorporation of fluorinated groups has proven to be an effective strategy for modifying molecular properties, including lipophilicity, solubility, electrophilicity, metabolic stability, and molecular conformation [15–19]. Therefore, transformation of the ubiquitous N–H bond in diaryl amines to an N–CF3 is pivotal to unlock new properties and functionalities in complex molecules, allowing to extend the boundaries of pharmaceutical and material innovation [20–24]. However, the conversion of N–H motifs to R2N–CF3 compounds remains insufficiently investigated [25,26].
Existing direct N–H trifluoromethylation methods, typically employing Togni and Ruppert-Prakash reagents, face challenges related to generality and specific substrate types, such as azoles [27–30], imines [31–33], certain amides [34,35], and selected dialkyl/alkylaryl amines [36]. A promising alternative involves stepwise sequences in functional group interconversion, gaining attention over the past decade through different retrosynthetic approaches [37–43]. A notable advancement was introduced by Schoenebeck, who developed a one-pot method for synthesizing trifluoromethyl amines (R2N–CF3) from N–H motif. Their method involved the formation of N-thiocarbonyl fluoride (R2N–CSF) intermediate with solid Me4N+SCF3- and the subsequent desulfurfluorination (Fig. 1B, right) [44,45]. Later on, Xiao and Jiang, as well as Yi independently realized the one-pot syntheses of secondary R2N–CF3 from free amines by utilizing the intermediacy of R2N–CSF [46–48]. Tlili’s recent work demonstrated a new synthetic method of R2N–CSF through the fluorinative desulfurization of CS2 by developing versatile SF5-based reagent [49,50]. However, these approaches usually benefit dialkyl and alkylaryl NH substrates with more nucleophilic and basic properties, such as cyclopentylamine (pKaH = 11.27), Me2NH (pKaH = 10.71) and PhMeNH (pKaH = 4.85) [51–53]. In contrast, diaryl amine derivatives present a challenge in these developed systems due to their electron-deficient and weakly basic N–H sites, exemplified by Ph2NH (pKaH = 0.79) [54]. Furthermore, transformations involving significantly less basic substrates like carbazoles (pKaH = −4.87) remain untapped [55]. Encouraged by Radosevich's recent works [56], we herein address the challenges on tertiary N–CF3 formation through diaryl amines by developing a synergistic organophosphorus(Ⅲ)-silane catalytic N-fluoro-thiocarbonylation (Fig. 1C). The mechanistic insight revealed that the reaction undergoes a PⅢ/PⅤ redox cycle. Hydrosilane serves two critical functions: (1) as a terminal reductant to cycle the PⅢ/PⅤ redox pair and (2) acting as an effective fluoride acceptor, thus enhancing the reaction between the thiophosgene intermediate and inert secondary amines. Notably, recently reported stability of R2N–CSF compounds in the presence of different amino acids or serums in PBS, allows for their further applications under near-physiological conditions [57].
We initiated the synthesis of thiocarbamoyl fluorides via PⅢ/PⅤ catalysis by reacting the challenging substrate carbazole 1a with TfCl 2 and phenylsilane (Table 1). The reaction afforded desired product (carbazole-CSF) in only trace amount at 40 ℃ in the presence of 0.25 equiv. of triphenylphosphine or tri(o-tolyl)phosphine in DCE, while tri-n-butylphosphine gave 67% yield (entries 1–3). Replacing the catalyst with a bulkier tricyclohexylphosphine (Cy3P) enhanced the yield to 95%, whereas reducing the catalyst loading to 0.1 equiv. decreased the yield to 78% (entries 4 and 5). Alternative solvents such as THF, acetone, DCE, and toluene resulted in lower yields (entries 6–9). Further studies revealed that the corresponding tricyclohexylphosphine oxide (Cy3P=O) also facilitated the formation of the desired product with a yield of 72%, indicating that the reaction proceeds via a PⅢ/PⅤ redox cycle (entry 10). Control experiments in the absence of organophosphorus(Ⅲ) catalyst or hydrosilane showed no reactivity, confirming the necessity of both components for this catalytic N-fluoro-thiocarbonylation of carbazole (entries 11 and 12). The structure of 3a was verified through X-ray diffraction analysis (CCDC: 2377182).
 DownLoad:
CSV
DownLoad:
CSV
 |
||||
| Entry | [P]cat. (eq.) | Silane | Solvent | Yield (%)b |
| 1 | Ph3P (0.25) | PhSiH3 | DCE | 2 |
| 2 | (o-tolyl)3P (0.25) | PhSiH3 | DCE | 1 |
| 3 | nBu3P (0.25) | PhSiH3 | DCE | 67 |
| 4 | Cy3P (0.25) | PhSiH3 | DCE | 95 |
| 5c | Cy3P (0.10) | PhSiH3 | DCE | 78 |
| 6 | Cy3P (0.25) | PhSiH3 | THF | 46 |
| 7 | Cy3P (0.25) | PhSiH3 | Acetone | 56 |
| 8 | Cy3P (0.25) | PhSiH3 | MeCN | 74 |
| 9 | Cy3P (0.25) | PhSiH3 | Toluene | 22 |
| 10d | Cy3P=O (0.25) | PhSiH3 | DCE | 72 |
| 11 | Cy3P (0.25) | None | DCE | 0 |
| 12 | None | PhSiH3 | DCE | 0 |
| a Reaction was carried out by using 1a (0.25 mmol), 2 (0.375 mmol, 1.5 equiv.), [P]cat. (0–0.25 equiv.), phenylsilane (0–3.0 equiv.), in solvent at 40 ℃ for 24 h under Ar. b Yields were determined by GC–MS using naphthalene as an internal standard. c For 36 h. d Phenylsilane (1.25 mmol, 5.0 equiv.) was used. |
||||
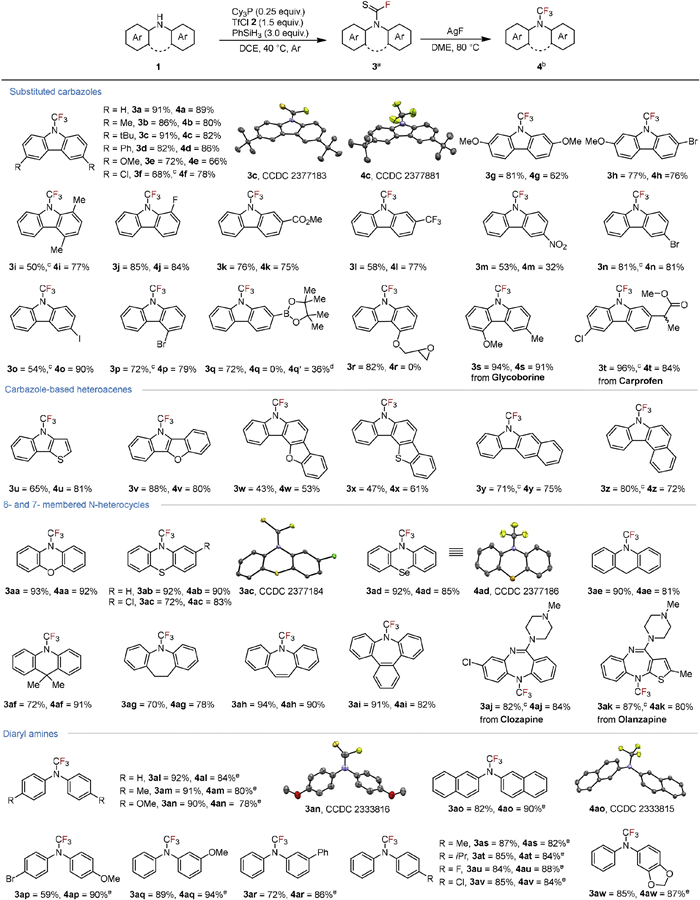
Having established optimized reaction conditions, we proceeded to explore the scope of sequential N-fluoro-thiocarbonylation and desulfurization-fluorination reactions using silver fluoride (Scheme 1). This protocol was effectively applied to carbazoles bearing various electron-rich, -neutral, or -deficient functional groups, such as alkyl, alkoxy, phenyl, halogen, nitro, ester, and trifluoromethyl groups, affording carbazole N–CSF and N–CF3 products (3a-3p and 4a-4p). Substrates featuring reducible functionalities, such as ester and nitro groups, successfully formed the desired products (3k, 3m, 4k, 4m). The tolerance of aryl halide, especially bromide and iodide, allows further elaboration of the N–CF3 products (3n-3p and 4n-4p). Remarkably, sensitive groups like epoxy and Bpin could be tolerated in the N-fluoro-thiocarbonylation stage (3q-3r), although they did not proceed to form trifluoromethylated products. Specifically, for transformation of 3q, a homocoupling of arylboronic ester occurred in the presence of silver salt, resulting in a trifluoromethyl carbazole dimer intermediate 4q′ (see Supporting information for details). These results encouraged us to examine carbazole-based heteroacenes, which are commonly utilized in organic light-emitting diodes (OLEDs) and organic field-effect transistors (OFETs) [58–60], were successfully converted into R2N–CSF and the following R2N–CF3 products (3u-3z and 4u-4z) in moderate to good yields. The low solubility of both N–H substrate and the corresponding products were probably attributed to the diminished yields, caused by the planarity of these molecules. Additionally, this protocol proved compatible with other NH-containing heterocycles such as phenoxazine, phenothiazine, phenoselenazine, dihydroacridine and dibenzo[b,f]azepine (3aa-3ai and 4aa-4ai). Furthermore, the more nucleophilic and basic structures based on N,N-diphenyl amines demonstrated robust reactivity as expected. Both symmetrical and asymmetrical diphenyl amines with either electron-rich or electron-poor substituents yielded the desired compounds efficiently (3al-3aw and 4al-4aw). To illustrate the practical application of this method in late-stage functionalization of complex scaffolds, natural products and drug molecules were involved, such as Glycoborine (3s and 4s), Carprofen methyl ester (3t and 4t), Clozapine (3ak and 4ak), and Olanzapine (Zyprexa). The reactions produced the desired R2N–CSF and R2N–CF3 compounds in good to excellent yields without altering the core structures of these molecules.
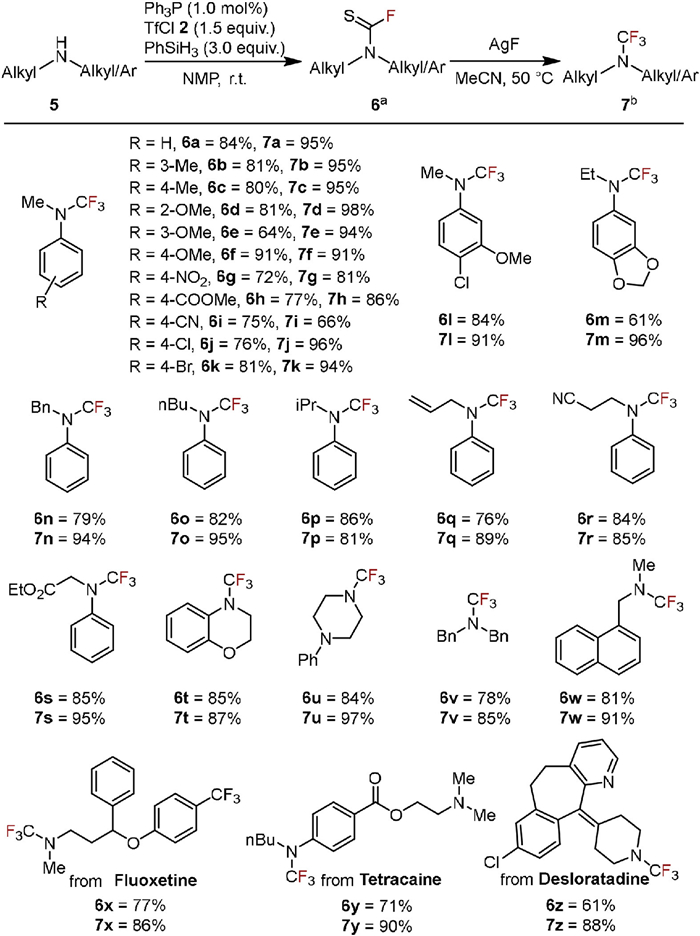
When employing more nucleophilic and basic dialkyl and arylalkyl amines, the PⅢ catalyst was effectively replaced with Ph3P at a reduced loading of 1 mol% in NMP at room temperature (Scheme 2). This adjustment allowed various alkylaryl and dialkyl amines to undergo N-fluoro-thiocarbonylation, yielding the corresponding products in good to excellent quantities while maintaining compatibility with various functional groups (6a-6w). Subsequent desulfurization-fluorination steps using AgF were efficiently carried out in acetonitrile (7a-7w). This process was then applied to pharmaceutical compounds such as Fluoxetine, Tetracaine, and Desloratadine, producing N-trifluoromethylated derivatives with satisfactory yields (6x-6z and 7x-7z).
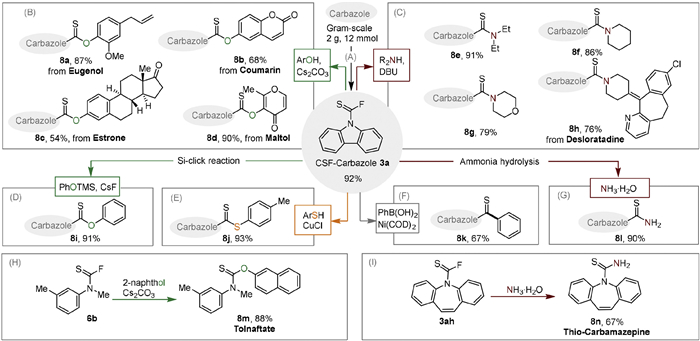
The unique structure of thiocarbamoyl fluorides not only facilitates the transformation to attractive N–CF3 compounds, but also serves as a versatile linker to connect different bio-active scaffold, leveraging fluoride's role as a leaving group (Scheme 3). To demonstrate the synthetic application of thiocarbamoyl fluorides, we initially scaled up the synthesis of carbazole-CSF 3a to 12.0 mmol, maintaining a high yield of 92% (Scheme 3A). Furthermore, the fluoride on 3a was effectively replaced with various phenols using Cs2CO3 in acetone. This substitution involved natural bioactive molecules such as Eugenol, endogenous hormones like Estrone, and other compounds including Coumarin and Maltol (Scheme 3B, 8a-8d). Additionally, carbamothioate 8i was synthesized through a Si-click reaction utilizing PhOTMS (Scheme 3D). Remarkably, the antifungal drug Tolnaftate 8m was afforded with an 88% yield starting from N-methyl-m-toludine (Scheme 3H). Employing aliphatic amines or ammonia as nucleophiles, including Desloratadine, led to the creation of several notable carbothioamides (8e-8h and 8l), and thio-carbamazepine 8n was synthesized from 3ah through the same reaction conditions (Schemes 3C, G and I). The carbamoyl fluoride proved reactive towards thiophenol in the presence of a catalytic amount of CuCl, facilitating its conversion to carbamodithioate 8j (Scheme 3E). Finally, an efficient nickel-catalyzed Suzuki cross-coupling reaction was utilized to produce thioamide 8k (Scheme 3F).
During the reaction, we observed gas formation, which was suggested as molecular hydrogen (δ(1H) = 4.65) (Fig. S1B in Supporting information). To explore further insight of the roles of organophosphorus(Ⅲ) and silicon species in the reaction pathway, a two-chamber reactor was employed. Initially, TfCl, Cy3P, and PhSiH3 were mixed in chamber 1, and trace amount of bridged product 10 was captured using pentamethylcyclopentadiene 9 in chamber 2. Meanwhile, the yield of 10 increased to 18% in the presence of Cy3P and PhSiH3 in chamber 2, indicating that the generation of thiocarbonyl difluoride was promoted by organophosphorus(Ⅲ) and hydrosilane (Scheme 4A). To further confirm the intermediacy of thiocarbonyl difluoride, we added carbazole in chamber 2 along with Cy3P and PhSiH3, resulting product 3a in an 89% yield (Scheme 4B). A slightly lower yield of 75% was obtained when replacing Cy3P with Cy3P=O. Additional control experiments without Cy3P or PhSiH3 did not produce the target product, underscoring their critical roles in the reaction of thiophosgene and the less nucleophilic diaryl amines. Further NMR spectroscopy analysis of the catalytic reaction mixture revealed that trifluoromethylsulfenyl chloride C (δ(19F) = −46.3) and chlorophosphonium salt F (δ(31P) = 105.4) were present in the reaction (Figs. S4 and S5 in Supporting information); HRMS-ESI analysis revealed peaks at m/z 381.1988 and 397.1700, which were proposed to be the trifluoromethyl thiophosphonium cation D and an adduct E of the phosphonium salt with thiocarbonyl difluoride, respectively (Fig. S6 in Supporting information).
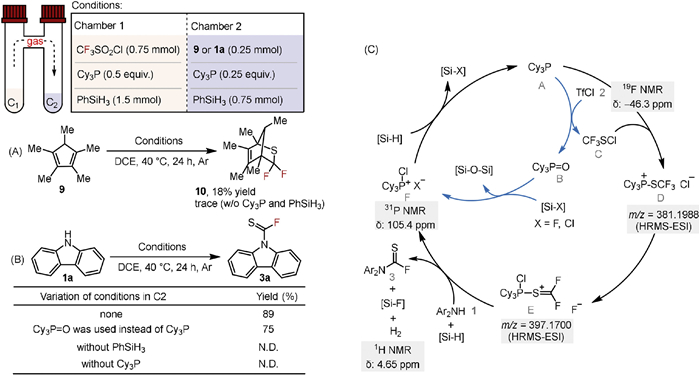
Based on the above results, we propose a plausible reaction pathway for the N-fluoro-thiocarbonylation step as depicted in Scheme 4C. The reaction initiates with sequential deoxygenation of TfCl 2 facilitated by PⅢ A. The reduced intermediate sulfenium ion C forms Lewis acid-base adduct with PⅢ [61], which then decomposes into E. The reaction of E with diarylamine leads the final product with the release of molecular hydrogen, supported by the high fluorophilicity of silicon species. This step is corroborated by the detection of a Si-F species [62], evidenced by 19F NMR detection of the reaction mixture (Fig. S4 in Supporting information). The fluorosilane species suggests an increase in the Lewis acidity at the silicon center, further enhancing its affinity for fluoride. In the other cycle, the resulting PⅤ=O B was speculated to convert into phosphonium cation F, facililated by halosilane due to its enhanced oxophilicity [63,64]. Finally, Si-H species reduce the released halophosphonium salt liberating PⅢ to complete the catalytic cycle.
In summary, we have disclosed catalytic N-fluoro-thiocarbonylation of secondary amines with trifluoromethanesulfonyl chloride and hydrosilane through a PⅢ/PⅤ couple, facilitating efficient conversion of N–H into R2N–CSF and leading to R2N–CF3 with silver fluoride. This work addresses the gap of diaryl amine derivatives, which pose a challenge in previous works due to their electron-deficient and weakly basic characteristics. The mechanistic research confirms the essential roles of the PⅢ/PⅤ catalyst and silane species. This procedure opens new and efficient pathways for the synthesis of structurally diverse tertiary N–CSF and N–CF3 compounds, allowing for various applications in chemistry and materials.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Dongke Zhang: Methodology, Investigation, Data curation. Li-Ao Ding: Validation, Investigation. Qiuyu Xiang: Methodology, Investigation. Zhuojun Li: Methodology, Investigation. Qian Wu: Writing – review & editing, Writing – original draft, Validation, Supervision, Project administration, Funding acquisition, Data curation, Conceptualization.
We acknowledged for the financial support from the National Natural Science Foundation of China (Nos. 22371133, 22171142, 22188101), the Fundamental Research Funds for the Central Universities, Nankai University (Nos. 63241203, 63253258). The authors thank Quan-wen Li and Hai-bin Song for the X-ray crystallography data collection and analysis.
Supplementary material associated with this article can be found, in the online version, at doi:
A.W. Schmidt, K.R. Reddy, H.J. Knölker, Chem. Rev. 112 (2012) 3193–3328. doi: 10.1021/cr200447s
K.U. Ingold, D.A. Pratt, Chem. Rev. 114 (2014) 9022–9046. doi: 10.1021/cr500226n
C. Sambiagio, S.P. Marsden, A.J. Blacker, P.C. McGowan, Chem. Soc. Rev. 43 (2014) 3525–3550. doi: 10.1039/C3CS60289C
P. Ruiz-Castillo, S.L. Buchwald, Chem. Rev. 116 (2016) 12564–12649. doi: 10.1021/acs.chemrev.6b00512
S. Tabassum, A.F. Zahoor, S. Ahmad, et al., Mol. Divers. 26 (2022) 647–689. doi: 10.1007/s11030-021-10195-6
A.D. Favia, D. Habrant, R. Scarpelli, et al., J. Med. Chem. 55 (2012) 8807–8826. doi: 10.1021/jm3011146
R. Shah, E.A. Haidasz, L. Valgimigli, D.A. Pratt, J. Am. Chem. Soc. 137 (2015) 2440–2443. doi: 10.1021/ja5124144
Z. Wang, H. Wang, J. Zhu, et al., ACS Appl. Mater. Interfaces 9 (2017) 21346–21354. doi: 10.1021/acsami.7b04987
C. Cerchia, R. Nasso, M. Mori, et al., J. Med. Chem. 62 (2019) 7089–7110. doi: 10.1021/acs.jmedchem.9b00632
H. Liu, Z. Liu, G. Li, et al., Angew. Chem. Int. Ed. 60 (2021) 12376–12380. doi: 10.1002/anie.202103187
P. Onnuch, K. Ramagonolla, R.Y. Liu, Science 383 (2024) 1019–1024. doi: 10.1126/science.adl5359
P. Cohen, Nat. Rev. Drug. Discov. 1 (2002) 309–315. doi: 10.1038/nrd773
A.M. Aronov, B. McClain, C.S. Moody, M. Murcko, J. Med. Chem. 51 (2008) 1214–1222. doi: 10.1021/jm701021b
Z.Z. Wang, X.X. Shi, G.Y. Huang, G.F. Hao, G.F. Yang, Trends Pharmacol. Sci. 42 (2021) 551–565. doi: 10.1016/j.tips.2021.04.001
E.P. Gillis, K.J. Eastman, M.D. Hill, D.J. Donnelly, N. Meanwell, J. Med. Chem. 58 (2015) 8315–8359. doi: 10.1021/acs.jmedchem.5b00258
Y. Zhou, J. Wang, Z. Gu, et al., Chem. Rev. 116 (2016) 422–518. doi: 10.1021/acs.chemrev.5b00392
Y. Ogawa, E. Tokunaga, O. Kobayashi, K. Hirai, N. Shibata, iScience 23 (2020) 101467. doi: 10.1016/j.isci.2020.101467
H. Chepliaka, J. Kollback, T. Quennesson, W. Czechtizky, R. Cox, J. Med. Chem. 63 (2020) 13076–13089. doi: 10.1021/acs.jmedchem.0c01457
Z. Lei, W. Chang, H. Guo, J. Feng, Z. Zhang, Molecules 28 (2023) 3012. doi: 10.3390/molecules28073012
S. Purser, P.R. Moore, S. Swallow, V. Gouverneur, Chem. Soc. Rev. 37 (2008) 320–330. doi: 10.1039/B610213C
N.A. Meanwell, J. Med. Chem. 54 (2011) 2529–2591. doi: 10.1021/jm1013693
P. Furet, V. Guagnano, R.A. Fairhurst, et al., Bioorg. Med. Chem. Lett. 23 (2013) 3741–3748. doi: 10.1016/j.bmcl.2013.05.007
A. Abula, Z. Xu, Z. Zhu, et al., J. Chem. Inf. Model. 60 (2020) 6242–6250. doi: 10.1021/acs.jcim.0c00898
P. Pattanayak, S. Nikhitha, D. Halder, B. Ghosh, T. Chatterjee, RSC Adv. 14 (2024) 18856–18870. doi: 10.1039/d4ra02856b
T. Milcent, B. Crousse, Chimie 21 (2018) 771–781.
B. Crousse, Chem. Rec. 23 (2023) e202300011. doi: 10.1002/tcr.202300011
K. Niedermann, N. Früh, E. Vinogradova, et al., Angew. Chem. Int. Ed. 50 (2011) 1059–1063. doi: 10.1002/anie.201006021
K. Niedermann, N. Früh, R. Senn, et al., Angew. Chem. Int. Ed. 51 (2012) 6511–6515. doi: 10.1002/anie.201201572
Z.E. Blastik, S. Voltrová, V. Matoušek, et al., Angew. Chem. Int. Ed. 56 (2017) 346–349. doi: 10.1002/anie.201609715
R.Z. Zhang, R.X. Zhang, S. Wang, et al., Angew. Chem. Int. Ed. 61 (2022) e202110749. doi: 10.1002/anie.202110749
F. Teng, J. Cheng, C. Bolm, Org. Lett. 17 (2015) 3166–3169. doi: 10.1021/acs.orglett.5b01537
G. Zheng, X. Ma, J. Li, D. Zhu, M. Wang, J. Org. Chem. 80 (2015) 8910–8915. doi: 10.1021/acs.joc.5b01468
C. Xu, X. Song, J. Guo, et al., Org. Lett. 20 (2018) 3933–3937. doi: 10.1021/acs.orglett.8b01510
Z. Zhang, J. He, L. Zhu, et al., Chin. J. Chem. 38 (2020) 924–928. doi: 10.1002/cjoc.202000132
S. Liu, Y. Huang, J. Wang, F.L. Qing, X.H. Xu, J. Am. Chem. Soc. 144 (2022) 1962–1970. doi: 10.1021/jacs.1c12467
T. Umemoto, K. Adachi, S. Ishihara, J. Org. Chem. 72 (2007) 6905–6917. doi: 10.1021/jo070896r
T. Scattolin, S. Bouayad-Gervais, F. Schoenebeck, Nature 573 (2019) 102–107. doi: 10.1038/s41586-019-1518-3
J. Liu, M.F.L. Parker, S. Wang, et al., Chem 7 (2021) 2245–2255. doi: 10.1016/j.chempr.2021.07.005
L. Wang, J. Wang, S. Ye, et al., Angew. Chem. Int. Ed. 61 (2022) e202212115. doi: 10.1002/anie.202212115
M. Tao, J. Qian, Z. Chen, et al., J. Org. Chem. 88 (2023) 15237–15248. doi: 10.1021/acs.joc.3c01740
Y. Yang, N. Saffon-Merceron, J.C. Vantourout, A. Tlili, Chem. Sci. 14 (2023) 3893–3898. doi: 10.1039/d2sc06542h
M. Spennacchio, M. Bernús, J. Stanić, et al., Science 385 (2024) 991–996. doi: 10.1126/science.adq2954
H. Song, Q. Wang, X. Wang, et al., CCS Chem. 7 (2025) 381–391. doi: 10.31635/ccschem.024.202404432
T. Scattolin, K. Deckers, F. Schoenebeck, Angew. Chem. Int. Ed. 56 (2017) 221–224. doi: 10.1002/anie.201609480
T. Scattolin, M. Pu, F. Schoenebeck, Chem. Eur. J. 24 (2018) 567–571. doi: 10.1002/chem.201705240
J. Yu, J.H. Lin, J.C. Xiao, Angew. Chem. Int. Ed. 56 (2017) 16669–16673. doi: 10.1002/anie.201710186
S. Liang, J. Wei, L. Jiang, et al., Chem. Commun. 55 (2019) 8536–8539. doi: 10.1039/c9cc03282g
J. Wei, S. Liang, L. Jiang, W. Yi, J. Org. Chem. 85 (2020) 12374–12381. doi: 10.1021/acs.joc.0c01634
K. Onida, L. Vanoye, A. Tlili, Eur. J. Org. Chem. 2019 (2019) 6106–6109. doi: 10.1002/ejoc.201901113
A. Taponard, T. Jarrosson, L. Khrouz, et al., Angew. Chem. Int. Ed. 61 (2022) e202204623.
N.F. Hall, M.R. Sprinkle, J. Am. Chem. Soc. 344 (1932) 3469–3485. doi: 10.1021/ja01348a001
L. Sooväli, T. Rodima, I. Kaljurand, et al., Org. Biomol. Chem. 4 (2006) 2100–2105. doi: 10.1039/B602797K
I. Kaljurand, R. Lilleorg, A. Murumaa, et al., J. Phys. Org. Chem. 26 (2013) 171–181. doi: 10.1002/poc.2956
I. Kaljurand, A. Kütt, L. Sooväli, et al., J. Org. Chem. 70 (2005) 1019–1028. doi: 10.1021/jo048252w
M. Balón, M.C. Carmona, M.A. Muñoz, J. Hidalgo, Tetrahedron 45 (1989) 7501–7504. doi: 10.1016/S0040-4020(01)89212-7
A. Ghosh, M. Lecomte, S.H. Kim-Lee, A.T. Radosevich, Angew. Chem. Int. Ed. 58 (2019) 2864–2869. doi: 10.1002/anie.201813919
C. Delobel, E. Chefdeville, F. Toulgoat, T. Billard, Adv. Synth. Catal. 366 (2024) 3474–3480. doi: 10.1002/adsc.202400412
R.K. Konidena, K.H. Lee, J.Y. Lee, W.P. Hong, Org. Electron. 70 (2019) 211–218. doi: 10.1016/j.orgel.2019.04.006
H. Srour, T.H. Doan, E.D. Silva, R.J. Whitby, B. Witulski, J. Mater. Chem. C 4 (2016) 6270–6279.
Z. Zheng, Q. Dong, L. Gou, J.H. Su, J. Huang, J. Mater. Chem. C 2 (2014) 9858–9865. doi: 10.1039/C4TC01965B
Y. Ouyang, X.H. Xu, F.L. Qing, Angew. Chem. Int. Ed. 58 (2019) 18508–18512. doi: 10.1002/anie.201911323
S.V. Basenko, L.E. Zelenkov, M.G. Voronkov, A.I. Albanov, Russ. J. Gen. Chem. 80 (2010) 242–244. doi: 10.1134/S107036321002009X
Q. Li, Y. Sun, M.X. Fu, J.H. Lin, J.C. Xiao, J. Org. Chem. 89 (2024) 16022–16027. doi: 10.1021/acs.joc.4c01847
M. You, Z. Zhang, C. Chen, et al., Green Chem. 27 (2025) 3743–3750. doi: 10.1039/d5gc00477b

Figure 1 The background and our study on N-fluoro-thiocarbonylation and the following N-trifluoromethylation of diaryl amines.

Scheme 1 Scope of diaryl amines for synthesizing thiocarbamoyl fluorides and the tertiary N–CF3 products. a Reaction conditions: substrate 1 (0.25 mmol), Cy3P (0.25 equiv.), TfCl 2 (1.5 equiv.), PhSiH3 (3.0 equiv.), DCE (1.0 mL), 40 ℃, 24 h, Ar, isolated yield. b Substrate 3 (0.25 mmol), AgF (5.0 equiv.), DME (1.0 mL), 80 ℃, 12 h, Ar, isolated yield. c 48 h. d 9,9′-Bis(trifluoromethyl)-9H, 9′H-2,2′-bicarbazole 4q′ was obtained. e Substrate 3 (0.2–0.5 mmol), AgF (4.5 equiv.), MeCN (1.5 mL), 50 ℃, 5 h. Please see Supporting information for experimental details.

Scheme 2 Scope of other secondary amine derivatives. a Secondary amine 5 (0.5 mmol), Ph3P (1.0 mol%), TfCl 2 (1.5 equiv.), PhSiH3 (3.0 equiv.), in NMP, r.t, 5 h, under air, isolated yield. b R2N–CSF 6 (0.3–0.5 mmol), AgF (4.5 equiv.), MeCN (1.5 mL), 50 ℃, 5 h, Ar, isolated yield.

Scheme 3 The synthetic application of thiocarbamoyl fluoride compounds. Please see Supporting information for experimental details.

Scheme 4 (A) Detection of thiophosgene with 9 using two-chamber reactor. (B) Control experiments with 3a using two-chamber reactor. (C) Proposed reaction pathway for the N-fluoro-thiocarbonylation stage in organophosphorus-silicon catalytic system. Chamber 1: TfCl 2 (0.75 mmol), Cy3P (0.5 equiv.), PhSiH3 (1.5 mmol), DCE (2.0 mL), 40 ℃, 24 h, Ar. Chamber 2: 9 or 2a (0.25 mmol), Cy3P (0.25 equiv.), PhSiH3 (0.75 mmol), DCE (1.0 mL), 40 ℃, 24 h, Ar, isolated yield. Please see Supporting information for experimental details.
Table 1. Reaction conditions optimization.a
|
||||
| Entry | [P]cat. (eq.) | Silane | Solvent | Yield (%)b |
| 1 | Ph3P (0.25) | PhSiH3 | DCE | 2 |
| 2 | (o-tolyl)3P (0.25) | PhSiH3 | DCE | 1 |
| 3 | nBu3P (0.25) | PhSiH3 | DCE | 67 |
| 4 | Cy3P (0.25) | PhSiH3 | DCE | 95 |
| 5c | Cy3P (0.10) | PhSiH3 | DCE | 78 |
| 6 | Cy3P (0.25) | PhSiH3 | THF | 46 |
| 7 | Cy3P (0.25) | PhSiH3 | Acetone | 56 |
| 8 | Cy3P (0.25) | PhSiH3 | MeCN | 74 |
| 9 | Cy3P (0.25) | PhSiH3 | Toluene | 22 |
| 10d | Cy3P=O (0.25) | PhSiH3 | DCE | 72 |
| 11 | Cy3P (0.25) | None | DCE | 0 |
| 12 | None | PhSiH3 | DCE | 0 |
| a Reaction was carried out by using 1a (0.25 mmol), 2 (0.375 mmol, 1.5 equiv.), [P]cat. (0–0.25 equiv.), phenylsilane (0–3.0 equiv.), in solvent at 40 ℃ for 24 h under Ar. b Yields were determined by GC–MS using naphthalene as an internal standard. c For 36 h. d Phenylsilane (1.25 mmol, 5.0 equiv.) was used. |
||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载: