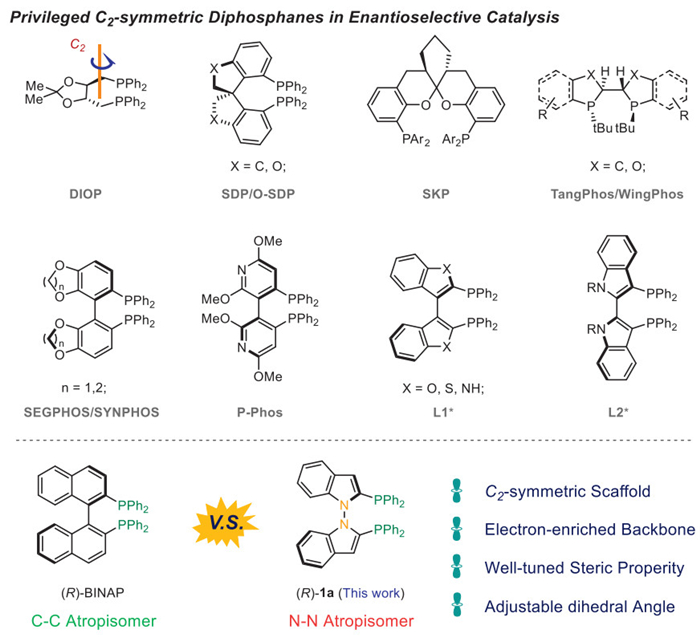
Scheme 1.
Selected privileged C2-symmetric diphosphanes.
C2-Symmetric N–N atropisomeric diphosphines: Synthesis and application in enantioselective dearomatization of heteroaryls
Xiao-Kai Li , Si-Hao Fu , Yi Yue , Rui-Jing Pang , Jia Feng , Ren-Rong Liu

Atropisomers with flexible structures and tunable properties have emerged as privileged structures in natural products, drug design, and asymmetric catalysis in recent decades [1–6]. Compared to conventional C–C/C–N atropisomers, N–N atropisomeric scaffolds have garnered increasing interest in the last few years due to their unique N–N axis [7,8]. In 2021, Lu and Liu independently conducted research on the synthesis of N–N atropisomers [9,10]. Subsequently, a series of protocols has been developed to enable their enantioselective synthesis. The desymmetrization of substrates with a prochiral N–N axis has proven to be an efficient strategy for producing N–N atropisomers [11–13]. For nonaromatic N–N atropisomers, N-functionalization offers a readily available route [14–19]. Enantioselective synthesis from hydrazine substrates can be achieved through de novo aza-heterocycle synthesis [20–30]. Additionally, various other strategies have been demonstrated for the synthesis of N–N atropisomers [31,32].
However, despite the unique electronic and tunable steric properties of N–N atropisomeric scaffolds, they have rarely been applied to the synthesis of chiral ligands or catalysts. In 1997, Sannicolò reported the synthesis of N–N bisbenzimidazole diphosphines, which offer a modulated electron-deficient backbone for diphosphines synthesis [33]. More recently, Shi [20] and our group [21] developed N–N atropisomeric-based catalysts/ligands, some of which have been successfully applied to asymmetric catalysis. The tunable steric and electronic properties of N–N atropisomers makes them desirable for catalyst/ligand design, although their application remains challenging.
Phosphines are the most used ligands bound to metal in enantioselective synthesis, particularly for transition metals. Among all phosphines, bidentate diphosphines help to lock potential coordinated sites of the metal, thus providing a definite binding pocket for stereoinduction [34–42]. In recent decades, a series of C2-symmetric diphosphine scaffolds have been developed and have contributed significantly to the most useful ligand library. Some selected C2-symmetric diphosphines are depicted in Scheme 1. DIOP, derived from natural product scaffolds, was developed by Kagan in 1971 [43,44]. Spirocyclic diphosphines SDP/O-SDP or SKP were independently invented by Zhou, Ding, and Zhang and successfully applied to enantioselective synthesis [45–50]. P-stereogenic ligands TangPhos/WingPhos and QuinoxP* have also emerged as promising chiral ligands in enantioselective catalysis [51–53]. Electron-enriched biaryl backbones have been successfully applied to construct chiral diphosphines including SEGPHOS, SYNPHOS, among others [54,55]. Diheteroaryl scaffold-based ligands including dipyridylphosphine ligands P-Phos and di-indoylphosphine ligands L1* and L2* bearing a chiral C–C axis, have been applied to enantioselective synthesis [56–59]. One of the most successful examples is 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP), which is widely used in asymmetric catalysis, especially in ruthenium-mediated asymmetric hydrogenation [60–63]. Herein, we developed a synthetic procedure for N–N atropisomer-based diphosphine 1 and successfully applied it to enantioselective dearomatization. The newly developed N–N atropisomeric diphosphines have properties comparable to BINAP. The first and most important ones are their essential electron-enriched backbones, which can properly coordinate to electron-deficient metal centers [64,65]. The second is their tunable steric properties, which can establish a library of N–N bisindolyl axially chiral ligands. The last is the newly formed ligands featuring an N–N bond axis, which can offer a different bite angle compared to other axially chiral diphosphines [66–71]. These varied angles may provide more possibilities in asymmetric catalysis.
We commenced our synthetic procedure with (2-nitrophenyl)methanol 2 and indoline 3, which are commercially available and cost as little as 0.05 $/g from Bide Pharmatech Ltd. (Scheme 2). A photo-induced N–N coupling produced aminoindole 4 [72], which was then converted to β-dibromo styrene 5 via a Wittig reaction with moderate yields. The subsequent intramolecular Ullmann coupling, catalyzed by copper and the BINOL system, smoothly facilitated the cyclization to yield the indolyl bromides 5, which could be readily installed with a diarylphosphine moiety to obtain 6 in satisfactory yields. With the diarylphosphine group as the directing group, the second diarylphosphine moiety can be installed at the 2′-position of indole, leading to the synthesis of racemic diphosphines 1.
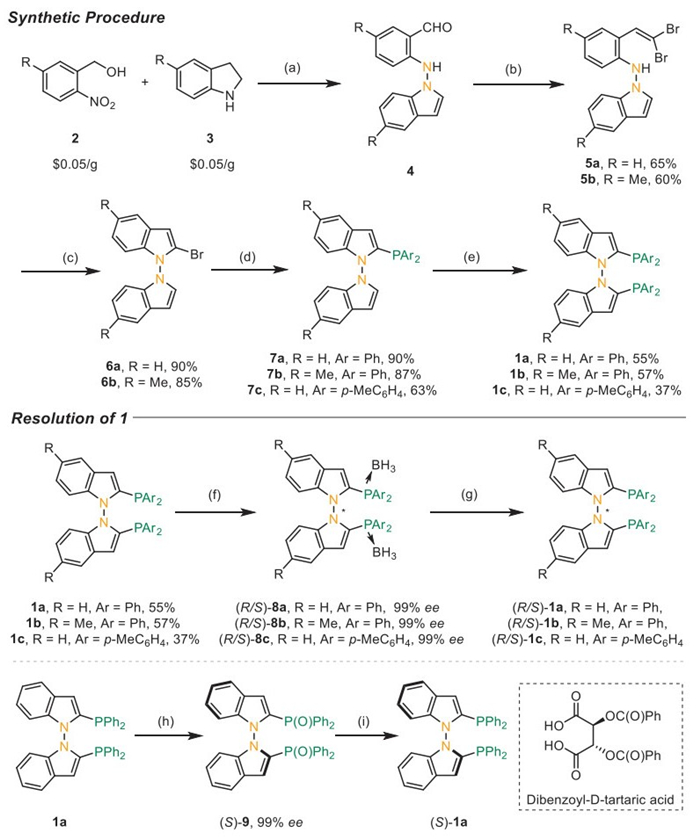
Two synthetic procedures were investigated to obtain the enantioenriched diphosphines. The first route involved converting 1 into diphosphine borates, which could then be separated using preparative chiral HPLC to yield enantiomers 8 with 99% ee. The final removal of the boron moiety with an organic base resulted in the synthesis of optically pure 1. Additionally, a kinetic resolution process was developed using diphosphate oxide obtained through the simple oxidation of diphosphine. By using dibenzoyl-D-tartaric acid as a chiral resolving agent, (S)-9 could be obtained with up to 99% ee, which could be easily reduced to diphosphine (S)-1a.
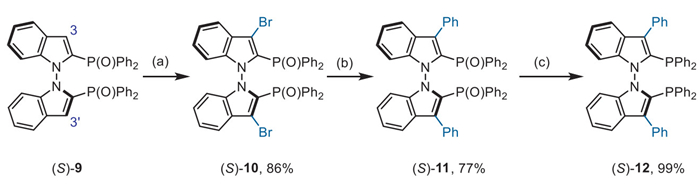
A synthetic route was developed to access 3,3′-substituted N–N atropisomeric diphosphanes (Scheme 3). The introduction of a bromine moiety was accomplished using KBr under oxidative conditions, resulting in the formation of dibromide (S)-10 in 86% yield. The subsequent Suzuki-Miyaura coupling proceeded smoothly to yield arylated compound (S)-11, which could then be readily reduced to compound (S)-12. Further modifications at the ortho position of the phosphorus moiety allow for enhanced tuning of steric hindrance and electronic properties.
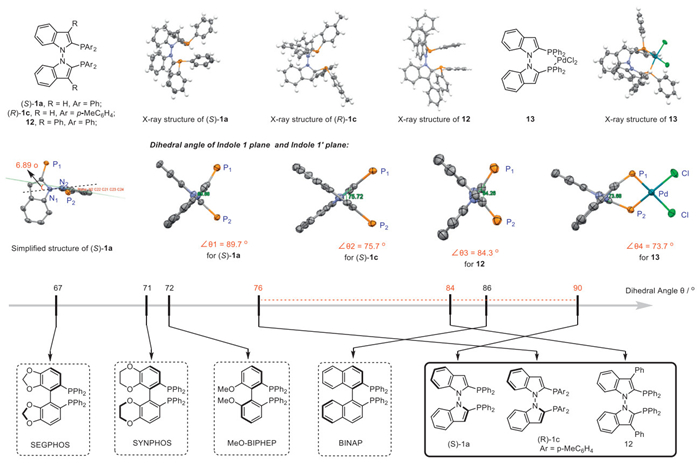
To further investigate the structure of the newly developed N–N atropisomeric diphosphine ligands, single crystals of (S)-1a, (R)-1c and 12 were cultivated and analyzed by X-ray diffraction (Fig. 1). Owing to their inherent properties and steric hindrance, the N–N axis exhibits a shorter distance of 1.383 Å compared to its C–C axis analogs [73,74], which connect two twisted indoles. From the simplified structure of (S)-1a, it is evident that the N1 atom deviates from the plane of the N2 installation by 6.89°, probably due to the repulsive effect of lone pair electrons on the N atom. Generally, the structure of (R)-1c and 12 is similar to that of (S)-1a in the solid state. A palladium complex was obtained from the reaction of rac-1a and PdCl2(MeCN)2, whose X-ray structure is shown below (Fig. 1). Considering the crucial role of the dihedral angle of biaryl diphosphines in enantioselective catalysis, the dihedral angles of the N–N atropisomeric ligands were measured in the solid state. For (S)-1a, the dihedral angle was found to be 89.7°, indicating that the second indole plane was almost perpendicular to the first indole plane. As the 3,3'-position was ocupied with phenyl groups, the dihedral angle was decreased slightly to 84.3°. When a methyl moiety was installed at the para position of the phenyl substituent, the dihedral angle decreased sharply to 75.7°, demonstrating excellent angle-tunable properties of the ligands. The trends of the variation of the dihedral angle was generally consistent with its BINAP anologues [75]. In the coordinating state, the dihedral angle of 13 could be further reduced to 73.7°. The novel N–N atropisomeric diphosphines exhibit excellent substituent-dependent tunable dihedral angles comparable to other useful electron-enriched diphosphines, suggesting potential applications in enantioselective catalysis. The racemization experiments were conducted with (S)-1a (99% ee), and no erosion of enantioselectivity was observed even heated in 150 ℃ for 72 h. The results showcase the newly developed N–N diphosphines bear a high configuration stability.
With the enantiopure N–N atropisomeric diphosphines in hand, we now aim to examine their stereoinduction abilities in enantioselective catalysis. Nonaromatic heterocycles featuring a stereocenter are valuable scaffolds in natural products and pharmaceuticals, the enantioselective synthesis of which is both challenging and appealing. Enantioselective dearomative functionalization including Rhodium-catalyzed dearomative arylation of pyridinium salts or quinolinium salts, represents a direct strategy to access these structures [76–83].
Under the catalysis of rhodium/N–N atropisomeric ligands 1 system, the enantioselective dearomative arylation of heterocycles proceeded smoothly with excellent enantioselectivities. When quinolinium salts were used, the addition of aryl boronic acid to facilitate enantioselective dearomative functionalization was conducted using [Rh(COD)Cl]2, (S)-1a, AgBF4, and K3PO4·3H2O in toluene. A series of substituted aryl boronic acids were applied to the protocol, yielding dihydroquinolines with excellent enantioselectivity (Scheme 4). When phenyl boronic acid was used, 15a was obtained in an 81% yield with 95% ee, which was as efficient as the Rh/(R)-BINAP system (79%, 95% ee) [79]. With ortho- or meta-substituted phenyl boronic acids, the enantioselective dearomative arylation proceeded smoothly, providing 15b and 15c in good yields with 93% ee and 97% ee, respectively. para-Substituted or 3,5-disubstituted phenyl boronic acids were also tested and yielded the desired products with excellent enantioselectivities (15d–15f).
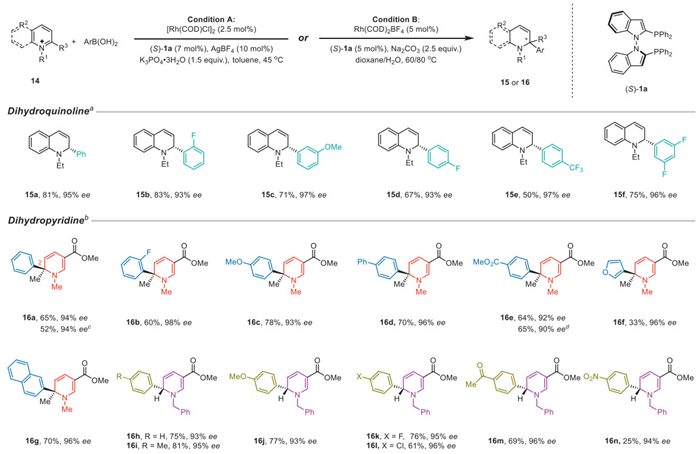
N-Alkylpyridinium salts were successfully applied to the methodology with conditions including Rh(COD)BF4, (S)-1a, and Na2CO3 in dioxane/H2O. When 2-methyl N-methylpyridinium salts were used as starting materials, the addition of phenyl boronic acid yielded 16a with a quaternary stereocenter in 65% yield with 94% ee. ortho-Substituted phenyl boronic acid was also used, resulting in the formation of 16b with a lower yield and 98% ee. Regardless of electron-rich or electron-deficient substituents at the para position of phenyl boronic acid, excellent enantioselectivities could be achieved (16c–16e). When (S)-12 was used instead of (S)-1a, 16e was obtained in 65% yield with a slight decreased enantioselectivity of 90%. A 3-furyl or 2-naphthyl moiety could also be installed with excellent enantioselectivities (16f–16g). N-Benzylpyridinium salts were found to be suitable substrates for enantioselective arylation, reacting with phenyl boronic acid to produce 16h in good yield with 93% ee. When para-methyl or para-methoxyl phenyl boronic acid was used, satisfactory enantioselectivities were obtained (16i–16j). para-Substituents of phenyl boronic acid including -F, -Cl, and -C(O)Me were tested, resulting in the formation of desired products with excellent enantioselectivities (16k–16m). para-Nitro substituted phenyl boronic acid was also found to be a suitable reactant, achieving excellent enantioselectivities, albeit with a 25% yield (16n).
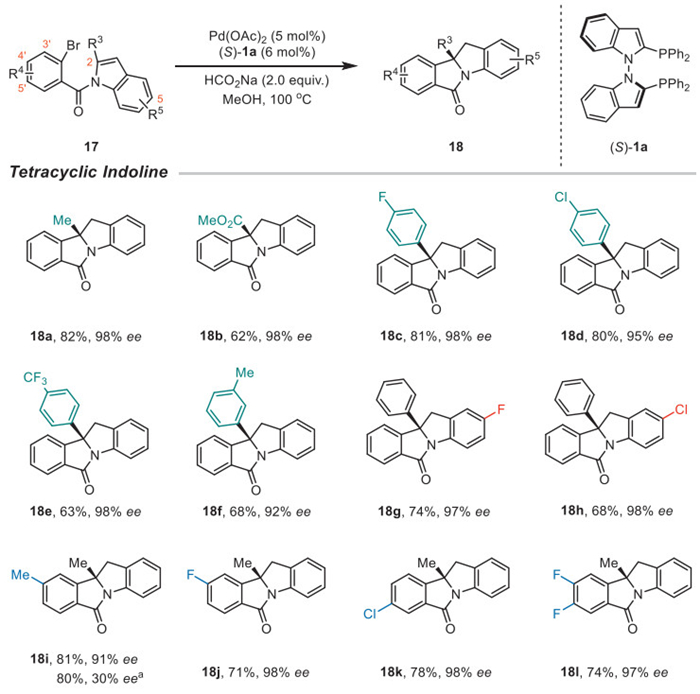
The palladium-catalyzed dearomative arylation of indoles provides an efficient method to access chiral indolines with a quaternary stereocenter [84–86]. The enantioselective dearomative arylation of 17 was carried out using Pd(OAc)2, (S)-1a, and HCO2Na in MeOH at 100 ℃, resulting in the formation of cyclized products 18 with excellent enantioselectivities (Scheme 5). When 2-methyl indoles were used as starting materials, the reductive arylation proceeded smoothly, yielding 18a in 82% yield with 98% ee. The result was comparative to the Pd/(R)-BINAP system (81%, 98% ee) reported by Jia et al. [84]. Additionally, 2- methyl ester indole was tested and yielded 18b with 98% ee. para-Halo substituted phenyl groups at the 2-position of indoles were used, providing satisfactory yields and enantioselectivities (18c–18d). para-CF3 or meta-Me substituted phenyl groups were tolerated and yielded products with over 92% ee (18e–18f). 5-F or 5-Cl substituted indoles were compatible with the protocol, exhibiting excellent stereoselectivities (18g–18h). Substituents at the 4′-position of the phenyl moiety, including -Me or -F, were also tolerated, resulting in cyclized products with excellent enantioselectivities (18i–18j). When the ortho substituted diphosphines (S)-12 was used, a sharp drop of enantioselectivity (30% ee) was got. The result indicates that the increased steric hinderance was not suitable for the procedure. Substrates with 5′-Cl or 4′, 5′-F2 yield 18k and 18l with 98% ee and 97% ee, respectively.
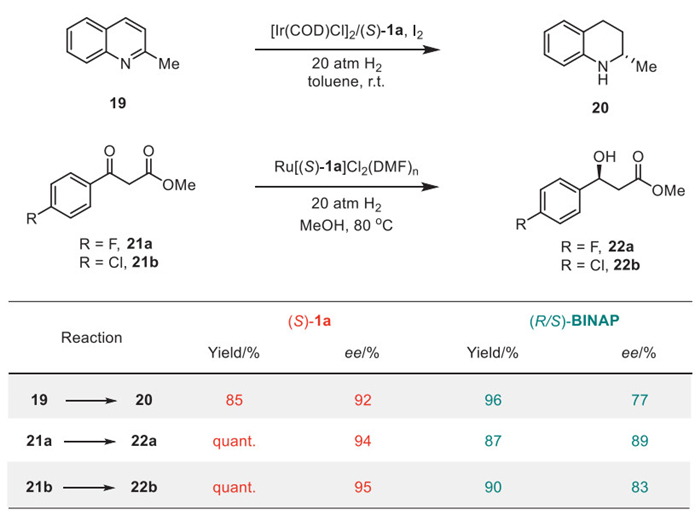
Furthermore, we examined the stereo-induction ability of the newly developed N–N atropisomeric diphosphines 1 in enantioselective hydrogenations of aromatic heterocycles or ethyl benzoylacetates (Scheme 6). We conducted an iridium-catalyzed enantioselective hydrogenation of quinoline using (S)-1a [86–88]. Tetrahydroquinoline 20 was obtained with an 85% yield and improved enantioselectivity compared to BINAP (92% ee vs. 77% ee) [88]. Next, an enantioselective ruthenium-catalyzed hydrogenation of β-keto ester 21 was conducted [89,90]. With the aid of (S)-1a, the enantioselective reduction proceeded smoothly to yield 22 with over 94% ee, demonstrating a comparable stereo-induction ability to BINAP [90].
In conclusion, we have successfully synthesized C2-symmetric N–N atropisomeric diphosphines in a few steps and obtained enantiopure diphosphines through preparative chiral HPLC or chiral resolving agent. Structural analysis of single crystals of the diphosphines and the palladium complex revealed that the newly developed ligands exhibit excellent substituent-dependent tunable dihedral angles that are comparable to those of other useful electron-enriched diphosphines. Furthermore, the diphosphines were used as chiral ligands in transition-metal catalyzed enantioselective dearomatization of heteroaryls, resulting in products with excellent enantioselectivities. Additionally, the diphosphines demonstrated remarkable stereoinduction abilities in enantioselective hydrogenations of aromatic heterocycles and β-keto esters.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Xiao-Kai Li: Methodology, Investigation. Si-Hao Fu: Methodology, Investigation, Formal analysis. Yi Yue: Methodology. Rui-Jing Pang: Methodology. Jia Feng: Writing – original draft, Project administration. Ren-Rong Liu: Writing – review & editing, Supervision, Project administration, Conceptualization.
We are grateful for the generous support of the National Natural Science Foundation of China (No. 22371152, 22271169), Natural Science Foundation of Shandong Province (Nos. ZR2023JQ006, ZR2024YQ015), Taishan Scholar Youth Expert Program in Shandong Province (No. tsqn 202312165) and Qingdao Natural Science Foundation (No. 23-2-1-244-zyyd-jch).
Supplementary material associated with this article can be found, in the online version, at doi:
G. Bringmann, T. Gulder, T.A.M. Gulder, M. Breuning, Chem. Rev. 111 (2011) 563–639. doi: 10.1021/cr100155e
J.K. Cheng, S.H. Xiang, S. Li, L. Ye, B. Tan, Chem. Rev. 121 (2021) 4805–4902. doi: 10.1021/acs.chemrev.0c01306
G.J. Mei, W.L. Koay, C.Y. Guan, Y. Lu, Chem 8 (2022) 1855–1893. doi: 10.1016/j.chempr.2022.04.011
B. Zilate, A. Castrogiovanni, C. Sparr, ACS Catal. 8 (2018) 2981–2988. doi: 10.1021/acscatal.7b04337
P. Rodríguez-Salamanca, R. Fernández, V. Hornillos, J.M. Lassaletta, Chem. Eur. J. 28 (2022) e202104442. doi: 10.1002/chem.202104442
C.X. Liu, W.W. Zhang, S.Y. Yin, Q. Gu, S.L. You, J. Am. Chem. Soc. 143 (2021) 14025–14040. doi: 10.1021/jacs.1c07635
G. Centonze, C. Portolani, P. Righi, G. Bencivenni, Angew. Chem. Int. Ed. 62 (2023) e202303966. doi: 10.1002/anie.202303966
T.Y. Song, R. Li, L. Huang, S.K. Jia, G.J. Mei, Chin. J. Org. Chem. 43 (2023) 1977–1990. doi: 10.6023/cjoc202212003
G.J. Mei, J.J. Wong, W. Zheng, et al., Chem 7 (2021) 2743–2757. doi: 10.1016/j.chempr.2021.07.013
X.M. Wang, P. Zhang, Q. Xu, et al., J. Am. Chem. Soc. 143 (2021) 15005–15010. doi: 10.1021/jacs.1c07741
Q. Xu, H. Zhang, F.B. Ge, et al., Org. Lett. 24 (2022) 3138–3143. doi: 10.1021/acs.orglett.2c00812
W. Yao, C.J. Lu, L.W. Zhan, et al., Angew. Chem. Int. Ed. 62 (2023) e202218871. doi: 10.1002/anie.202218871
W.T. Wang, S. Zhang, W. Lin, et al., Org. Chem. Front. 11 (2024) 3308–3319. doi: 10.1039/d4qo00294f
W. Lin, Q. Zhao, Y. Li, et al., Chem. Sci. 13 (2022) 141–148. doi: 10.1039/d1sc05360d
M. Pan, Y.B. Shao, Q. Zhao, X. Li, Org. Lett. 24 (2022) 374–378. doi: 10.1021/acs.orglett.1c04028
T.J. Han, C.Y. Guan, N. Li, et al., iScience 26 (2023) 107978. doi: 10.1016/j.isci.2023.107978
C. Song, C. Pang, Y. Deng, et al., ACS Catal. 14 (2024) 6926–6935. doi: 10.1021/acscatal.4c00720
Z. Huang, Y. Xu, W. Lin, et al., Org. Chem. Front. 11 (2024) 5437–5442. doi: 10.1039/d4qo01056f
X. Wang, S.J. Wang, X. Xin, et al., Chem. Sci. 15 (2024) 13240–13249. doi: 10.1039/d4sc03190c
K.W. Chen, Z.H. Chen, S. Yang, et al., Angew. Chem. Int. Ed. 61 (2022) e202116829. doi: 10.1002/anie.202116829
P. Zhang, Q. Xu, X.M. Wang, et al., Angew. Chem. Int. Ed. 61 (2022) e202212101. doi: 10.1002/anie.202212101
L.Y. Pu, Y.J. Zhang, W. Liu, F. Teng, Chem. Commun. 58 (2022) 13131–13134. doi: 10.1039/d2cc05172a
Y. Gao, L.Y. Wang, T. Zhang, B.M. Yang, Y. Zhao, Angew. Chem. Int. Ed. 61 (2022) e202200371. doi: 10.1002/anie.202200371
Y. Wei, F. Sun, G. Li, et al., Org. Lett. 26 (2024) 2343–2348. doi: 10.1021/acs.orglett.3c03280
X. Zhu, H. Wu, Y. Wang, et al., Chem. Sci. 14 (2023) 8564–8569. doi: 10.1039/d3sc02800c
Z.H. Chen, T.Z. Li, N.Y. Wang, et al., Angew. Chem. Int. Ed. 62 (2023) e202300419. doi: 10.1002/anie.202300419
S.S. Ranganathappa, B.S. Dehury, G.K. Singh, S. Shee, A.T. Biju, ACS Catal. 14 (2024) 6965–6972. doi: 10.1021/acscatal.4c00513
J. Wang, D. Pan, F. Wang, et al., Sci. Adv. 10 (2024) eado4489. doi: 10.1126/sciadv.ado4489
Q. Huang, Y. Li, C. Yang, et al., Org. Chem. Front. 11 (2024) 726–734. doi: 10.1039/d3qo01877f
V. Hutskalova, C. Sparr, Synthesis 55 (2023) 1770–1782. doi: 10.1055/a-1993-6899
S.Y. Yin, Q. Zhou, C.X. Liu, Q. Gu, S.L. You, Angew. Chem. Int. Ed. 62 (2023) e202305067. doi: 10.1002/anie.202305067
T.T. Wang, J. Cao, X. Li, Org. Lett. 26 (2024) 6179–6184. doi: 10.1021/acs.orglett.4c02031
T. Benincori, E. Brenna, F. Sannicolo, et al., Organomet. Chem. 529 (1997) 445–453. doi: 10.1016/S0022-328X(96)06682-X
A.L. Clevenger, R.M. Stolley, J. Aderibigbe, J. Louie, Chem. Rev. 120 (2020) 6124–6196. doi: 10.1021/acs.chemrev.9b00682
M.J. Burk, J. Am. Chem. Soc. 113 (1991) 8518–8519. doi: 10.1021/ja00022a047
C.C. Pai, C.W. Lin, C.C. Lin, et al., J. Am. Chem. Soc. 122 (2000) 11513–11514. doi: 10.1021/ja000163n
S. Duprat de Paule, S. Jeulin, V. Ratovelomanana-Vidal, et al., Org. Process Res. Dev. 7 (2003) 399–406. doi: 10.1021/op034016w
T. Imamoto, K. Sugita, K. Yoshida, J. Am. Chem. Soc. 127 (2005) 11934–11935. doi: 10.1021/ja053458f
D. Liu, F. Xie, X. Zhao, W. Zhang, Tetrahedron 64 (2008) 3561–3566. doi: 10.1016/j.tet.2008.01.108
W. Zhou, X. Su, M. Tao, et al., Angew. Chem. Int. Ed. 54 (2015) 14853–14857. doi: 10.1002/anie.201508108
Y. Xu, Y. Luo, J. Ye, D. Liu, W. Zhang, J. Am. Chem. Soc. 145 (2023) 21176–21182. doi: 10.1021/jacs.3c07509
K.B. Gan, R.L. Zhong, Z.W. Zhang, F.Y. Kwong, J. Am. Chem. Soc. 144 (2022) 14864–14873. doi: 10.1021/jacs.2c06240
T.P. Dang, H.B. Kagan, J. Chem. Soc., Chem. Commun. (1971) 481.
W. Dumont, J.C. Poulin, P.D. Tuan, H.B. Kagan, J. Am. Chem. Soc. 95 (1973) 8295–8299. doi: 10.1021/ja00806a015
J.H. Xie, L.X. Wang, Y. Fu, et al., J. Am. Chem. Soc. 125 (2003) 4404–4405. doi: 10.1021/ja029907i
X. Wang, P. Guo, Z. Han, et al., J. Am. Chem. Soc. 136 (2014) 405–411. doi: 10.1021/ja410707q
X. Wang, P. Guo, X. Wang, Z. Wang, K. Ding, Adv. Synth. Catal. 355 (2013) 2900–2907. doi: 10.1002/adsc.201300380
X. Wang, F. Meng, Y. Wang, et al., Angew. Chem. Int. Ed. 51 (2012) 9276–9282. doi: 10.1002/anie.201204925
X. Wang, Z. Han, Z. Wang, K. Ding, Angew. Chem. Int. Ed. 51 (2012) 936–940. doi: 10.1002/anie.201106488
G.Q. Chen, J.M. Huang, B.J. Lin, et al., CCS Chem. 2 (2020) 468–477. doi: 10.31635/ccschem.020.202000176
W. Tang, X. Zhang, Angew. Chem. Int. Ed. 41 (2002) 1612–1614. doi: 10.1002/1521-3773(20020503)41:9<1612::AID-ANIE1612>3.0.CO;2-H
F. Wan, W. Tang, Chin. J. Chem. 39 (2021) 954–968. doi: 10.1002/cjoc.202000605
G. Liu, X. Liu, Z. Cai, et al., Angew. Chem. Int. Ed. 52 (2013) 4235–4238. doi: 10.1002/anie.201300646
T. Saito, T. Yokozawa, T. Ishizaki, et al., Adv. Synth. Catal. 343 (2001) 264–267. doi: 10.1002/1615-4169(20010330)343:3<264::AID-ADSC264>3.0.CO;2-T
S. Duprat de Paule, S. Jeulin, V. Ratovelomanana-Vidal, et al., Tetrahedron Lett. 44 (2003) 823–826. doi: 10.1016/S0040-4039(02)02637-0
T. Baumann, R. Brückner, Angew. Chem. Int. Ed. 58 (2019) 4714–4719. doi: 10.1002/anie.201806294
U. Berens, J.M. Brown, J. Long, R. Selke, Tetrahedron: Asymmetry 7 (1996) 285–292. doi: 10.1016/0957-4166(95)00447-5
T. Benincori, E. Brenna, F. Sannicolò, et al., J. Org. Chem. 61 (1996) 6244–6251. doi: 10.1021/jo960211f
T. Benincori, O. Piccolo, S. Rizzo, F. Sannicoló, J. Org. Chem. 65 (2000) 8340–8347. doi: 10.1021/jo001207d
A. Miyashita, A. Yasuda, H. Takaya, et al., J. Am. Chem. Soc. 102 (1980) 7932–7934. doi: 10.1021/ja00547a020
A. Miyashita, H. Takaya, R. Noyori, ACS Symp. Ser. 185 (1982) 274–277.
A. Miyashita, H. Takaya, T. Souchi, R. Noyori, Tetrahedron 40 (1984) 1245–1253. doi: 10.1016/S0040-4020(01)82411-X
H. Takaya, S. Akutagawa, R. Noyori, Org. Synth. 67 (1989) 20–32. doi: 10.15227/orgsyn.067.0020
M. Murata, T. Morimoto, K. Achiwa, Synlett (1991) 827–829.
T. Benincori, O. Piccolo, S. Rizzo, F. Sannicolò, J. Org. Chem. 65 (2000) 8340–8347. doi: 10.1021/jo001207d
M.N. Birkholz, Z. Freixa, P.W.N.M. van Leeuwen, Chem. Soc. Rev. 38 (2009) 1099–1118.
D.J. Durand, N. Fey, Chem. Rev. 119 (2019) 6561–6594. doi: 10.1021/acs.chemrev.8b00588
Y. Xu, Y. Luo, J. Ye, et al., J. Am. Chem. Soc. 144 (2022) 20078–20089. doi: 10.1021/jacs.2c09266
Y. Xu, D. Liu, Y. Deng, Y. Zhou, W. Zhang, Angew. Chem. Int. Ed. 60 (2021) 23602–23607. doi: 10.1002/anie.202110286
S. Jeulin, S. Duprat de Paule, V. Ratovelomanana-Vidal, et al., Angew. Chem. Int. Ed. 43 (2004) 320–325. doi: 10.1002/anie.200352453
S.D. de Paule, S. Jeulin, V. Ratovelomanana-Vidal, et al., Eur. J. Org. Chem. (2003) 1931–1941.
Y. Ou, T. Yang, N. Tang, et al., Org. Lett. 23 (2021) 6417–6422. doi: 10.1021/acs.orglett.1c02227
M.B. Smith, J. March, Advanced Organic Chemistry, Wiley & Sons, Inc., Hoboken, New Jersey, 2006.
J. Feng, C.J. Lu, R.R. Liu, Acc. Chem. Res. 56 (2023) 2537–2554. doi: 10.1021/acs.accounts.3c00419
M. Berthod, G. Mignani, G. Woodward, M. Lemaire, Chem. Rev. 105 (2005) 1801–1836. doi: 10.1021/cr040652w
M. Escolano, D. Gaviña, G. Alzuet-Piña, et al., Chem. Rev. 124 (2024) 1122–1246. doi: 10.1021/acs.chemrev.3c00625
C. Nadeau, S. Aly, K. Belyk, J. Am. Chem. Soc. 133 (2011) 2878–2880. doi: 10.1021/ja111540g
D.J. Robinson, S.P. Spurlin, J.D. Gorden, R.R. Karimov, ACS Catal. 10 (2020) 51–55. doi: 10.1021/acscatal.9b03874
Y. Wang, Y. Liu, D. Zhang, et al., Angew. Chem. Int. Ed. 55 (2016) 3776–3780. doi: 10.1002/anie.201511663
K.G. Ortiz, J.J. Dotson, D.J. Robinson, M.S. Sigman, R.R. Karimov, J. Am. Chem. Soc. 145 (2023) 11781–11788. doi: 10.1021/jacs.3c03048
Y. Xiao, Z.Y. Zhao, S. Kemper, E. Irran, M. Oestreich, Angew. Chem. Int. Ed. 63 (2024) e202407056. doi: 10.1002/anie.202407056
S. Somprasong, M.C. Reis, S.R. Harutyunyan, Angew. Chem. Int. Ed. 62 (2023) e202217328. doi: 10.1002/anie.202217328
X. Yan, L. Ge, M.C. Reis, S.R. Harutyunyan, J. Am. Chem. Soc. 142 (2020) 20247–20256. doi: 10.1021/jacs.0c09974
C. Shen, R.R. Liu, R.J. Fan, et al., J. Am. Chem. Soc. 137 (2015) 4936–4939. doi: 10.1021/jacs.5b01705
W.Y. Zhang, H.C. Wang, Y. Wang, C. Zheng, S.L. You, J. Am. Chem. Soc. 145 (2023) 10314–10321. doi: 10.1021/jacs.3c01994
X. Yu, C. Zheng, S.L. You, J. Am. Chem. Soc. 146 (2024) 25878–25887. doi: 10.1021/jacs.4c09814
Z.W. Li, T.L. Wang, Y.M. He, et al., Org. Lett. 10 (2008) 5265–5268. doi: 10.1021/ol802016w
L.S. Zheng, F. Wang, X.Y. Ye, G.Q. Chen, X. Zhang, Org. Lett. 22 (2020) 8882–8887. doi: 10.1021/acs.orglett.0c03261
R. Noyori, T. Ohkuma, M. Kitamura, H. Takaya, J. Am. Chem. Soc. 109 (1987) 5856–5858. doi: 10.1021/ja00253a051
V. Ratovelomanana-Vidal, C. Girard, R. Touati, et al., Adv. Synth. Catal. 345 (2003) 261–274. doi: 10.1002/adsc.200390021

Scheme 2 Synthetic approach toward C2-symmetric N–N atropisomeric diphosphanes. Reaction conditions: (a) TEAI (1.0 equiv.), AcOH (1.0 equiv.) in DCM at room temperature with a 24 W UV LEDs (385−405 nm) irradiation under air, (b) CBr4 (2.0 equiv.), PPh3 (2.0 equiv.), Zn (8.0 equiv.) in DCM at 0 ℃ for 12 h; (c) CuI (20 mol%), rac-BINOL (40 mol%), K2CO3 (2.0 equiv.) in toluene at 100 ℃ for 12 h; (d) PAr2Cl (1.5 equiv.), nBuLi (1.4 equiv.) in THF at −78 ℃ for 12 h; (e) PAr2Cl (1.5 equiv.), LDA (1.5 equiv.), LiCl (12 equiv.) in THF at −78 ℃ for 24 h; (f) (i) BH3.THF (4.0 equiv.), THF at room temperature for 10 min; (ii) chiral preparative HPLC, conditions: CHIRALPAK IG, Eluent: Hexane/DCM 70:30, Flow rate: 1.0 mL/min; (g) Et2NH (20.0 equiv.) at room temperature for 10 min; (h) (i) H2O2 (4.0 equiv.) in DCE at room temperature; (ii) Dibenzoyl-D-tartaric acid (1.1 equiv.) in Et2O/DCM; (i) HSiCl3 (5.0 equiv.), Et3N (5.5 equiv.) in toluene at 120 ℃ for 10 h.

Scheme 3 Further modification at the ortho position of the phosphorous moiety. Reaction conditions: (a) KBr (2.5 equiv.), Oxone (2.0 equiv.) in DCE at 70 ℃ for 18 h; (b) Pd(PPh3)4 (10 mol%), Na2CO3 (2.0 equiv.) in a mixture of toluene/EtOH/H2O at 110 ℃; (c) HSiCl3 (5.0 equiv.), Et3N (5.5 equiv.) in toluene at 120 ℃ for 10 h.

Scheme 4 Enantioselective dearomative arylation of pyridinium salts and quinolinium salts. a Reaction conditions: 14 (0.1 mmol), ArB(OH)2 (0.2 mmol), [Rh(COD)Cl]2 (2.5 mol%), (S)-1a (7.0 mol%), AgBF4 (10 mol%), and K3PO4·3H2O (1.5 equiv.) in toluene (3 mL) at 45 ℃ for 12 h. b Reaction conditions: 14 (0.1 mmol), ArB(OH)2 (0.25 mmol), Rh(COD)BF4 (5 mol%), (S)-1a (5 mol%), and Na2CO3 (2.0 equiv.) in solvent at 60/80 ℃, for N-methylpyridinium salts: in dioxane (0.5 mL) and water (0.125 mL) at 80 ℃ for 2 h; for N-benzylpyridinium salts: in dioxane (10 mL/g) and water (1.0 mL/g) at 60 ℃ for 3 h. c (S)-1a was acidized with diluted HCl. d (S)-12 was used instead of (S)-1a.

Scheme 5 Enantioselective dearomative cyclization of indoles. Reaction conditions: 17 (0.1 mmol), Pd(OAc)2 (5 mol%), (S)-1a (6 mol%), and HCO2Na (2.0 equiv.) in MeOH (1.0 mL) at 100 ℃ for 10–16 h. a (S)-12 was used instead of (S)-1a.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:


 下载:
下载: