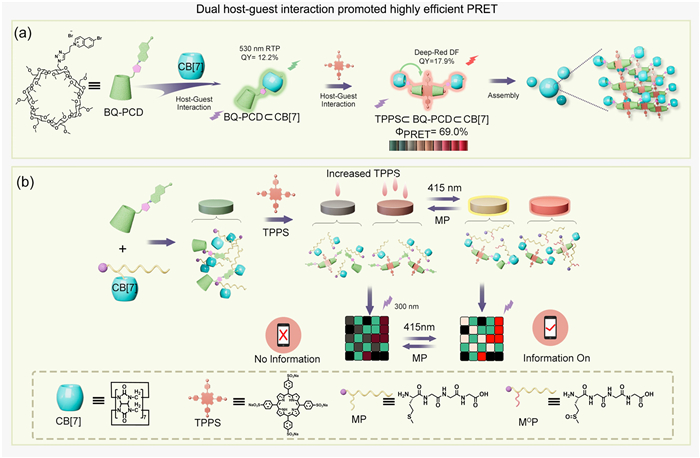
Scheme 1.
Schematic illustration of (a) multicolor delayed luminescence supramolecular assembly and (b) multicolor information encryption driven by the porphyrin-photosensitization.
Cucurbit[7]uril-confined cascade assembly of cyclodextrin phosphor derivative achieving multicolor delayed luminescence for information encryption
Jie Niu , Xuan Wu , Jie Yu , Zhuo Lei , Ying-Ming Zhang , Li-Hua Wang , Yu Liu

Non-covalent supramolecular cascade assembly [1] has become a research hotspot for constructing delayed fluorescence materials via cascade energy transfer [2–5]. Particularly, macrocycle-based cascade assemblies not only enhance photophysical properties through macrocyclic confinement [6–9] but also improve energy transfer efficiency through spatial confinement assembly. Consequently, tremendous emission-tunable luminescence systems have been developed for bioimaging [10,11], information encoding [12,13], smart optical devices [14], and so on. In these systems, water-soluble cyclodextrins (CDs) [15] and cucurbit[n]urils (CB[n]) [16] have been widely explored for encapsulating various functional guest molecules, enhancing phosphorescence emission and achieving efficient phosphorescence resonance energy transfer (PRET) for long-lived luminescence with large red-shift [17,18]. The rims of CDs are rich in hydroxyl groups, enabling them to encapsulate anionic guests into their cavities [19], while CBs preferentially bind cationic guests via ion-dipole interactions [20,21]. Even though these two kinds of macrocycles have different host-guest properties [22,23], they could improve the luminescent properties of encapsulated guests through a similar confinement mechanism, which could restrict molecular movement, suppress non-radiative transitions, promote intersystem crossing efficiency, and reduce oxygen-induced quenching in solution, for highly efficient room-temperature phosphorescence (RTP) [24,25]. For example, Yang, Tang, Li, and co-workers reported that β-CD could bind anionic p-biphenylboronic acid, exhibiting a long-lived phorsphorescence in aqueous solution, which could be applied for information encryption [26]. Liu et al. reported a thermally activated reversible RTP supramolecular assembly by encapsulating anionic terephthalic acid into β-CD cavity, which was further mixed with polyvinyl alcohol, achieving a long-lived multicolor luminescent film for information encoding and smart materials [27]. The strong binding constant between CB and cationic luminophore guests could also improve photoluminescence performance, Ma et al. reported that CB[8] could bind cationic 4-(4-bromophenyl)pyridinium derivatives to enhance RTP by forming a 2:2 host-guest complex, enabling multicolor luminescence for bioimaging with varying amounts of CB[8] [28]. Ni and Liu et al. recently comprehensively reviewed CB-mediated cationic guest confinement-derived supramolecular organic luminescence for bioimaging and multi-luminescent materials [29]. Benefitting from the different host-guest properties of these two kinds of macrocycles, the dual macrocyclic confinement effect in one system could be easily realized by encapsulating different luminophores into respective cavities [30]. Nevertheless, the simultaneous utilization of CB and CD to bind functional substrates and induce pure organic RTP through spatial cascade confinement assembly for multicolor delayed luminescence has rarely been reported, to the best of our knowledge.
Herein, we wish to report a delayed multicolor luminescence supramolecular assembly constructed by CB[7], 6-bromoisoquinolinium-modified permethylated cyclodextrin (BQ-PCD), and tetra(4-sulfonatophenyl)porphyrin (TPPS) via cascade confinement assembly (Scheme 1). BQ (6-bromoisoquinolinium) in BQ-PCD could be firstly encapsulated by CB[7], leading to RTP emission at 530 nm with phosphorescence quantum yields (QY) of 12.2% and lifetime of 132.1 µs. And the PCD (permethylated cyclodextrin) in BQ-PCD could bind TPPS with a high binding constant without dissociating the BQ-PCD$\subset$CB[7] complex. Benefiting from the dual host-guest interactions, the distance between phosphorescent donor (BQ) and fluorescent acceptor (TPPS) was significantly reduced, resulting in delayed luminescence at 645 and 710 nm through high efficient PRET process, as well as high delayed fluorescence QY (17.9%). Meanwhile, the multicolor luminescence could be achieved by adding different amounts of TPPS to BQ-PCD$\subset$CB[7] solution, in which the visible color would change from green to yellow, orange and eventually to red. The methionine-containing peptide was incorporated as the competitive guest due to its higher binding constant with CB[7] and oxidation stimuli-responsiveness via porphyrin photosensitization, enabling reversibility through guest competitive binding, which was successfully applied in information encoding and encryption. This research offers a novel PRET approach for multicolor delayed luminescence via macrocyclic cascade confinement assembly.
Phosphor 6-bromoisoquinolinium-modified permethylated cyclodextrin (BQ-PCD) was designed and synthesized via “click” reaction between azide-functionalized permethyl β-cyclodextrin (N3-PCD) and alkynyl modified 6-bromoisoquinolinium derivative (BQ-yne) (Scheme S1 in Supporting information). Due to the existence of dual binding sites (BQ and PCD motif), the BQ-PCD could act as the guest molecule for CBs, as well as host molecule for TPPS. First, the host-guest complexation between CB[7] and BQ-PCD was studied by NMR spectroscopy (Fig. 1a). The proton signals assigned to Ha-f in 6-bromoisoquinolinium shifted upfield upon the addition of 0.5 equiv. CB[7] into BQ-PCD solution, presenting a slow exchange within NMR timescale and suggesting the encapsulation within the CB[7] cavity. Meanwhile, the triazole proton (Hg) shifted from 7.93 ppm to 8.22 ppm, possibly due to hydrogen bonding with CB[7] carbonyl groups, indicating its position outside the cavity. And the signals of unbound species disappeared at the molar ratio of 1:1 (CB[7]/[BQ-PCD]).
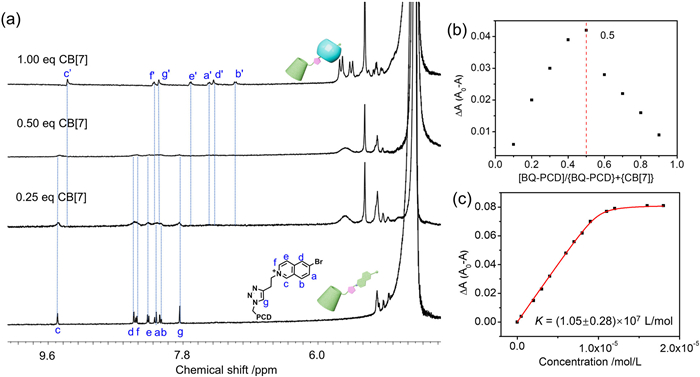
Then, the UV–vis, mass and NMR spectroscopies were employed to further certificate the host-guest binding modes between BQ-PCD and CB[7]. From the Job plot of BQ-PCD and CB[7] complex (Fig. S10 in Supporting information), an inflection point at a molar ratio of 0.5 ([BQ-PCD]/([BQ-PCD] + [CB[7]]) could be observed (Fig. 1b), indicating a 1:1 binding stoichiometry between CB[7] and BQ-PCD. The mass spectrum also confirmed the 1:1 binding stoichiometry between CB[7] and BQ-PCD. As shown in Fig. S11 (Supporting information), the peak at m/z 2864.0422 ([BQ-PCD+CB[7]-Br]+) could be observed. Followingly, the binding constant was also determined by UV–vis titration experiment. As shown in Fig. S12 (Supporting information), the characteristic absorption peak of BQ-PCD at 243 nm decreased gradually upon the addition of CB[7], and then reached the equilibrium at the molar ratio of 1.4 ([CB[7]]/[BQ-PCD]). Accordingly, the binding constant (K) was determined to be (1.05 ± 0.28) × 107 L/mol by analyzing the sequential changes in absorption intensity at 243 nm of BQ-PCD through a nonlinear least-squares curve fitting method (Fig. 1c). The Hill equation was also employed to quantitatively evaluate the molecular recognition behavior between BQ-PCD and CB[7] (Fig. S13 in Supporting information). The logK value in CB[7] complexe was determined to be (6.97 ± 0.15). The Hill coefficient was determined to be 1.38, indicating the 1:1 stoichiometry for the BQ-PCD$\subset$CB[7]. These data were well consistent with the UV–vis titration results.
After verifying the binding behavior between BQ-PCD and CB[7], their photo-physical properties in aqueous solution were investigated. Initially, the photoluminescence spectrum of BQ-PCD exhibited two emission peaks at 377 nm and 530 nm, with average lifetimes of 5.3 ns and 14.3 ns from time-resolved decay profiles (Fig. S14 in Supporting information), respectively. Upon gradual addition of CB[7] to BQ-PCD solution, the 377 nm emission peak gradually decreased, while the 530 nm peak remarkably increased, stabilizing at 1.4 equiv. of CB[7] (Fig. 2a). Moreover, only the emission peak at 530 nm was observed in the delayed spectrum (delay time = 0.05 ms), indicating the short lifetime emission at 377 nm and the long-lived feature at 530 nm. The delayed spectra also provided the similar results, in which an enhanced emission at 530 nm could be observed upon CB[7] addition, and stabilized at 1.4 equiv. CB[7] (Fig. 2b). Moreover, the significant overlap between the normalized photoluminescence spectrum and gated emission spectrum at 530 nm further corroborated that CB[7] induced long-lived emission of BQ-PCD. In the complex of BQ-PCD$\subset$CB[7], the lifetime at 377 nm remained as 5.5 ns determined by lifetime decay profile (Fig. S15 in Supporting information), while the lifetime at 530 nm increased to 132.1 µs (Fig. 2c). And the phosphorescence QY of BQ-PCD was 0.2% (Fig. S16 in Supporting information), which remarkably increased to 12.2% upon complexation with CB[7] (Fig. S17 in Supporting information). These phenomena may be caused by that the CB[7] can restrict the rotation of BQ and shield it from being quenched by dissolved oxygen, thereby significantly improving the phosphorescence quantum yield and lifetime of BQ-PCD.
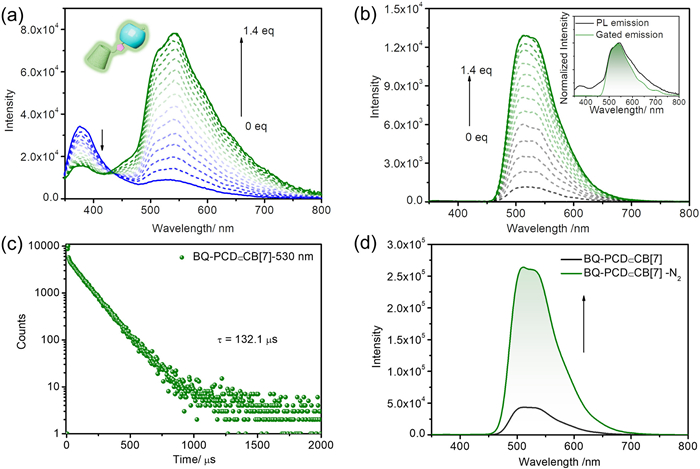
Subsequently, the bubling of N2 into BQ-PCD$\subset$CB[7] solution led to the enhancement of 530 nm emission intensity (Fig. 2d), which was attributed to the suppression of the triplet electron quenching by oxygen, confirming its phosphorescence nature. Temperature-dependent emission studies further supported this conclusion. With the temperature decreased from 330 K to 77 K, the emission intensity of 530 nm steadily increased in BQ-PCD$\subset$CB[7] system (Figs. S18 and S19 in Supporting information). Moreover, the temperature-dependent decay curves showed a significant lifetime enhancement at 77 K (Figs. S20 and S21 in Supporting information), whose values increased from 14.3 ns to 7.3 ms (BQ-PCD), 132.1 µs to 7.2 ms (BQ-PCD$\subset$CB[7]), respectively. These results collectively demonstrated that benefiting from macrocyclic confinement assembly of CB[7], the activated phosphorescence emission with improved QY and prolonged life-time would be achieved.
PRET, as an efficient method to construct long-lived emission materials with large red-shift, requires not only an excellent overlap between the emission spectrum of the donor and the excitation spectrum of the acceptor, but also the close spatial distance between them. TPPS, as a commercially available red fluorescence dye, exhibits the distinctive absorption peaks of the Soret band at 411 nm and the Q-band between 450 nm and 700 nm, and the absorption peaks of the Q-band overlapped well with the phosphorescence emission spectrum of BQ-PCD$\subset$CB[7], making it as the ideal candidate for the RTP energy transfer acceptor. Meantime, the strong binding affinity between permethylated β-cyclodextrin and TPPS [31] would lead to the short spatial distance between the phosphorescent donor BQ-PCD$\subset$CB[7] and the acceptor TPPS, which would facilitate the RTP energy transfer process in the host-guest complex between BQ-PCD$\subset$CB[7] and TPPS. Therefore, it could be envisioned that the efficient long-lived emission system could be conveniently constructed by the dual host-guest interactions among TPPS, BQ-PCD and CB[7].
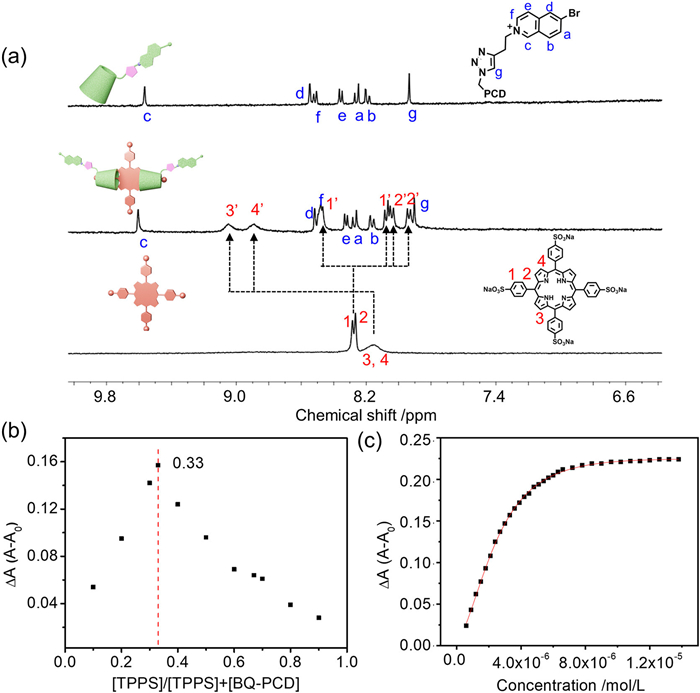
Initially, the binding behavior between TPPS and BQ-PCD was investigated through NMR and UV–vis spectroscopies. As depicted in Fig. 3a and Fig. S22 (Supporting information), upon the addition of BQ-PCD, the protons of TPPS split into two sets, indicating the formation of host-guest complex between PCD and TPPS. Subsequently, the UV–vis titration profiles indicated the absorption peak at 411 nm of TPPS gradually increased and redshifted to 413 nm upon the addition of BQ-PCD (Fig. S23 in Supporting information), along with a progressive rise in the Q-band between 450 and 700 nm. The Job plot exhibited a maximum value at a molar ratio of 0.33 (Fig. 3b and Fig. S24 Supporting information), indicating the stoichiometric binding ratio between TPPS and BQ-PCD was 1:2, which was consistent with previous report [32]. Furthermore, by nonlinear curve fitting the changes of absorption intensity at 413 nm, the binding constants between BQ-PCD and TPPS were determined as K1 = 2.93 × 105 L/mol and K2 = 3.03 × 106 L/mol (Fig. 3c). To further confirm the occurrence of dual host-guest interaction, the NMR spectroscopy was conducted. As shown in Fig. S25 (Supporting information), the signals assigned to the encapsulated TPPS in TPPS$\subset$BQ-PCD complex exhibited little change upon the addition of CB[7]. Furthermore, the protons of BQ motif exhibited obvious upfield shifts compared with those in TPPS$\subset$BQ-PCD, indicating the formation of TPPS$\subset$BQ-PCD$\subset$CB[7] host-guest complex. These results indicated the dual host-guest complexation could be successfully constructed, which would be essential for realizing the cascade confinement effect of dual macrocycles in one single system.
Due to the excellent overlap between the long-lived phosphorescent emission of BQ-PCD$\subset$CB[7] and the Q-band of TPPS (Fig. 4a), the efficient PRET was proposed through the closer spatial distance via the dual host-guest complexation. As the control experiments, time-gated emission spectroscopy confirmed TPPS exhibited negligible delayed fluorescence under 413 and 300 nm excitation (Figs. S26 and S27 in Supporting information), and the photoluminescence spectra of TPPS show only a slight increase upon adding BQ-PCD (Fig. S28 in Supporting information). Meantime, the time-gated luminescence spectra of TPPS$\subset$BQ-PCD$\subset$CB[7] under 413 nm excitation were recorded (Fig. S29 in Supporting information), no significant enhancement at 645 or 710 nm could be observed. According to the time-resolved decay curves under 450 nm excitation, the lifetimes were determined as follows: TPPS, 8.7 ns (645 nm), 6.7 ns (710 nm) (Fig. S30 in Supporting information); TPPS$\subset$BQ-PCD$\subset$CB[7], 13.5 ns (645 nm), 13.6 ns (710 nm) (Fig. S31 in Supporting information), respectively. Therefore, the TPPS itself exhibited no long-lived emission properties under 413 or 300 nm excitation, and no long-lived emission in TPPS$\subset$BQ-PCD$\subset$CB[7] would be achieved by directly activating TPPS.
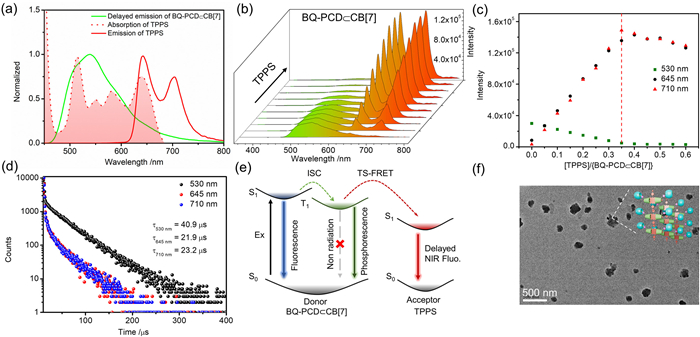
Followingly, the PRET process was investigated under the irradiation at 300 nm. As shown in Figs. 4b and c, accompanying with the addition of TPPS into BQ-PCD$\subset$CB[7], the phosphorescence at 530 nm gradually decreased, while an emergence of emission peaks at 645 nm and 710 nm in the gated emission spectra (delay time = 0.05 ms). The gated emission peaks at 645 nm and 710 nm were consistent with the characteristic fluorescence peaks of free TPPS, indicating the successful RTP energy transfer from BQ motif to TPPS, resulting in long-lived luminescence. With the addition of 0.5 equiv. TPPS into BQ-PCD$\subset$CB[7], the phosphorescence emission at 530 nm was nearly quenched, indicating the highly efficient energy transfer to TPPS. The time-resolved decay curves (Fig. 4d) revealed that the phosphorescence lifetime at 530 nm decreased from 132.1 µs to 40.9 µs in TPPS$\subset$BQ-PCD$\subset$CB[7]. Additionally, the lifetimes at 645 and 710 nm were determined to be 21.9 and 23.2 µs in TPPS$\subset$BQ-PCD$\subset$CB[7], confirming the long-lived characteristics. These findings suggested the triplet-to-singlet Förster resonance energy transfer (TS-FRET), and further corroborated the occurrence of PRET process (Fig. 4e). The energy transfer efficiency (ΦPRET) was further calculated by the lifetime changes of donor complexes, giving the ΦPRET value of 69.0% from BQ-PCD$\subset$CB[7] to TPPS. Meantime, the delayed fluorescence QY of TPPS$\subset$BQ-PCD$\subset$CB[7] (Fig. S32 in Supporting information) was increased to 17.9%.
Similarly, the bubbling of N2 into the TPPS$\subset$BQ-PCD$\subset$CB[7] solution led to enhancement of the emissions at 530, 645, and 710 nm (Fig. S33 in Supporting information), which could suppress the triplet electron quenching by oxygen for better RTP performance, further confirming the PRET process from phosphor to TPPS. Time-resolved decay analysis (Figs. S34 and S35 in Supporting information) revealed a further increase in lifetimes of 645 and 710 nm upon N₂ bubbling, reaching 34.7 and 34.0 µs in CB[7] complexes, indicating the oxygen sensitivity of long-lived emission. These results clearly indicated an efficient phosphorescence energy transfer occurred due to the dual host-guest complexation mediated self-assembly for the well-overlap in phosphorescence emission spectrum of donor and absorbance spectrum of acceptor, as well as the closer distance between them.
To further validate the dual host-guest complexation mediated efficient PRET, a series of control experiments were performed. Firstly, time-gated luminescence spectra of adding TPPS into BQ-PCD without CB[7] (Fig. S36 in Supporting information) showed no detectable emission. Moreover, the introduction of CB[7] into TPPS$\subset$BQ-PCD led to the emergence of delayed fluorescence at 645 nm and 710 nm (Fig. S37 in Supporting information), indicating the vital roles of CB[7] in activating RTP for the further PRET process. Additionally, BQ-3C [33] was synthesized as a reference to investigate the crucial role of cyclodextrin in the PRET process. Even though 530 nm phosphorescence emission decreased upon adding TPPS to BQ-3C$\subset$CB[7], delayed emission at 645 nm and 710 nm could not be observed (Fig. S38 in Supporting information), resulting from the random dispersion in solution, which would lead to relatively long distance between donor and acceptor, then prevent efficient energy transfer. The modification of phosphor onto PCD could afford dual binding sites in one compound, in which the dual host-guest interactions would active the efficient RTP and reduce the spatial distance between phosphor donor and fluorescence acceptor for highly efficient PRET. Moreover, the transmission electronic microscopy (TEM) was employed to investigate their self-assembly morphologies. The 1:1 binding stoichiometric ratio between BQ-PCD and CB[7] would lead to the spherical nanoparticles in the presence of TPPS (Fig. 4f).
Owing to the dynamic nature of host-guest interactions, stimuli-responsive cascade assemblies can be achieved through the addition of competitive guest, offering promising applications in information encryption, bioimaging, and sensing. CB[7] has been reported to encapsulate the N-terminal methionine side chain with moderate binding constant stronger than BQ motif [34], therefore the methionine-containing peptide (MGGG, MP) was introduced as the competitor. Moreover, the thioether motif undergoes oxidation to sulfoxide and sulfone under oxidative conditions [35,36], whose binding constants with CB[7] might be remarkably weakened due to the electronegativity of sulfoxide or sulfone. Considering the photosensitizing properties of porphyrin, an oxidation-driven stimuli-responsive delayed multicolor fluorescence cascade system could be realized by incorporation MP with TPPS$\subset$BQ-PCD$\subset$CB[7], through the competitive binding between MP and BQ-PCD$\subset$CB[7], followed by porphyrin-sensitization mediated disassembly of MP$\subset$CB[7].
First, the binding behavior of MP and oxidized sulfoxide peptide (MOGGG, MOP) with CB[7], was studied through NMR and isothermal titration calorimetry (ITC). From the 1H NMR spectra (Fig. S39 in Supporting information), the signals of Met in MP underwent upfield-shift upon the addition of CB[7], indicating the encapsulation of Met residue into CB[7] cavity. And the protons of Met residue in MOP showed weak upfield-shift and broaden effect in the presence of CB[7], demonstrating the weak binding behavior. As expected, MP exhibited a strong 1:1 binding with CB[7] through the “one-set-of-binding-sites” model, yielding the binding constant of (2.88 ± 0.27) × 106 L/mol (Fig. S40a in Supporting information). However, the binding constant for the sulfoxide form (MOP) and CB[7] significantly decreased to (9.64 ± 1.39) × 104 L/mol (Fig. S40b in Supporting information). Additionally, the binding constant between BQ-PCD and CB[7] was also determined by ITC, whose value was (1.42 ± 0.10) × 106 L/mol (Fig. S40c in Supporting information). Based on those obtained binding constants (Table S1 in Supporting information), it could be anticipated the introduction of MP to TPPS$\subset$BQ-PCD$\subset$CB[7] assembly would lead to the competitive binding complex of MP$\subset$CB[7], which would quench the phosphorescence of BQ-PCD and the delayed fluorescence through the disassembly of BQ-PCD$\subset$CB[7]. After being oxidized, the MP$\subset$CB[7] complex would be destroyed to recover the inclusion of BQ-PCD$\subset$CB[7] as well as delayed luminescence.
As expected, in the delayed spectra of BQ-PCD$\subset$CB[7], the phosphorescence emission at 530 nm gradually decreased with the addition of methionine peptide (Fig. S41 in Supporting information). Similarly, the phosphorescence at 530 nm and delayed fluorescence at 645 nm and 710 nm decreased with the addition of methionine peptide in the TPPS$\subset$BQ-PCD$\subset$CB[7] solution (Fig. S42 in Supporting information). Thus, methionine peptide indeed quenched the phosphorescence and PRET process through competitive binding process. Moreover, the introduction of the oxidized forms of sulfoxide peptides (Fig. S43a in Supporting information) and sulfone (Fig. S43b in Supporting information) into TPPS$\subset$BQ-PCD$\subset$CB[7] resulted in a slight decrease in the delayed fluorescence at 645 and 710 nm. The above results perfectly certificated our anticipation the MP would switch off the PRET delayed fluorescence in CB[7] system. Moreover, the weak competitive binding process between MOP and CB[7] encouraged us to explore the recovery of RTP of BQ-PCD and followed PRET delayed fluorescence through the porphyrin-sensitization process.
In the mixed solution of TPPS$\subset$BQ-PCD$\subset$CB[7] ([TPPS]:[BQ-PCD]:[CB[7]] = 1:2:2) and MP, a gradual increase of phosphorescence at 530 nm, as well as delayed fluorescence at 645 nm and 710 nm could be observed in the delayed fluorescence spectra, upon visible light irradiation (415 nm), reaching equilibrium after irradiation for 18 min (Fig. 5a). Moreover, this porphyrin-sensitization induced disassembly process could also be realized within low TPPS content. The similar phenomena could also be observed within 0.2 equiv., or even 0.05 equiv. TPPS (Figs. S44 and S45 in Supporting information). As comparison, there was no obvious enhancement in phosphorescence at 530 nm in the solution containing BQ-PCD$\subset$CB[7] and MP under the light irradiation (415 nm) for 40 min (Fig. S46 in Supporting information). Remarkably, this revsersible process could be achieved by the addition of MP and followed by light irradiation at 415 nm (Fig. S47 in Supporting information). To further confirm the oxidized form of MP, the liquid chromatography-mass spectrometry (LCMS) was employed. The light irradiation of TPPS/MP for 20 min would result in a new peak with the retention time of 0.50 min, and a new m/z peak with an increase by 16, indicating the conversion from MP to MOP in the photo-sensitization process (Fig. S48 in Supporting information). Therefore, the TPPS could act as the efficient photo-sensitizer to disassemble MP$\subset$CB[7], leading to the restoration of phosphorescence and multicolor delayed luminescence. Hence, we have successfully established a competitive binding-mediated reversible PRET system for delayed multicolor luminescence.
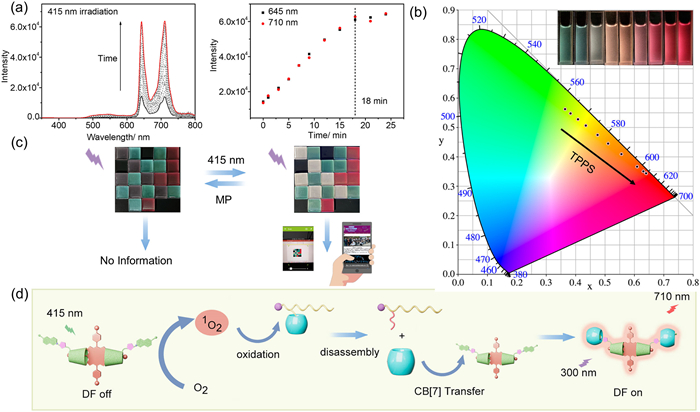
The multicolor luminescence system could be achieved by adding different amounts of TPPS to BQ-PCD$\subset$CB[7] solution, in which the visible color would change from green to yellow, orange and eventually to red under 300 nm irradiation (Fig. 5b). The CIE chromaticity diagram also depicted the same change tendency, with coordinates shifting from (0.36, 0.56) to (0.64, 0.34). Accompanying with the above MP-mediated competitive binding process and TPPS-photosensitization process, the multicolor TPPS$\subset$BQ-PCD$\subset$CB[7] assembly would be applied for information encryption through a 3D color code (Fig. 5c), in which the stored information behind the specific sequence of three-color modules could be read by software scanning. Three module colors, the green BQ-PCD$\subset$CB[7] (1/1), light yellow TPPS$\subset$BQ-PCD$\subset$CB[7] (0.05/1/1), and red TPPS$\subset$BQ-PCD$\subset$CB[7] (0.3/1/1), were poured into square containers, from which the combination color could be observed under 300 nm irradiation, separately. After being arranged in a specific sequence of the three color modules, a specific website was stored with the assistance of the COLORCODE app. The addition of MP to the light-yellow solution TPPS$\subset$BQ-PCD$\subset$CB[7] (0.05/1/1) and the red solution TPPS$\subset$BQ-PCD$\subset$CB[7] (0.3/1/1) caused the quenching in red and light-yellow color due to the competitive binding process, which made the encoded information unreadable. And upon the irradiation of 415 nm for 20 min, TPPS would produce 1O2 to oxidize the MP, leading to the recovery of RTP of BQ-PCD and delayed fluorescence of TPPS, making the stored information readable once again (Fig. 5d). Therefore, the simultaneous binding of functional substrates by CD and CB in our work could not only enhance PRET to realize multicolor photoluminescence, but also achieve the reversible luminescence for information encoding mediated by competitive binding process, which may hold promise in the fabrication of advanced anti-counterfeit materials.
As can be concluded from the above results, the BQ-PCD with dual binding sites displayed the multi host-guest interactions with CB[7] and TPPS. The BQ motif could be encapsulated into the cavity of CB[7], leading to the efficient RTP at 530 nm. And the PCD could include the fluorescence dye (TPPS) to reduce the distance between phosphor donor (BQ) and fluorescence acceptor (TPPS), as well as improve the luminescence behavior of TPPS, which could result in the long-lived fluorescence in TPPS$\subset$BQ-PCD$\subset$CB[7] complex with high PRET efficiency and high delayed fluorescence QY (17.9%). Meantime, due to the 1O2 generation ability of TPPS under light irradiation, the TPPS concentration-dependent reversible multicolor luminescence assembly was also achieved by reconstructed assembly of porphyrin-photosensitized methionine peptides$\subset$CB[7] complex, which was successfully applied in information encryption. Thus, the present work provided a convenient approach to construct efficient PRET system through cascade supramolecular assembly to realize long-lived multicolor emission, which could expand the potential application of supramolecular chemistry in optical materials.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Jie Niu: Writing – review & editing, Writing – original draft, Visualization, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Xuan Wu: Writing – review & editing, Writing – original draft, Methodology, Funding acquisition, Conceptualization. Jie Yu: Writing – review & editing, Methodology, Funding acquisition, Conceptualization. Zhuo Lei: Methodology, Investigation, Formal analysis. Ying-Ming Zhang: Writing – review & editing, Resources, Project administration, Funding acquisition. Li-Hua Wang: Software, Resources, Methodology. Yu Liu: Writing – review & editing, Supervision, Resources, Funding acquisition.
This work was financially supported by the National Natural Science Foundation of China (Nos. 22171148, 22371148, 22131008, 22101280, and 22201142), the Natural Science Foundation of Tianjin (No. 21JCZDJC00310), Haihe Laboratory of Sustainable Chemical Transformation, and the Fundamental Research Funds for the Central Universities (Nankai University).
Supplementary material associated with this article can be found, in the online version, at doi:
Y.Y. Liu, X.Y. Yu, Y.C. Pan, et al., Sci. China Chem. 67 (2024) 1397–1441. doi: 10.1007/s11426-024-1971-4
X.Y. Dai, M. Huo, Y. Liu, Nat. Rev. Chem. 7 (2023) 854–874. doi: 10.1038/s41570-023-00555-1
Y. Zhang, X. Chen, J. Xu, et al., J. Am. Chem. Soc. 144 (2022) 6107–6117. doi: 10.1021/jacs.2c02076
T. He, W.J. Guo, Y.Z. Chen, et al., Aggregate 4 (2023) e250. doi: 10.1002/agt2.250
Z. Wu, H. Qian, X. Li, et al., Chin. Chem. Lett. 35 (2024) 108829. doi: 10.1016/j.cclet.2023.108829
D. Xia, P. Wang, X. Ji, et al., Chem. Rev. 120 (2020) 6070–6123. doi: 10.1021/acs.chemrev.9b00839
Z. Liu, W. Lin, Y. Liu, Acc. Chem. Res. 55 (2022) 3417–3429. doi: 10.1021/acs.accounts.2c00462
J. Jiao, J. Yu, X. Tian, et al., Chin. Chem. Lett. 36 (2025) 111026. doi: 10.1016/j.cclet.2025.111026
M. Zuo, T. Li, H. Feng, et al., Small 20 (2024) 2306746. doi: 10.1002/smll.202306746
Q. Wang, J. Fan, X. Bian, et al., Chin. Chem. Lett. 33 (2022) 1979–1982. doi: 10.1016/j.cclet.2021.10.040
J. Yu, J. Niu, J. Yue, L.H. Wang, Y. Liu, ACS Nano 17 (2023) 19349–19358. doi: 10.1021/acsnano.3c06697
Q. Zhang, Z. Chang, W. Han, et al., Adv. Optical Mater. 12 (2024) 2400231. doi: 10.1002/adom.202400231
D. Chen, C. Bao, L. Zhang, et al., Adv. Funct. Mater. 34 (2024) 2314093.
K.R. Zhang, M. Hu, J. Luo, et al., Chin. Chem. Lett. 33 (2022) 1505–1510. doi: 10.1016/j.cclet.2021.08.072
Z. Liu, Y. Liu, Chem. Soc. Rev. 51 (2022) 4786–4827. doi: 10.1039/d1cs00821h
S.J. Barrow, S. Kasera, M.J. Rowland, et al., Chem. Rev. 115 (2015) 12320–12406. doi: 10.1021/acs.chemrev.5b00341
F. Lin, H. Wang, Y. Cao, et al., Adv. Mater. 34 (2022) 2108333. doi: 10.1002/adma.202108333
S. Kuila, K.V. Rao, S. Garain, et al., Angew. Chem. Int. Ed. 57 (2018) 17115–17119. doi: 10.1002/anie.201810823
A. Douhal, Chem. Rev. 104 (2004) 1955–1976.
X.L. Ni, X. Xiao, H. Cong, et al., Chem. Soc. Rev. 42 (2013) 9480–9508. doi: 10.1039/c3cs60261c
Y. Lu, Z. Yu, X. Yang, et al., Chin. Chem. Lett. 34 (2023) 108040.
K. Kano, R. Nishiyabu, T. Asada, Y. Kuroda, J. Am. Chem. Soc. 124 (2002) 9937–9944.
K. Liu, Y. Liu, Y. Yao, et al., Angew. Chem. Int. Ed. 52 (2013) 8285–8289. doi: 10.1002/anie.201303387
D. Li, F. Lu, J. Wang, et al., J. Am. Chem. Soc. 140 (2018) 1916–1923. doi: 10.1021/jacs.7b12800
J. Yu, J. Niu, X. Xu, Y. Liu, Adv. Sci. 11 (2024) 2408107.
D. Li, Z. Liu, M. Fang, et al., ACS Nano 17 (2023) 12895–12902. doi: 10.1021/acsnano.3c04971
X. Zhou, X. Zhao, X. Bai, Q. Cheng, Y. Liu, Adv. Funct. Mater. 34 (2024) 2400898.
J. Wang, Z. Huang, X. Ma, H. Tian, Angew. Chem. Int. Ed. 59 (2020) 9928–9933. doi: 10.1002/anie.201914513
H. Nie, Z. Wei, X.L. Ni, Y. Liu, Chem. Rev. 122 (2022) 9032–9077. doi: 10.1021/acs.chemrev.1c01050
F.F. Shen, Y. Chen, X. Dai, et al., Chem. Sci. 12 (2021) 1851–1857. doi: 10.1039/d0sc05343k
A. Nakagami, Q. Mao, G. Gouhier, et al., ChemBioChem 22 (2021) 3190–3198. doi: 10.1002/cbic.202100380
J. Niu, Y.H. Liu, W. Xu, et al., Chem. Commun. 59 (2023) 4680–4683. doi: 10.1039/d3cc00735a
S. Banu, K. Singh, S. Tyagi, et al., Org. Biomol. Chem. 19 (2021) 9433–9438. doi: 10.1039/d1ob01865e
A.R. Urbach, V. Ramalingam, Isr. J. Chem. 51 (2011) 664–678. doi: 10.1002/ijch.201100035
J. Niu, J. Yu, X. Wu, et al., Chem. Sci. 15 (2024) 13779–13787. doi: 10.1039/d4sc04261a
N. Song, Z. Zhou, Y. Song, et al., Nano Today 38 (2021) 101198.

Scheme 1 Schematic illustration of (a) multicolor delayed luminescence supramolecular assembly and (b) multicolor information encryption driven by the porphyrin-photosensitization.

Figure 1 (a) Partial 1H NMR (400 MHz, D2O, 298 K) spectra of BQ-PCD and CB[7] ([BQ-PCD] = 1 mmol/L, [CB[7]] = 0–1 mmol/L). (b) Job plot showing 1:1 stoichiometry complexation between BQ-PCD and CB[7] at 243 nm ([BQ-PCD] + [CB[7]] = 10 µmol/L). (c) Nonlinear curve fitting of absorption intensity at 243 nm of BQ-PCD versus CB[7] concentration ([BQ-PCD] = 10 µmol/L, [CB[7]] = 0–18 µmol/L).

Figure 2 (a) Photoluminescence spectra of BQ-PCD upon adding CB[7] ([BQ-PCD] = 10 µmol/L, [CB[7]] = 0–14 µmol/L). (b) Gated emission spectra (delay time = 0.05 ms) of BQ-PCD upon adding CB[7] ([BQ-PCD] = 10 µmol/L, [CB[7]] = 0–14 µmol/L, λex = 300 nm), (insert) normalized photoluminescence and gated emission spectra (delay time = 0.05 ms) of BQ-PCD$\subset$CB[7]. (c) Time-resolved photoluminescence decay spectrum of BQ-PCD$\subset$CB[7] at 530 nm. (d) Gated emission spectra of BQ-PCD$\subset$CB[7] with pump N2 (delay time = 0.05 ms, [BQ-PCD] = [CB[7]] = 10 µmol/L, λex = 300 nm).

Figure 3 (a) Partial spectra 1H NMR (400 MHz, D2O, 298 K) of TPPS and BQ-PCD ([TPPS] = 1 mmol/L, [BQ-PCD] = 2 mmol/L). (b) Job plot showing 1:2 stoichiometry complexation between TPPS and BQ-PCD at 413 nm ([TPPS] + [BQ-PCD] = 6 µmol/L). (c) Nonlinear curve fitting of absorption at 413 nm of TPPS versus BQ-PCD concentration ([TPPS] = 5 µmol/L, [BQ-PCD] = 0–15 µmol/L).

Figure 4 (a) Normalized phosphorescence emission spectra of BQ-PCD$\subset$CB[7], and normalized absorption and emission spectra of TPPS. (b) Phosphorescence emission spectra and (c) emission intensity changes at 530, 645, 710 nm of BQ-PCD$\subset$CB[7] upon adding TPPS (delay time = 0.05 ms, [BQ-PCD] = [CB[7]] = 10 µmol/L, [TPPS] = 0–6 µmol/L, λex = 300 nm). (d) Time-resolved photoluminescence decay spectra at 530, 645, 710 nm of TPPS$\subset$BQ-PCD$\subset$CB[7]. (e) Diagram of possible mechanism for RTP energy transfer process (ISC = intersystem crossing, TS-FRET = triplet-to-singlet FRET). (f) TEM images of TPPS$\subset$BQ-PCD$\subset$CB[7].

Figure 5 (a) Delayed spectra (delay = 0.05 ms) of MP/TPPS$\subset$BQ-PCD$\subset$CB[7] upon irradiation with 415 nm light (0–25 min) and emission intensity changes at 645 and 710 nm. (b) 1931 CIE chromaticity diagram of adding different amount of TPPS to BQ-PCD$\subset$CB[7], and (inset) photographic images of TPPS$\subset$BQ-PCD$\subset$CB[7] upon 300 nm light. (c) Photographs of 3D code encryption and oxidation stimulus-responsive 3D code transformation and corresponding information (website) was read by smartphone software (under 300 nm irradiation). (d) Schematic diagram of porphyrin-photosensitization induced methionine oxidation and host-guest competitive binding.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
 下载:
下载: