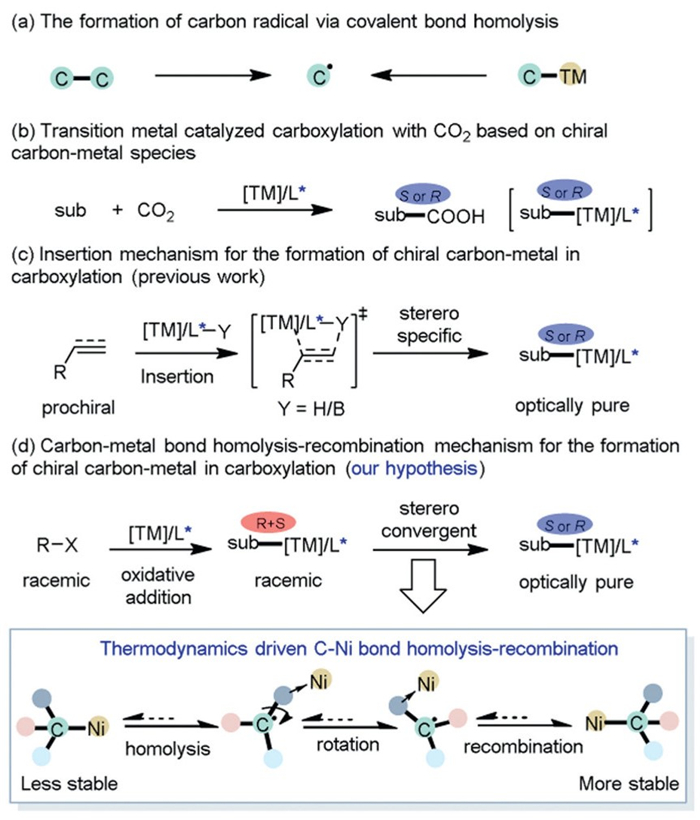
Scheme 1.
The formation of chiral carbon-metal species in transition metal-catalyzed carboxylation with CO2. TM = transition metal, L = ligand, sub = substrate.
Carbon–metal bond homolysis-recombination enabling enantioconvergent carboxylation with CO2: A theoretical study
Qi Zhou , Cefei Zhang , Hui-Lin Luo , Chuan-Xi Nie , Changwei Hu , Jian-Heng Ye , Zhishan Su , Li-Li Liao , Da-Gang Yu

Radicals are exceptionally reactive intermediates that readily engage with a wide array of organic molecules [1–4]. Their defining characteristic lies in their rapid reactivity, often occurring at diffusion-limited rates. While this property enables efficient substrate activation, it also presents challenges in achieving selective control, particularly in the context of enantioselective transformations. Among these, the carbon radical has been established as a crucial intermediate for C–C bond formation, attracting significant attention [5–7]. Covalent bond homolysis has been demonstrated as a versatile method for carbon radical generation (Scheme 1a) [8–16]. Homolytic cleavage of a C–C bond by high-temperature thermolysis or ultraviolet irradiation has been employed to generate radical pairs [8,9]. Similarly, homolytic cleavage of a carbon-metal bond in alkyl metal compounds may take place, making it prone to generating transient radical species [10–15]. Especially when the metals exhibit spin diversity, such as Fe [11], Co [12], and Ni [13,14] in the early transition metals of the fourth period, the phenomenon becomes particularly noteworthy. The generation of transient radical species via carbon–metal bond homolysis is highly valuable, as it can be strategically leveraged to facilitate selective and efficient radical-type transformations in conjunction with transition metal catalysis [16,17].
Recently, transition metals, including Ni, Cu, Rh, and Pd, catalyzed asymmetric carboxylation with carbon dioxide (CO2) have attracted much attention as a complementary method to construct important chiral carboxylic acids (Scheme 1b) [18–22]. These transformations usually involve the generation of an optically pure enantiomeric carbon-metal species as the key intermediate for CO2 transformation and enantioselectivity control. From a theoretical perspective, π-bond insertion of a prochiral substrate into a metal-hydride/boron bond stereospecifically generates an optically pure enantiomeric carbon-metal species efficiently [23–26]. In this case, substrates such as alkenes and alkynes can participate in transition-metal catalyzed asymmetric carboxylation with CO2 pioneered by Mori [27], Mikami [28], Yu [26,29–34], Tang, and Zhou [35,36] groups, by constructing alkyl- or alkenyl–metal species that can be directly transformed into various central or axial chiral carboxylic acids (Scheme 1c).
In addition to the π-bond insertion strategy which is effective for both alkenes and alkynes in generating carbon-metal species, an alternative approach known as oxidative addition facilitates the formation of carbon-metal species from electrophiles such as alkyl (pseudo)halides [30,37–39]. Nevertheless, oxidative addition of a racemic alkyl (pseudo)halide to a low-valent transition metal usually results in the generation of a racemic carbon-metal species. One potential way to obtain optically pure enantiomeric carbon-metal species may involve the homolysis of the carbon-metal bond. The homolytic process is inherently thermodynamically endergonic, resulting in the formation of a transient radical. The transient nature and inherent reactivity of the radical facilitate the dynamic rotation along the C–C• bond achieving an isomerization process. After recombination with metal species, another stereo-configurated carbon-metal species can be generated. This means upon generation of racemic carbon-metal species, one enantiomer may convert into another more thermodynamically stable one. We propose that applying this protocol to organo (pseudo)halides could provide chiral carboxylated products by the generation of optically pure enantiomeric carbon–metal species. Herein, we report the theoretical study on enantioconvergent carboxylation with CO2 based on carbon-metal bond homolysis/recombination model (Scheme 1d).
To address the above issues, we conducted theoretical investigations and chose Ni-catalyzed enantioconvergent carboxylation of alkyl ammonium salts with CO2 reported by Li’s research group as the model reaction (Scheme 2a) [40]. Their experimental exploration showed that this carboxylation could be achieved in the presence of a Ni catalyst, the chiral ligand Me–SBpy, and manganese (Mn) powder under one atmosphere of CO2. It was found that the excellent yields and enantioselectivities were almost obtained for two separate reactions whether using (S)- or (R)-alkyl ammonium salt 1a, indicating a racemization step before generating a chiral alkyl Ni complex. This study aims to verify the carbon-metal bond homolysis/recombination model for Ni-catalyzed enantioconvergent carboxylation with CO2, providing direction for the rational design of Ni-catalyzed enantioconvergent carboxylation reactions, which are attractive for synthetic applications.
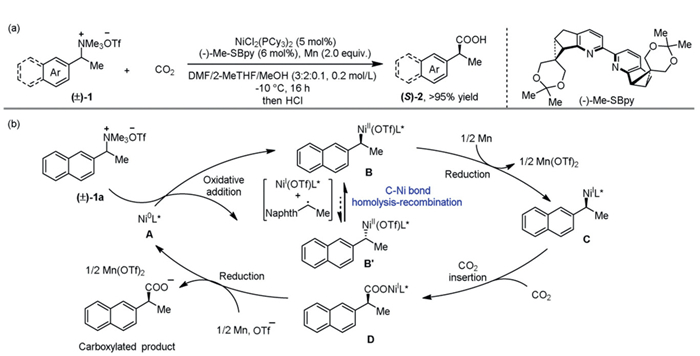
On the basis of literature [40,41] and our proposed model, we proposed the mechanistic cycle for the Ni-catalyzed enantioconvergent carboxylation of alkyl ammonium salts with CO2, as shown in Scheme 2b. In this process, Ni(0) complex is considered to the active catalytic species. Oxidative addition of the racemic ammonium salt 1a to Ni(0) complex A generates benzylic Ni(Ⅱ) intermediates B and B’ as two enantiomers. Then, C–Ni bond homolysis-recombination occurs, facilitating the formation of a thermodynamically stable enantiomer. The resulting optically pure enantiomeric Ni(Ⅱ) complex could be reduced by Mn(0) to generate Ni(Ⅰ) species C. This is followed by the insertion of CO2 into the C–Ni(Ⅰ) bond, leading to the formation of Ni(Ⅰ) carboxylate intermediate D. Further reduction then regenerates the Ni(0) species A and releases the carboxylated product.
All structures in this study were optimized using the Gaussian 09 package [42], employing the B3LYP-D3(BJ) [43,44] functional in the N,N-dimethylformamide (DMF) solvent. The relative effective core potential (ECP) in combination with the LanL2DZ [45–47] basis set was employed for Ni and Mn atoms, whereas the 6–31G(d) basis set [48] was assigned to all remaining atoms. Thermodynamic energy corrections were obtained at 298 K. Frequency analysis was conducted to confirm that the optimized geometries corresponded to a minimum (with no imaginary frequencies) or a transition state (with exactly one imaginary frequency). The solvent effect was evaluated using the self-consistent reaction field (SCRF) method and the SMD model [49]. Intrinsic reaction coordinate (IRC) [50] calculations were performed to verify the transition state (TS) associated with the corresponding minimum on the potential energy surface. Single-point energy calculations were carried out at the B3LYP-D3(BJ)/[SDD, 6–311+G(d,p)](SMD, DMF) level of theory [51,52]. Optimized molecular structures were visualized with CYLview (version 2.0) [53]. To gain further insight into the electron properties of key species in the reaction, natural bond orbital (NBO) analysis was carried out at the same level of theory as the structural optimization [54,55]. The extended transition state-natural orbitals for chemical valence (ETS-NOCV) [56–59] analysis was performed with Multiwfn 3.8 (dev) software [60], and the corresponding results were visualized using VMD software [61].
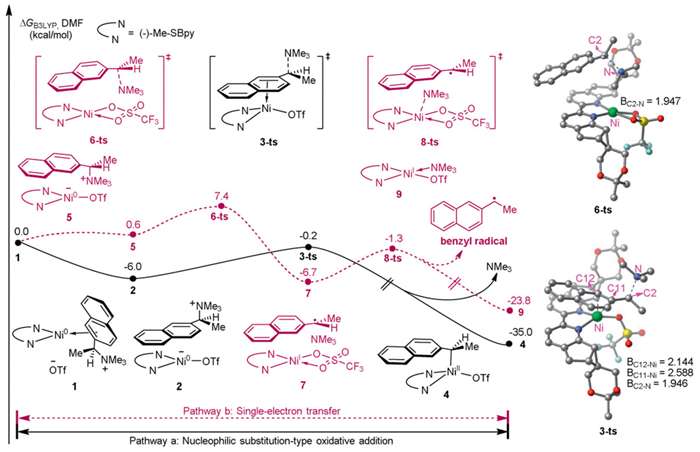
A detailed density functional theory (DFT) calculation was performed to investigate the mechanism of the Ni-catalyzed enantioconvergent carboxylation, which was initiated with oxidative addition. As shown in Fig. 1, the Ni(0) complex 1 generated by coordination with (S)-ammonium salt 1a was considered as the zero point of the free energy profiles. Owing to the features of the Ni(0) complex and the alkyl (pseudo)halides [62–64], two possible pathways for the oxidative addition of C(benzyl)–N bond were proposed and investigated, including nucleophilic substitution-type C–N oxidative addition (pathway a) and single-electron transfer (pathway b) [65]. We failed to locate three membered-ring oxidative addition transition state, likely due to steric hindrance. First, we calculated the activation free energy for the nucleophilic substitution-type C(benzyl)–N bond oxidative addition with the Ni(0) complex to deliver benzyl-Ni(Ⅱ) complex 4 (pathway a). This nucleophilic substitution step via transition state 3-ts, featuring a triplet state at the Ni center, has an energy barrier of 5.8 kcal/mol. Geometry analysis shows that the length of C2–N, C11–Ni, C12–Ni bonds in 3-ts are 1.946, 2.588, and 2.144 Å, respectively, indicating a coordination of Ni center to naphthalene group. Pathway b involves a radicophilic Ni(0) center [63], which undergoes single-electron transfer to form a benzyl radical through the breaking of the C(benzyl)–N bond. The relative free energy of 6-ts is calculated as 7.4 kcal/mol. Geometry analysis shows that the length of C2–N bond in 6-ts is 1.947 Å, and the spin population of Ni atom and the total benzyl group is −0.907 and 0.562, respectively. The relative free energy of transition state 3-ts is 7.6 kcal/mol lower than that of transition state 6-ts. Therefore, nucleophilic substitution-type oxidative addition via 3-ts is the dominant pathway according to our computational results, being exoergic by 35.0 kcal/mol for the generation of (R)-benzylic Ni(Ⅱ) intermediate 4 (Figs. S2 in Supporting information).
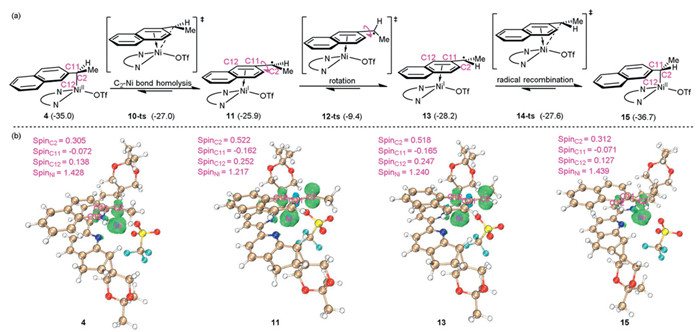
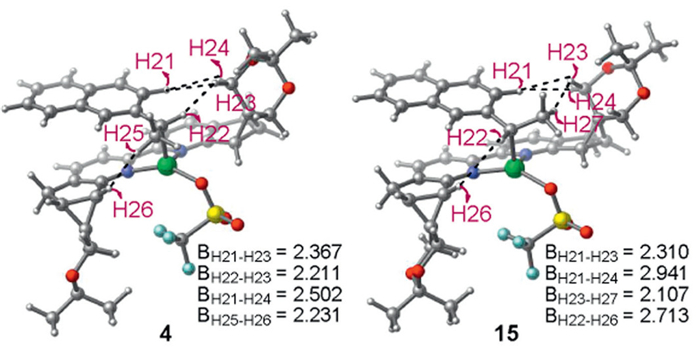
We then investigated how the enantiomeric carbon-metal species was formed. Our calculations using (S)-ammonium salt 1a ligated with Ni(0) species demonstrate the generation of benzyl Ni(Ⅱ) complex 4 via configuration reversal nucleophilic substitution-type oxidative addition transition state 3-ts. In experimental observations, TEMPO-benzyl adduct and benzyl radical homocoupling side product were detected in the reaction system mechanistic studies, indicating the formation of a benzyl radical. We proposed that the homolysis of C2–Ni bond in 4 could form benzyl radical and Ni(Ⅰ) species. This process via aryl-ring coordinated C–Ni bond homolysis transition state 10-ts requires an energy barrier of 8.0 kcal/mol, leading to the formation of Ni(Ⅰ) species 11. Geometry analysis shows that the lengths of C2–Ni, C11–Ni and C12–Ni bonds in 11 are 3.091, 2.575 and 2.153 Å, respectively, indicating an η2-coordination of Ni center (Fig. S4c in Supporting information). Moreover, spin population analysis of 11 shows spin densities of 0.522, −0.162, 0.252 and 1.217 on the C2, C11, C12 and Ni atoms, respectively, indicating the radical character of benzyl fragment. Furthermore, we located the transition states 12-ts associated with the C–C bond rotation in intermediate 11. The energy barrier via 12-ts is 16.5 kcal/mol, indicating that the process is kinetically feasible (Fig. S7 in Supporting information). These results mean C–C• bond rotation can take place, establishing a pre-equilibrium between the 13 and 11 species [66]. The endergonic process makes it difficult for the transient radical to be released, thus enabling rapid recombination and aromatization between the benzyl radical and Ni(Ⅰ) center. This step generates benzyl Ni(Ⅱ) complex 15 via aryl-ring coordinated recombination transition state 14-ts forming C–Ni bond, which has an energy barrier of 0.6 kcal/mol. Both geometry and spin population analysis reveal the radical character of benzyl fragment in 11 and 13, indicating the possible homolysis and radical recombination. Since 15 is 1.7 kcal/mol more stable than 4, this makes the process of C2–Ni bond homolysis-recombination thermodynamically favorable and benzyl Ni(Ⅱ) complex 15 is regarded as the major one in the reaction system (Fig. 2). This benzyl radical involved transformation can also be supported by the concept of radical buffering because of the high stability of benzyl radical [67]. By comparing the H···H distances between the substrates and ligands of intermediates 4 and 15 (Fig. 3), we observed that the distance of H12···H24 in 4 is 2.502 Å, whereas in 15, the distance of H21···H24 is 2.941 Å. Additionally, the distance of H25···H26 in 4 is 2.231 Å, while in 15, the distance of H22···H26 is 2.713 Å. These results suggest the H–H repulsion between the substrate and chiral ligand in 4 and 15 may be responsible for stability difference.
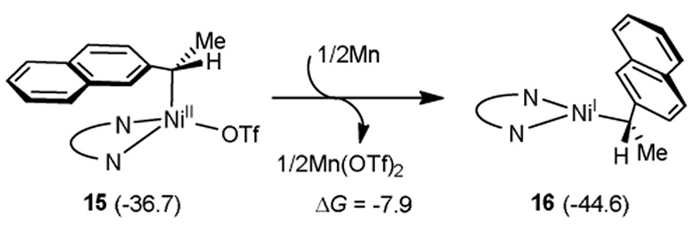
Ni(Ⅱ) complex 15 can undergo one-electron reduction by Mn(0) (Fig. 4). The Gibbs free energy change (∆G) is −7.9 kcal/mol, indicating that one-electron reduction readily occurs [68].
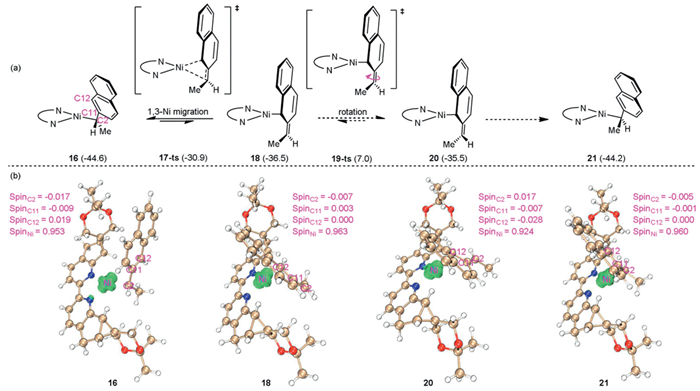
We also investigated the cleavage process of C2–Ni bond in Ni(Ⅰ) species 16. This process proceeds via a 1,3-Ni migration transition state 17-ts with an activation free energy of 13.7 kcal/mol, leading to the formation of Ni(Ⅰ) species 18. Geometry analysis shows that the lengths of C2–Ni, C2–C11 and C12–Ni bonds in 18 are 3.573, 1.370 and 2.062 Å, respectively, indicating an η1-bonding of Ni center (Fig. S5c in Supporting information). Moreover, spin population analysis of 18 shows spin densities of −0.007, 0.003, 0.000 and 0.963 on the C2, C11, C12 and Ni atoms, respectively, which suggests a negotiable radical characteristics of benzyl fragment. These computational results show a significant double bond characteristic of C2–C11 bond in 18, thus hampering the rotation of C2–C11 bond. As a result, the corresponding transition state 19-ts requires an energy barrier of 43.5 kcal/mol, suggesting an unlikely rotation process. Therefore, the stero-isomerization process involving the cleavage of C2–Ni bond in Ni(Ⅰ) species 16 followed by bond rotation would not occur (Fig. 5).
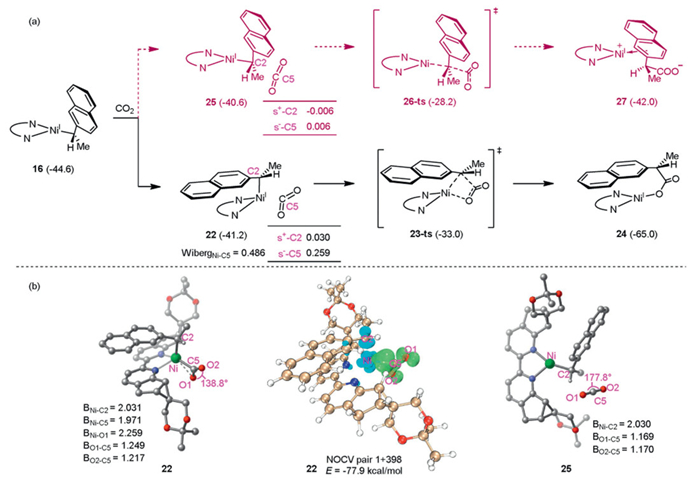
The next step is the carboxylation of 16 with CO2 for the formation of C–C bond, as shown in the Fig. 6a. In this process, two pathways, including stereoretentive inner-sphere and stereoinvertive outer-sphere pathway were considered [69]. In inner-sphere pathway, approaching of CO2 generates an η2-coordinated intermediate 22 with a Ni···CO2 distance of 1.971 Å and the Wiberg bond order of 0.486, indicating a chelation-assisted CO2 insertion process. ETS-NOCV analysis reveals a significant back-donation interaction between the dx2-y2 orbital of Ni atom and the unoccupied π* orbital of CO2 (E = −77.9 kcal/mol), effectively stabilizing the Ni–CO2 complex and contributing to the activation of CO2 as well (Fig. 6b). As a result, the C5–O bond in 22 is significantly weakened, accompanied by a decrease in O–C–O bond angle from 180° in free CO2 to 139°. One C=O bond of CO2 which coordinates with the Ni center becomes somewhat longer to 1.249 Å than that of free CO2 (1.169 Å). This activation effect also enhances the condensed local softness (s-) [70] of the C5 atom in CO2, facilitating subsequent C–C bond formation. Thereafter, carboxylation occurs via a four-membered ring transition state 23-ts, generating Ni(Ⅰ) carboxylate species 24 with the energy barrier of 8.2 kcal/mol. We also investigated the carboxylation proceeds in an outer-sphere pathway. In this case, a molecular complex 25 might be formed, in which CO2 has no significant interaction with metal center, suggesting a nucleophilic attack on CO2 by the backside of the C2 atom in 16. A slight structural deformation is observed in CO2 fragment, with the O–C–O bond angle changing to 177° and two C=O bonds of CO2 (1.169 Å, 1.170 Å) being similar to that of free CO2 (1.169 Å). It means CO2 was less activated, resulting in the carboxylation via an outer-sphere linear transition state 26-ts has a higher activation free energy (12.4 kcal/mol) than that via inner-sphere transition state 23-ts. Therefore, the outer-sphere pathway can be excluded.
Based on the results above, a comprehensive pathway for the Ni-catalyzed enantioconvergent carboxylation with CO2 was proposed, with the corresponding energy profiles shown in Fig. 7. The Ni(0)-L catalyst ligated with racemic 1a, generates 1/28. The following nucleophilic substitution-type oxidative addition takes place via transition state 3-ts/30-ts. After releasing NMe3, benzyl Ni(Ⅱ) complex 4/15 was thermodynamically favorably formed. Since 15 is 1.7 kcal/mol more stable than 4, this makes the process of C(benzyl)–Ni bond homolysis-recombination thermodynamically favorable, enabling the transformation of 4 into 15 as the major one in the reaction system. Once the benzyl Ni(Ⅱ) species 15 is generated, it can be easily reduced to low-valent Ni(Ⅰ) species 16 in the presence of excess Mn powder. Subsequent CO2 insertion into the Ni(Ⅰ)–C(benzyl) bond of 16 affords the (S)-isomeric product, which is consistent with experimental observation. This step via inner-sphere transition state 23-ts requires an activation free energy of 8.2 kcal/mol. After carboxylation, reduction, dissociation of the carboxylate (1), and the coordination of the substrate 1a occur to regenerate the Ni(0) intermediate 1.
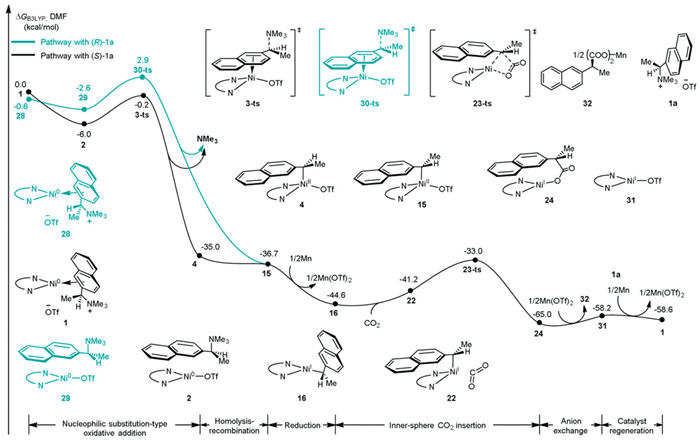
In summary, a carbon–metal bond homolysis/recombination model for Ni-catalyzed enantioconvergent carboxylation of benzyl ammonium salt with CO2 was proposed and validated. The key step of C(benzyl)–N bond activation by Ni(0) complex is stereoinvertive nucleophilic substitution-type oxidative addition, resulting in the formation of benzyl Ni(Ⅱ) complex. Then the C–Ni(Ⅱ) bond homolysis, rotation and recombination might occur giving a more thermodynamically stable stereoisomeric benzyl Ni(Ⅱ) complex. Subsequent reduction and CO2 insertion through stereoretentive inner-sphere pathway give the chiral carboxylated product. This model achieves catalytic carboxylation of benzyl (pseudo)halides with CO2 in high enantioselectivity. The comprehensive mechanistic investigation establishes fundamental principles governing the enantioconvergent catalytic carboxylation of benzyl (pseudo)halides with CO2, highlighting the potential utility of carbon–metal bond homolysis-recombination protocols in asymmetric reductive cross-coupling reactions. Future experimental and computational studies will further expand the scope and explore the generality of this mechanism.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Qi Zhou: Writing – original draft, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Cefei Zhang: Writing – original draft, Validation, Investigation, Formal analysis. Hui-Lin Luo: Writing – original draft, Investigation, Formal analysis. Chuan-Xi Nie: Writing – original draft, Investigation, Formal analysis. Changwei Hu: Writing – review & editing, Supervision, Project administration, Formal analysis. Jian-Heng Ye: Writing – review & editing, Supervision, Formal analysis. Zhishan Su: Writing – review & editing, Supervision, Project administration, Funding acquisition, Formal analysis. Li-Li Liao: Writing – review & editing, Supervision, Project administration, Funding acquisition, Formal analysis. Da-Gang Yu: Writing – review & editing, Supervision, Project administration.
This work was supported by the National Key R&D Program of China (No. 2024YFA1509703), National Natural Science Foundation of China (Nos. 22473081 and 22201027), Fundamental Research Funds from Sichuan University (No. 2020SCUNL102), Sichuan Science and Technology Program (No. 2025ZNSFSC0911), Sichuan University Interdisciplinary Innovation Fund, the Open Research Fund of State Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Nanjing University. We thank Y.-Y. Gui from Sichuan Normal University for valuable discussion and help.
Supplementary material associated with this article can be found, in the online version, at doi:
M.P. Plesniak, H.M. Huang, D.J. Procter, Nat. Rev. Chem. 1 (2017) 0077–0092. doi: 10.1038/s41570-017-0077
C.P. Jasperse, D.P. Curran, T.L. Fevig, Chem. Rev. 91 (1991) 1237–1286. doi: 10.1021/cr00006a006
M. Yan, J.C. Lo, J.T. Edwards, P.S. Baran, J. Am. Chem. Soc. 138 (2016) 12692–12714. doi: 10.1021/jacs.6b08856
S.P. Pitre, N.A. Weires, L.E. Overman, J. Am. Chem. Soc. 141 (2019) 2800–2813. doi: 10.1021/jacs.8b11790
K. Liu, M. Schwenzer, A. Studer, ACS Catal. 12 (2022) 11984–11999. doi: 10.1021/acscatal.2c03996
M. Tomanik, I.T. Hsu, S.B. Herzon, Angew. Chem. Int. Ed. 60 (2021) 1116–1150. doi: 10.1002/anie.201913645
L. Li, L.L. Liao, D.G. Yu, et al., Chin. Chem. Lett. 35 (2024) 110104. doi: 10.1016/j.cclet.2024.110104
S.P. Morcillo, Angew. Chem. Int. Ed. 58 (2019) 14044–14054. doi: 10.1002/anie.201905218
P. Sivaguru, Z. Wang, G. Zanoni, X. Bi, Chem. Soc. Rev. 48 (2019) 2615–2656. doi: 10.1039/c8cs00386f
S.W.M. Crossley, C. Obradors, R.M. Martinez, R.A. Shenvi, Chem. Rev. 116 (2016) 8912–9000. doi: 10.1021/acs.chemrev.6b00334
S.M. Bhutto, R.X. Hooper, P.L. Holland, et al., Chem. Sci. 15 (2024) 3485–3494. doi: 10.1039/d3sc05939a
Y.F. Sun, B.C. Wang, Z. Lu, et al., ACS Catal. 14 (2024) 8405–8413. doi: 10.1021/acscatal.4c02438
J.B. Diccianni, T. Diao, Trends. Chem. 1 (2019) 830–844. doi: 10.1016/j.trechm.2019.08.004
C.S. Zhang, B.B. Zhang, Z.X. Wang, et al., Chem. Sci. 13 (2022) 3728–3739. doi: 10.1039/d1sc05605k
R. Poli, Comptes Rendus. Chimie 24 (2021) 147–175. doi: 10.5802/crchim.73
G.Y. Han, P.F. Su, X.Z. Shu, et al., Nat. Catal. 7 (2024) 12–20.
D. Wu, W.Y. Kong, G.Y. Yin, et al., Nat. Catal. 6 (2023) 1030–1041. doi: 10.1038/s41929-023-01032-0
N. Kielland, C.J. Whiteoak, A.W. Kleij, Adv. Synth. Catal. 355 (2013) 2115–2138. doi: 10.1002/adsc.201300422
D. Ghosh, S. Dabas, S. Subramanian, et al., Catal. Sci. Technol. 15 (2025) 618–646. doi: 10.1039/d4cy01005a
C.K. Ran, X.W. Chen, D.G. Yu, et al., Sci. China Chem. 63 (2020) 1336–1351. doi: 10.1007/s11426-020-9788-2
Y. Shi, B.W. Pan, F. Zhou, et al., Org. Biomol. Chem. 18 (2020) 8597–8619. doi: 10.1039/d0ob01905d
X. Guo, Y. Wang, J.B. Xia, et al., Chin. J. Org. Chem. 40 (2020) 2208–2220. doi: 10.6023/cjoc202002032
D. García-Lopez, L. Pavlovic, K.H. Hopmann, Organometallics 39 (2020) 1339–1347. doi: 10.1021/acs.organomet.0c00090
L. Pavlovic, M. Pettersen, K.H. Hopmann, et al., Eur. J. Org. Chem. (2021) 663–670 2021. doi: 10.1002/ejoc.202001469
M. Pettersen, K.H. Hopmann, A. Bayer, et al., Adv. Synth. Catal. 366 (2024) 2976–2986. doi: 10.1002/adsc.202301285
X.W. Chen, Y. Lan, D.G. Yu, et al., J. Am. Chem. Soc. 141 (2019) 18825–18835. doi: 10.1021/jacs.9b09721
M. Takimoto, Y. Nakamura, K. Kimura, M. Mori, J. Am. Chem. Soc. 126 (2004) 5956–5957. doi: 10.1021/ja049506y
S. Kawashima, K. Aikawa, K. Mikami, Eur. J. Org. Chem. (2016) 3166–3170 2016. doi: 10.1002/ejoc.201600338
X.W. Chen, J.P. Yue, D.G. Yu, et al., Angew. Chem. Int. Ed. 60 (2021) 14068–14075. doi: 10.1002/anie.202102769
X.W. Chen, J.H. Ye, D.G. Yu, et al., Angew. Chem. Int. Ed. 63 (2024) e202403401. doi: 10.1002/anie.202403401
Q. Hu, D.G. Yu, C. Guo, et al., J. Am. Chem. Soc. 146 (2024) 14864–14874. doi: 10.1021/jacs.4c04211
C. Li, X.W. Chen, Y. Lan, D.G. Yu, et al., Angew. Chem. Int. Ed. 64 (2025) e202413305. doi: 10.1002/anie.202413305
Y.Y. Gui, N.F. Hu, D.G. Yu, et al., J. Am. Chem. Soc. 139 (2017) 17011–17014. doi: 10.1021/jacs.7b10149
Y.Y. Gui, J.H. Ye, D.G. Yu, et al., J. Am. Chem. Soc. 146 (2024) 2919–2927. doi: 10.1021/jacs.3c14146
L. Zhou, L. Li, Y. Tang, et al., J. Am. Chem. Soc. 146 (2024) 18823–18830. doi: 10.1021/jacs.4c05217
S.D. Zhang, L.P. Li, Y. Tang, et al., J. Am. Chem. Soc. 146 (2024) 2888–2894. doi: 10.1021/jacs.3c12720
B.H. Ye, L. Su, J.W. Liu, et al., Angew. Chem. Int. Ed. 64 (2025) e202413949. doi: 10.1002/anie.202413949
Y.Y. Gui, X.W. Chen, J.P. Yue, D.G. Yu, Chem. Synth. 4 (2024) 66.
Y.Y. Gui, X.W. Chen, D.G. Yu, Chin. J. Org. Chem. 44 (2024) 1038–1040. doi: 10.6023/cjoc202400015
L.H. Wang, T. Li, P.F. Li, et al., Angew. Chem. Int. Ed. 61 (2022) e202213943. doi: 10.1002/anie.202213943
T. Moragas, M. Gaydou, R. Martin, Angew. Chem. Int. Ed. 55 (2016) 5053–5057. doi: 10.1002/anie.201600697
M.J. Frisch, G.W. Trucks, H.B. Schlegel, et al., Gaussian 09, Gaussian, Inc., Wallingford, CT, USA, 2013 Revision D.01.
P.J. Stephens, F.J. Devlin, C.F. Chabalowski, M.J. Frisch, J. Phys. Chem. 98 (1994) 11623–11627. doi: 10.1021/j100096a001
S. Grimme, S. Ehrlich, L. Goerigk, J. Comput. Chem. 32 (2011) 1456–1465. doi: 10.1002/jcc.21759
P.J. Hay, W.R. Wadt, J. Chem. Phys. 82 (1985) 270–283. doi: 10.1063/1.448799
W.R. Wadt, P.J. Hay, J. Chem. Phys. 82 (1985) 284–298. doi: 10.1063/1.448800
P.J. Hay, W.R. Wadt, J. Chem. Phys. 82 (1985) 299–310. doi: 10.1063/1.448975
M.M. Francl, W.J. Pietro, M.S. Gordon, J. Chem. Phys. 77 (1982) 3654–3665. doi: 10.1063/1.444267
A.V. Marenich, C.J. Cramer, D.G. Truhlar, J. Phys. Chem. B 113 (2009) 6378–6396. doi: 10.1021/jp810292n
C. Gonzalez, H.B. Schlegel, J. Chem. Phys. 90 (1989) 2154–2161. doi: 10.1063/1.456010
M. Dolg, U. Wedig, H. Stoll, H. Preuss, J. Chem. Phys. 86 (1987) 866–872. doi: 10.1063/1.452288
R. Krishnan, J.S. Binkley, R. Seeger, J.A. Pople, J. Chem. Phys. 72 (1980) 650–654. doi: 10.1063/1.438955
C.Y. Legault, 2020 (
A.E. Reed, F. Weinhold, J. Chem. Phys. 78 (1983) 4066–4073. doi: 10.1063/1.445134
J.P. Foster, F. Weinhold, J. Am. Chem. Soc. 102 (1980) 7211–7218. doi: 10.1021/ja00544a007
T. Ziegler, A. Rauk, Theor. Chim. Acta 46 (1977) 1–10.
M. Mitoraj, A. Michalak, J. Mol. Model. 13 (2007) 347–355. doi: 10.1007/s00894-006-0149-4
A. Michalak, M. Mitoraj, T. Ziegler, J. Phys. Chem. A 112 (2008) 1933–1939. doi: 10.1021/jp075460u
M.P. Mitoraj, A. Michalak, T. Ziegler, J. Chem. Theory. Comput. 5 (2009) 962–975. doi: 10.1021/ct800503d
T. Lu, F. Chen, J. Comput. Chem. 33 (2012) 580–592. doi: 10.1002/jcc.22885
W. Humphrey, A. Dalke, K. Schulten, J. Mol. Graph. 14 (1996) 33–38.
T. Moragas, A. Correa, R. Martin, Chem. Eur. J. 20 (2014) 8242–8258. doi: 10.1002/chem.201402509
R.G. Tian, L.Y. Wang, L.Z. Zheng, S.K. Tian, ACS Catal. 14 (2024) 5039–5046. doi: 10.1021/acscatal.4c00504
Q. Lin, Y. Fu, P. Liu, T.N. Diao, J. Am. Chem. Soc. 143 (2021) 14196–14206. doi: 10.1021/jacs.1c05255
J. Liu, Y. Yang, K. Ouyang, W.X. Zhang, Green Synth. Catal. 2 (2021) 87–122. doi: 10.1117/12.2604794
S. Lee, H. Ki, H. Ihee, et al., Nat. Commun. 16 (2025) 1969.
M.H. Xu, Y.B. Li, H. Wang, F. Glorius, X.T. Qi, Angew. Chem. Int. Ed. (2025) e202500522.
H. Ryu, B. Pudasaini, M.H. Baik, et al., Chem. Sci. 13 (2022) 10707–10714. doi: 10.1039/d2sc01650h
S.Q. Zhang, X. Hong, Acc. Chem. Res. 54 (2021) 2158–2171. doi: 10.1021/acs.accounts.1c00050
R.K. Roy, S. Krishnamurti, P. Geerlings, S. Pal, J. Phys. Chem. A 102 (1998) 3746–3755.

Scheme 1 The formation of chiral carbon-metal species in transition metal-catalyzed carboxylation with CO2. TM = transition metal, L = ligand, sub = substrate.

Scheme 2 (a) Model reaction. (b) Possible mechanism for the Ni-catalyzed enantioconvergent carboxylation of alkyl ammonium salts with CO2. Naphth = Naphthyl group.

Figure 1 Energy profiles for the C(benzyl)–N bond activation with a Ni(0) complex via nucleophilic substitution-type oxidative addition and single-electron transfer (all energies are in kcal/mol) and optimized geometries of 3-ts and 6-ts (all distances are given in angstrom).

Figure 2 (a) Pathway of C2–Ni(Ⅱ) bond homolysis-recombination for the formation of optically pure enantiomeric carbon-metal species (all energies are in kcal/mol). (b) Spin population analysis of intermediates and transition states calculated using Multiwfn 3.8 (dev) software.

Figure 3 Optimized geometries of 4 and 15, and the H···H distance between the substrate and ligand in 4 and 15 (all distances are given in angstroms).

Figure 5 (a) Process of C2–Ni(Ⅰ) bond cleavage in Ni(Ⅰ) species 16 (all energies are in kcal/mol). (b) Spin population analysis of intermediates and transition states calculated using Multiwfn 3.8 (dev) software.

Figure 6 (a) Comparison of inner-sphere and outer-sphere reaction mechanisms for CO2 (all energies are in kcal/mol). (b) Optimized geometries of 22 and 25 (all distances are given in angstrom).

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:


 下载:
下载: