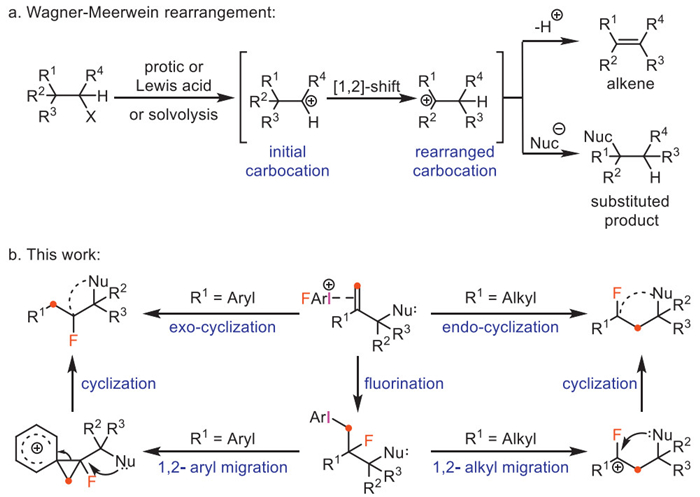
Scheme 1.
(a) Wagner−Meerwein rearrangement. (b) Monofluoroiodane(Ⅲ) reagent mediated Wagner−Meerwein rearrangement (this work).

Wagner−Meerwein reaction, as a classical carbocation rearrangement reaction, is of great significance in organic synthesis and biosynthetic chemistry [1]. The contemporary description of Wagner−Meerwein rearrangements includes 1,2-cationic migrations of alkyl and aryl groups as well as hydride (Scheme 1a) [2]. It is widely known that hypervalent iodine reagents, as a class of non-metallic oxidants, owing to their electrophilic nature and excellent leaving-group ability, have been widely used in various types of organo-oxidative rearrangement reactions [3–7]. These particular oxidative rearrangement reactions include [1,2]-migration [8–12], Hofmann rearrangement [13–15], Beckmann rearrangement [16,17], Curtius rearrangement [18], ring contraction [19–23]/expansion [24–26], and [3,3]-sigmatropic/iodonium−Claisen rearrangement [27,28]. Although some elegant Wagner−Meerwein reactions induced by hypervalent fluoroiodane(Ⅲ) reagents have been reported [29–35], these reactions can only lead to [1,2]-aryl migrated fluorinated products. Wagner−Meerwein rearrangement fluorination of [1,2]-alkyl or aryl group migration simultaneously mediated by hypervalent fluoroiodane(Ⅲ) reagent has rarely been reported to date, which we attribute to the formidable challenge involved in controlling the selective rearrangement of the highly active carbocation intermediates [36,37]. Recently, the elegant study represented by asymmetric 1,3-difluorinative Wagner–Meerwein rearrangement of β-substituted styrene derivatives [38] has been developed, and two different mechanisms are operative. Lately, Zhang group reported a novel hypervalent fluoroiodane(Ⅲ) reagent 1, which is an air- and moisture-stable compound and has been successfully used for metal-free carbon–carbon bond scission and intramolecular ring expansion fluorination of unactivated cyclopropanes [26]. Another appealing advantage is that reagent 1 could be prepared simply by oxy-fluorination of corresponding aryl iodides with commercially available silver difluoride, avoiding the use of multi-step ligand exchange reactions [39]. We wondered whether Wagner−Meerwein rearrangement fluorination could be achieved by reagent 1. We hypothesized that if nucleophilic substituents were present on the gem-disubstituted alkenes derivatives, Wagner−Meerwein rearrangement fluorination of [1,2]-alkyl or aryl group migration and intramolecular cyclization might be achieved, and the reaction might form quaternary carbon centers containing C(sp3)−F bond with excellent chemo-selectivity (Scheme 1b). The construction of C(sp3)−F bond is an important yet challenging task in organic synthesis. Recently, Feng’s group reported sulfone as a traceless activating group for constructing the C–F quaternary stereocenter [40]. It is worth noting that many marketed best-selling drugs featuring C(sp3)−F quaternary carbon centers, such as fludrocortisone [41], fluticasone propionate [42], and sofosbuvir [43].
In the initial reactions, we employed allylic gem-disubstituted alkenes with tethered amide groups as substrates in the presence of fluoroiodane(Ⅲ) reagent 1 as fluorine source in anhydrous dichloromethane (Table 1). We selected N-(2-methyl-3-phenylbut-3-en-2-yl)benzamide 2a as a model substrate for the optimization of the reaction conditions. The reaction of 2a and 1 (1.5 equiv.) afforded 5-benzyl-5-fluoro-4,4-dimethyl-2-phenyl-4,5-dihydrooxazole (3a, 27% yield) in the presence of 4 Å molecular sieve (25 mg) in anhydrous dichloromethane at room temperature for 12 h (Table 1, entry 1). Encouraged by this result, we carried out experiments to improve the chemical yield of 3a. The reaction yield was increased to 74% when AgBF4 (1.0 equiv.) was used (Table 1, entry 2). Next, we studied the solvent effect on the reaction. With the use of AgBF4 (1.0 equiv.), anhydrous dichloromethane was replaced by anhydrous acetonitrile, affording 3a with a low yield of 58% (Table 1, entry 3). The lower yield was obtained by using HBF4 (1.0 equiv.) instead of AgBF4 (1.0 equiv.) (Table 1, entry 4). To our delight, decreasing the amount of AgBF4 from 1.0 to 0.1 equiv. increased the yield of 3a to 79% (Table 1, entry 5). In addition, AgBF4 proved to be the best catalyst, as Zn(BF4)2 and BF3·OEt2 gave the amounts of 3a in 65% and 50% yield, respectively (Table 1, entries 6 and 7). When 1.2 equiv. reagent 1 were used, the yield of 3a was reduced to 61% yield (Table 1, entry 8). The yields slightly decreased when the reaction was performed under the air atmosphere (Table 1, entry 9).
 DownLoad:
CSV
DownLoad:
CSV
 |
|||||
| Entry | Solvent | 1 (equiv.) | Lewis acid (equiv.) | Time (h) | 3a yield (%)b |
| 1 | DCM | 1.5 | - | 12 | 27 |
| 2 | DCM | 1.5 | AgBF4 (1) | 4 | 74 |
| 3 | MeCN | 1.5 | AgBF4 (1) | 4 | 58 |
| 4 | DCM | 1.5 | HBF4 (1) | 2.5 | 23 |
| 5 | DCM | 1.5 | AgBF4 (10%) | 4 | 79 |
| 6 | DCM | 1.5 | Zn(BF4)2 (10%) | 4 | 65 |
| 7 | DCM | 1.5 | BF3·OEt2 (10%) | 4 | 50 |
| 8 | DCM | 1.2 | AgBF4 (10%) | 4 | 61 |
| 9c | DCM | 1.5 | AgBF4 (10%) | 6 | 73 |
| a Reaction conditions: allylic gem-disubstituted alkene 2a (0.1 mmol, 1.0 equiv.), fluoroiodane 1 (1.2−1.5 equiv.), a Lewis acid, and 4 Å molecular sieves (25 mg) were stirred in 1 mL of solvent at room temperature. b Yield after purification by column chromatography. c Under air atmosphere. |
|||||
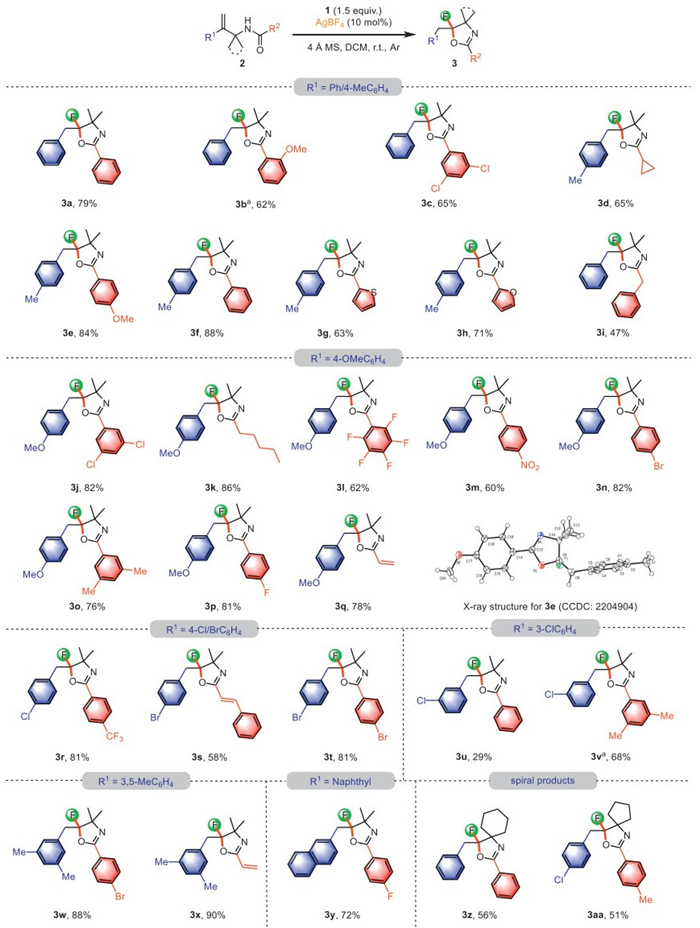
With the optimized conditions in hand (Table 1, entry 5), we were next intrigued to explore the generality and functional group compatibility of the developed protocol. As shown in Scheme 2, the ortho-methoxy substituted 2b could also undergo this transformation to deliver the desired products 3b. The substrate 2c containing 3,5-dichloro substituent also proceeded smoothly to give 3c in 65% yield. Subsequently, we maintained R1 as 4-MeC6H4 and screened a variety of substrates. Pleasantly, when R2 group was cyclopropyl (2d), 4-OMeC6H4 (2e), phenyl (2f), 2-thienyl (2g), or 2-furanyl (2h), the reaction exhibited high activities, delivering the corresponding products 3d−3h in 63%−88% yields. When R2 was benzyl, we obtained a 47% yield of the desired product 3i for 4 h at room temperature. Besides, the configuration of 3e was unambiguously determined by the X-ray crystallographic analysis. Also, when R1 was 4-OMeC6H4, the substrates bearing 3,5-ClC6H3 (2j), n-pentyl (2k), perfluorophenyl (2l), 4-NO2C6H4 (2m), 4-BrC6H4 (2n), 3,5-MeC6H3 (2o), 4-FC6H4 (2p) as well as vinyl (2q) were all compatible with this protocol. Noteworthy, the present transformation worked well when there was an electron-withdrawing substituent (chlorine or bromine) in the para-position of the phenyl ring, and the desired products 3r−3t were formed in the good to excellent yields. Specifically, when chlorine atom was present in the meta- position of the phenyl ring, the substrate 2u reacted slowly at room temperature to form the target product 3u in 29% yield, with 30% of the starting material recovered. Of note, for the substrate 2v, the desired product 3v was isolated in 68% yield by increasing the reaction temperature to 40 ℃. When R1 was 3,5-MeC6H3, two substrates bearing 4-BrC6H4 (2w) as well as vinyl (2x) were all compatible in the present reaction, giving the desired products 3w and 3x in 88% and 90% yield, respectively. In the case where R1 is a naphthyl group, the substrate 2y also exhibited good reactivity, giving the product 3y in 72% yield. Moreover, the substrates containing a cycloalkyl framework were found to be suitable for this system, affording spiral products 3z and 3aa in good yields.
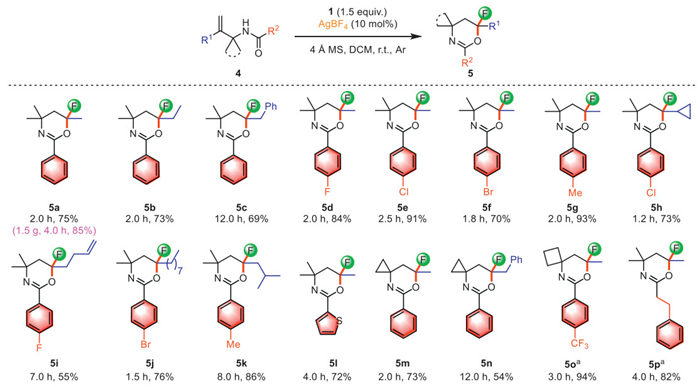
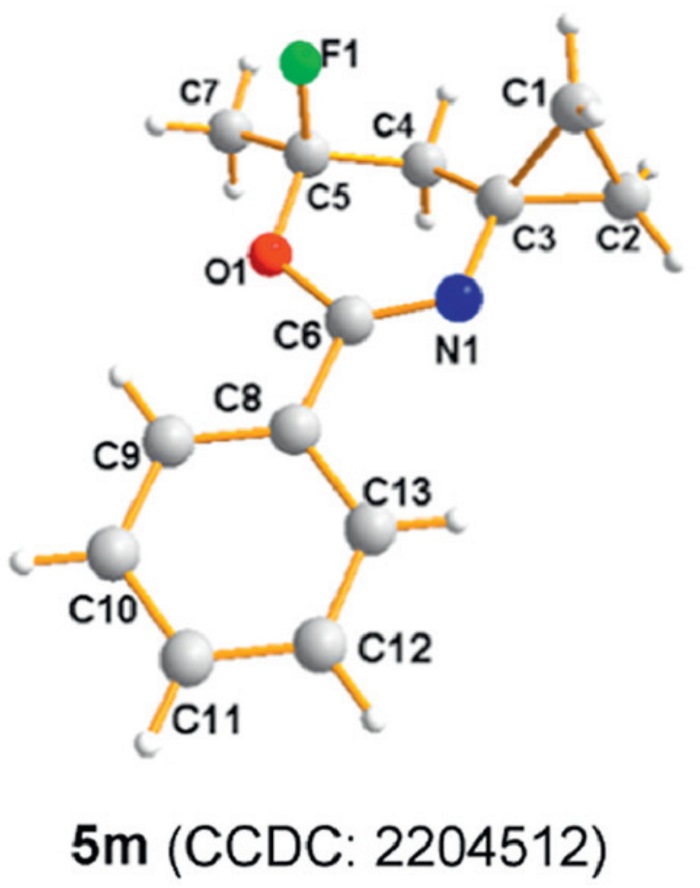
Next, we chose N-(2,3-dimethylbut-3-en-2-yl)benzamide 4a as a substrate to explore its reaction with reagent 1. To our surprise, reaction of 4a and 1 (1.5 equiv.) afforded 6-fluoro-4,4,6-trimethyl-2-phenyl-5,6-dihydro-4H-1,3-oxazine (5a, 75% yield) in the presence of AgBF4 (0.1 equiv.) and 4 Å molecular sieves (37 mg). In this reaction, we did not detect the five-membered oxazole ring fluorinated product, but obtained the six-membered oxazine ring fluorinated one, showing the excellent chemoselectivity of the reaction. We speculated that the reaction may have followed a different migration pathway. This finding inspired us to expand the scope of the discovered reaction to synthesize other six-membered oxazine ring fluorinated compounds, and the experimental results are summarized in Scheme 3. Substrates 4b and 4c with diverse alkyl groups (Et and Bn) as R1 group have also been proven to be suitable substrates, which participated in the reaction smoothly to produce products 5b and 5c in 73% and 69% yields, respectively. When R1 in the substrate was methyl group (4d-4g), the reaction conditions provided reliable good yields; in most cases, the aromatic ring of R2 with different para substituents had no significant effect on the product yields. Importantly, this rearrangement fluorination protocol was compatible with a variety of alkyl substituents, including cyclopropyl (4h), 1-butylenyl (4i), n-heptyl (4j), and isobutyl with large steric hindrance (4k), the reaction exhibited high activities, delivering the corresponding products 5h−5k in 55%−86% yields. When the substrate containing heteroaromatic substituent (4l) the reaction proceeded smoothly, delivering the corresponding product 5l in a 72% yield. Moreover, the substrates containing a cycloalkyl framework were found to be suitable for this system, affording spiral ring products 5m−5o in 54%−94% yields. Furthermore, the structure of 5m was identified by X-ray diffraction (Fig. 1). Also, when R2 was ethylbenzene, the substrate 4p was compatible with this protocol and the desired product 5p was obtained in 82% yield. To assess the scalability of the reaction, a gram-scale synthesis was conducted under standard conditions, affording product 5a (1.5 g) in 85% isolated yield.
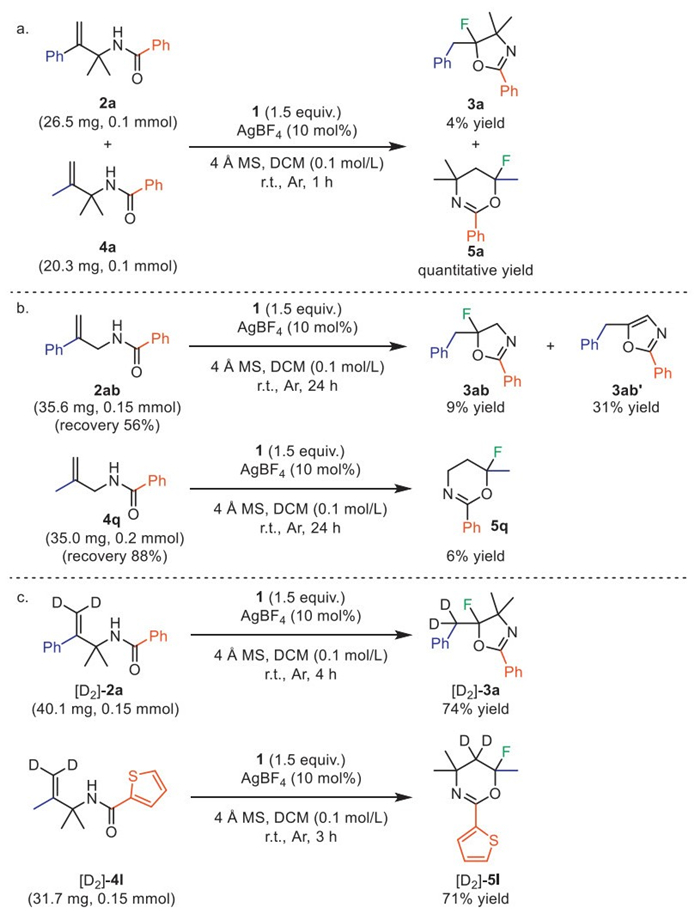
To gain insight into the mechanism of fluoroiodane(Ⅲ) reagent 1 mediated Wagner–Meerwein rearrangement fluorination, we conducted an array of control experiments (Scheme 4). To understand the difference in reaction rate between the above two reactions, we reacted 2a and 4a (the molar ratio of substrate 2a to 4a is 1:1) with fluoroiodane(Ⅲ) reagent 1 under the standard reaction conditions (Scheme 4a). This competitive reaction resulted in a yield of only 4% for 3a, compared to a quantitative yield for 5a, thus indicating that the generation of 5a is faster than that of 3a, which provides a clue for the subsequent theoretical calculation investigation for the reaction mechanism. To demonstrate the importance of the dimethyl moiety of the substrates as an essential auxiliary for the success, substrate 2ab without the dimethyl moiety was tested. The desired fluorinated product 3ab was obtained in only 9% yield and 56% of the starting material 2ab was recovered after 24 h, while the elimination by-product 3ab’ was formed in 31% yield (Scheme 4b). Similarly, when 4q was subjected to the standard reaction conditions, the desired fluorinated cyclized product was formed in only 6% yield after 24 h, with 88% of 4q remaining unreacted. These results obtained demonstrate the indispensability of the dimethyl moiety for the success of the reaction (Scheme 4b). Next, a series of H/D exchange and deuterium-labelling experiments were performed to probe the manner of the disconnection and formation of chemical bonds during the reaction (Scheme 4c). When [D2]-2a reacted with 1 in the presence of AgBF4 under the standard reaction conditions, a 74% yield of the compound [D2]-3a was obtained in a very clean reaction. The analysis of product [D2]-3a revealed that the isotopically labelled atoms are exclusively located in the benzylic position of 3a, clearly demonstrating the 1,2-shift of the aryl group in substrate 2a. Furthermore, when [D2]-4l was treated with 1, we obtained a 1,2-shift of the alkyl group deuterated product [D2]-5l in a 71% yield.
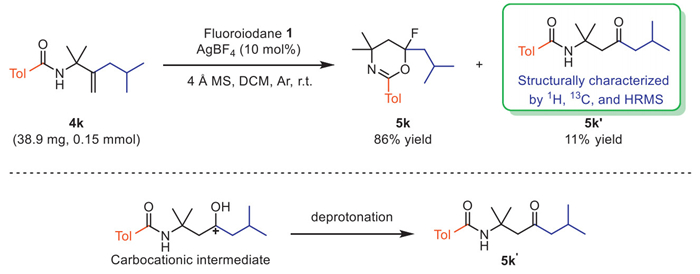
Interestingly, under standard reaction conditions, we successfully separated the by-product 5k’ by reacting the substrate 4k with 1. The formation of 5k’ could be due to a deprotonation reaction of a tertiary carbocation intermediate. Analysis of the by-product 5k’ revealed that trace water in the system competed with fluorine anion for nucleophilic reaction, then 1,2-alkyl migration occurred, and the deprotonation could afford 5k’ (Scheme 5). This experiment illustrated that the present reaction might proceed via a fluorination–migration–cyclization process.
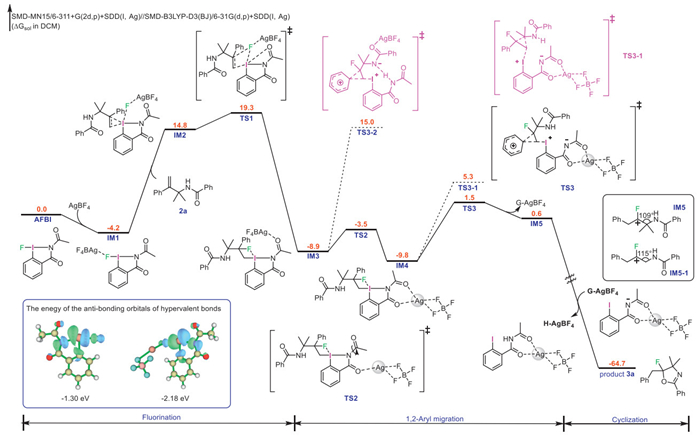
To further understand the mechanism of the above AFBI mediated cyclofluorination reaction of 2a, we performed the density functional theory (DFT) calculations on the reaction of substrate 2a and AFBI, as well as the reaction of 4a and AFBI, and the corresponding calculated Gibbs free energy profiles are depicted in Figs. 2 and 3, respectively. Regarding to the reaction of 2a and AFBI, the calculation result shows that the reaction is initiated by the formation of IM1 from AFBI and catalyst AgBF4 following a “F−coordination” activation mode [44,45] with the decrease of energy by 4.2 kcal/mol. To illustrate the enhancing effect of AgBF4 on the reactivity of AFBI, we conducted the electronic structure calculations to compare the energy of the respective anti-bonding orbital of the 3c-4e hypervalent bonds of AFBI and IM1. The presence of AgBF4 reduces the energy of the anti-bonding orbital of AFBI from -1.30 eV to -2.18 eV. This observation demonstrates that, with the aid of AgBF4, IM1 is capable of receiving electrons with greater ease than AFBI itself. Then, IM1 undergoes an electrophilic addition to the olefinic double bond in the substrate 2a to give the cyclic iodonium intermediate IM2. Subsequently, the fluoride anion attacks the most substituted carbon of the olefinic double bond to form IM3, which must overcome an activation energy barrier of 23.5 kcal/mol, making this the rate-determining step in the reaction. From IM2 to IM3, Ag(Ⅰ) stabilises the fluoride anion generated by the coordination of the olefinic double bond with the iodine(Ⅲ) centre, thus promoting the formation of C-F bond. With the help of AgBF4, the benziodazolone skeleton undergoes a conformational change due to the rotation of the N-Ac bond in IM3, leading to the formation of IM4 with an energy barrier of 5.4 kcal/mol. In IM4, Ag(Ⅰ) forms a stable 18-electrons complex with BF4− and two oxygen atoms on the benziodazolone skeleton. The phenyl migration occurs via a three-membered ring transition state TS3, which requires overcoming an activation energy barrier of 11.3 kcal/mol relative to IM4. In particular, the four-coordination mode for Ag(Ⅰ) reduces the energy by 13.5 kcal/mol compared to that of the three-coordinated transition state TS3-2. In TS3, Ag(Ⅰ) catalyst weakens the hypervalent iodine bond and enhances the leaving group ability of aryl iodine by coordinating with oxygen atoms of two carbonyl groups. This migration step results in the formation of the carbocation intermediate IM5 and an Ag(Ⅰ) complex G-AgBF4 with N-acetylbenziodazolone ligand. The competing [1,2]-aminoalkyl migration must overcome an energy barrier of 15.1 kcal/mol, which is much higher than that required for [1,2]-phenyl migration (11.3 kcal/mol), indicating that the [1,2]-phenyl migration is more favourable than [1,2]-aminoalkyl migration. Finally, IM5 forms a more thermodynamically favourable cyclization product 3a, which is exothermic by 64.7 kcal/mol. The effect of gem-dimethyl group can be explained by the Thorpe-Ingold effect, which suggests that the introduction of gem-dimethyl groups onto the carbon chains of substrates can facilitate the cyclization reactions by reducing the bond angles of the carbon chains. In IM5, the bond angle of C-C-N is 109°, whereas in the absence of the gem-dimethyl group, the angle of C-C-N is 115° in IM5-1, which suggests that the incorporation of the dimethyl group leads to a smaller angle of C-C-N in IM5, thus facilitating the cyclization process.
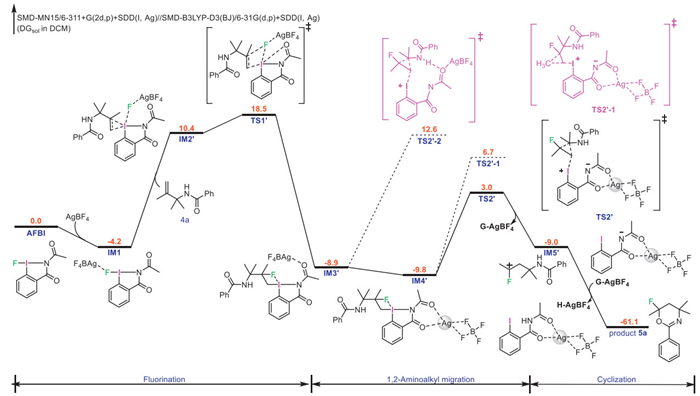
For another reaction (Fig. 3), it starts with the electrophilic addition of IM1 to olefinic double bond in the substrate 4a, yielding the cyclic iodonium intermediate IM2’. Then, the nucleophilic attack of fluoride anion occurs, with an activation energy barrier of 22.7 kcal/mol relative to IM1, to give IM3’; this process is the rate-limiting step in the reaction. The activation energy barrier of the reaction between 4a and AFBI (22.7 kcal/mol) is lower than that of the reaction between 2a and AFBI (23.5 kcal/mol), which is consistent with the results of the competition experiments (Scheme 4a). To explore the origin of the different chemoselectivity of two reactions, we calculated the energy barriers of the [1,2]-aminoalkyl migration and [1,2]-methyl migration for substrate 4a. The [1,2]-aminoalkyl migration occurs to give the carbocation intermediate IM5’ with the release of an Ag(Ⅰ) complex G-AgBF4, which needs to overcome an activation energy barrier of 12.8 kcal/mol relative to IM4’. However, the [1,2]-methyl migration has to overcome an activation energy barrier of 16.5 kcal/mol, illustrating that the [1,2]-aminoalkyl migration is more favorable than [1,2]-methyl migration. Finally, IM5’ forms a six-membered ring product with an intramolecular cyclization, which is exothermic by 61.1 kcal/mol. In conclusion, the expected order of the migration aptitude in AFBI-mediated cyclofluorination reactions of gem-disubstituted alkenes is aryl > aminoalkyl > methyl.
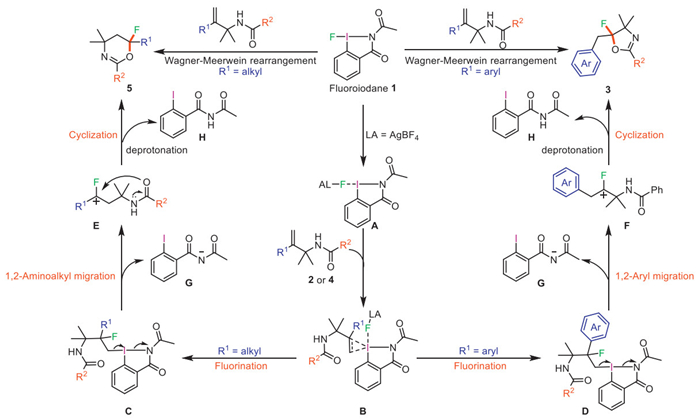
On the basis of these results and previous reports, we assumed the reaction pathway, in particular, to clarify the reaction sequence: the fluoroiodane(Ⅲ) reagent 1 is activated by the catalyst AgBF4 to form intermediate A, in which the fluorine is coordinated to AgBF4. Then, intermediate A undergoes an electrophilic addition to the alkene in gem-disubstituted alkenes 2 or 4 to give the cyclic iodonium intermediate B. An intramolecular nucleophilic attack of the fluoride occurs at the more-substituted carbon atom because it is better able to stabilize the partial positive charge and form hypervalent iodine(Ⅲ) intermediate C and D with the iodoaryl group acting as an excellent leaving group. When R1 is aryl, the cationic intermediate F is produced and imide anion G is released. Finally, an intramolecular cyclization occurs, resulting in 3 with fluorination at the quaternary center, along with the release of the N-acetyl-2-iodobenzamide H. The compound H has been isolated and characterized using ¹H NMR and ¹³C NMR. When R1 is alkyl, the migration rearrangement generated a tertiary carbocation intermediate E. Subsequently, tertiary carbocation intermediate E undergoes intramolecular nucleophilic attack of the oxygen atom of the amide group to generate product 5, along with the release of N-acetyl-2-iodobenzamide H (Fig. 4).
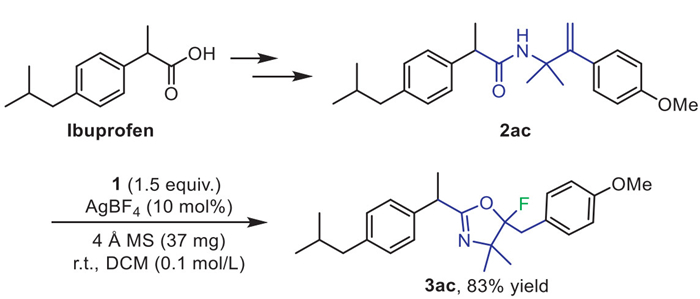
To demonstrate the synthetic utility of this novel method, we used it as the key step in the further conversion of ibuprofen (Scheme 6) [46]. Specifically, we prepared compound 2ac from ibuprofen, a commercial drug for antipyretic and analgesic, and then we carried out the reaction of 2ac with 1 and AgBF4 in DCM, which proceeded smoothly to afford 3ac in 83% yield.
In summary, we presented a mild Wagner−Meerwein rearrangement fluorination for the synthesis of highly functionalized five/six-membered heterocycles containing a tertiary carbon−fluorine bond. Interestingly, the tunable five/six-membered heterocycles selectivity is achieved by intramolecular Wagner−Meerwein rearrangement fluorination via a judicious choice of the group R1 attached to the C−C double bond. Mechanistic studies including control experiments and isolation of by-product indicated that the reaction follows a fluorination/1,2-aryl or alkyl migration/cyclization cascade reaction, which opens a new pathway of fluorinated heterocycles. The catalyst AgBF4 plays three roles in the reaction: (a) activates the fluoroiodane(Ⅲ) reagent AFBI through an “F-coordination” mode, (b) stabilises the fluoride anion in the fluorination process, and (c) promotes the rearrangement process by coordinating to two oxygen atoms on the benziodazolone skeleton in a tetra-coordinated manner. This new system features simple operation, broad substrate scope, high chemoselectivity, and excellent yields. Further development of Wagner−Meerwein rearrangement fluorination reactions using such a strategy and their applications for drug discovery is in progress.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Jing Ren: Writing – original draft, Investigation. Feng-Huan Du: Writing – original draft, Investigation. Xiaowei Chen: Investigation. Chi Zhang: Writing – review & editing, Project administration, Funding acquisition.
Supplementary material associated with this article can be found, in the online version, at doi:
X.M. Zhang, Y.Q. Tu, F.M. Zhang, Z.H. Chen, S.H. Wang, Chem. Soc. Rev. 46 (2017) 2272–2305.
R.R. Naredla, D.A. Klumpp, Chem. Rev. 113 (2013) 6905–6948. doi: 10.1021/cr4001385
F.V. Singh, T. Wirth, Synthesis 45 (2013) 2499–2511. doi: 10.1055/s-0033-1339679
B. Zhang, X. Li, B. Guo, Y. Du, Chem. Commun. 56 (2020) 14119–14136. doi: 10.1039/d0cc05354f
S.E. Wengryniuk, S. Canesi, Rearrangements and Fragmentations Mediated by Hypervalent Iodine Reagents, PATAI’S Chemistry of Functional Groups, John Wiley & Sons, Ltd, Chichester, UK, 2018, pp. 665–706.
Z. Yang, Y. Cheng, B. Zhang, et al., Chin. J. Org. Chem. 42 (2022) 3456–3505. doi: 10.6023/cjoc202206039
A. Yoshimura, V.V. Zhdankin, Chem. Rev. 124 (2024) 11108–11186. doi: 10.1021/acs.chemrev.4c00303
J. Qurban, M. Elsherbini, T. Wirth, J. Org. Chem. 82 (2017) 11872–11876. doi: 10.1021/acs.joc.7b01571
C.B. Pandey, T. Azaz, R.S. Verma, M. Mishra, J.L. Jat, B. Tiwari, J. Org. Chem. 85 (2020) 10175–10181. doi: 10.1021/acs.joc.0c00347
J. Zhang, A. Jalil, J. He, et al., Chem. Commun. 57 (2021) 7426–7429. doi: 10.1039/d1cc03110d
Q. Wang, M. Biosca, F. Himo, K.J. Szabó, Angew. Chem. Int. Ed. 60 (2021) 26327–26331. doi: 10.1002/anie.202109461
S. Manna, D. Aich, S. Hazra, S. Khandelwal, S. Panda, Chem. Sci. 15 (2024) 4989–4995. doi: 10.1039/d3sc06918d
N. Okamoto, Y. Miwa, H. Minami, K. Takeda, R. Yanada, Angew. Chem. Int. Ed. 48 (2009) 9693–9696. doi: 10.1002/anie.200904960
K. Miyamoto, Y. Sakai, S. Goda, M. Ochiai, Chem. Commun. 48 (2012) 982–984. doi: 10.1039/C2CC16360H
A. Yoshimura, K.R. Middleton, M.W. Luedtke, C. Zhu, V.V. Zhdankin, J. Org. Chem. 77 (2012) 11399–11404. doi: 10.1021/jo302375m
R. Oishi, K. Segi, H. Hamamoto, et al., Synlett 29 (2018) 1465–1468. doi: 10.1055/s-0037-1609686
T. Maegawa, R. Oishi, A. Maekawa, et al., Synthesis 54 (2022) 4095–4103. doi: 10.1055/a-1835-2188
X.Q. Li, W.K. Wang, C. Zhang, Adv. Synth. Catal. 351 (2009) 2342–2350. doi: 10.1002/adsc.200900428
L.F. Silva, F.A. Siqueira, E.C. Pedrozo, F.Y.M. Vieira, A.C. Doriguetto, Org. Lett. 9 (2007) 1433–1436. doi: 10.1021/ol070027o
M.S. Yusubov, G.A. Zholobova, I.L. Filimonova, K.W. Chi, Russ. Chem. Bull. Int. Ed. 53 (2004) 1735–1742. doi: 10.1007/s11172-005-0027-8
M. Kameyama, F.A. Siqueira, M. Gracia-Mijares, L.F. Silva, M.T.A. Silva, Molecules 16 (2011) 9421–9438. doi: 10.3390/molecules16119421
Y.C. Han, Y.D. Zhang, Q. Jia, J. Cui, C. Zhang, Org. Lett. 19 (2017) 5300–5303. doi: 10.1021/acs.orglett.7b02479
L.F. Silva, Molecules 11 (2006) 421–434. doi: 10.3390/11060421
L.F. Silva, R.S. Vasconcelos, M.A. Nogueira, Org. Lett. 10 (2008) 1017–1020. doi: 10.1021/ol800048f
T. Abo, M. Sawaguchi, H. Senboku, S. Hara, Molecules 10 (2005) 183–189. doi: 10.3390/10010183
J. Ren, F.H. Du, M.C. Jia, et al., Angew. Chem. Int. Ed. 60 (2021) 24171–24178. doi: 10.1002/anie.202108589
W.W. Chen, A.B. Cuenca, A. Shafir, Angew. Chem. Int. Ed. 59 (2020) 16294–16309. doi: 10.1002/anie.201908418
J. Tian, F. Luo, Q. Zhang, et al., J. Am. Chem. Soc. 142 (2020) 6884–6890. doi: 10.1021/jacs.0c00783
W. Carpenter, J. Org. Chem. 31 (1966) 2688–2689. doi: 10.1021/jo01346a512
N.O. Ilchenko, B.O.A. Tasch, K.J. Szabó, Angew. Chem. 126 (2014) 13111–13115. doi: 10.1002/ange.201408812
G.C. Geary, E.G. Hope, A.M. Stuart, Angew. Chem. Int. Ed. 54 (2015) 14911–14914. doi: 10.1002/anie.201507790
A. Andries-Ulmer, C. Brunner, J. Rehbein, T. Gulder, J. Am. Chem. Soc. 140 (2018) 13034–13041. doi: 10.1021/jacs.8b08350
W.X. Lv, Q. Li, J.L. Li, et al., Angew. Chem. Int. Ed. 57 (2018) 16544–16548. doi: 10.1002/anie.201810204
Z. Zhao, L. Racicot, G.K. Murphy, Angew. Chem. Int. Ed. 56 (2017) 11620–11623. doi: 10.1002/anie.201706798
Z. Zhao, A.J. To, G.K. Murphy, Chem. Commun. 55 (2019) 14821–14824. doi: 10.1039/c9cc08310c
E.V. Anslyn, D.A. Dougherty, Modern Physical Organic Chemistry, University Science Books, Sausalito, 2006, p. 658.
K.B. Wiberg, P.R. Rablen, J. Org. Chem. 85 (2020) 11741–11749. doi: 10.1021/acs.joc.0c01469
H.A. Sharma, K.M. Mennie, E.E. Kwan, E.N. Jacobsen, J. Am. Chem. Soc. 142 (2020) 16090–16096. doi: 10.1021/jacs.0c08150
J. Ren, M.C. Jia, F.H. Du, C. Zhang, Chin. Chem. Lett. 33 (2022) 4834–4837. doi: 10.1016/j.cclet.2022.01.070
J. Wu, L. Fu, R. Diao, et al., Green Synth. Catal. 6 (2025) 96–100.
J. Fried, E.F. Sabo, J. Am. Chem. Soc. 76 (1954) 1455–1456. doi: 10.1021/ja01634a101
P.M.A. Calverley, J.A. Anderson, B. Celli, et al., N. Engl. J. Med. 356 (2007) 775–789. doi: 10.1056/NEJMoa063070
G.M. Keating, A. Vaidya, Drugs 74 (2014) 273–282. doi: 10.1007/s40265-014-0179-7
B. Zhou, T. Yan, X.S. Xue, J.P. Cheng, Org. Lett. 18 (2016) 6128–6131. doi: 10.1021/acs.orglett.6b03134
B. Zhou, X.S. Xue, J.P. Cheng, Tetrahedron Lett. 58 (2017) 1287–1291.
M.W. Ha, S.M. Paek, Molecules 26 (2021) 4792. doi: 10.3390/molecules26164792

Scheme 1 (a) Wagner−Meerwein rearrangement. (b) Monofluoroiodane(Ⅲ) reagent mediated Wagner−Meerwein rearrangement (this work).

Scheme 2 Substrate scope for the 1,2-aryl migration. Reaction condition: allylic gem-disubstituted alkenes 2 (0.15 mmol, 1.0 equiv.), fluoroiodane 1 (0.225 mmol, 1.5 equiv.), AgBF4 (0.015 mmol, 10 mol%), 4 Å molecular sieves (37 mg) were stirred in dry DCM (0.1 mol/L) at room temperature. Yield after purification by column chromatography. a Reaction was conducted at 40 ℃.

Scheme 3 Substrate scope for the 1,2-alkyl migration. Reaction condition: allylic gem-disubstituted alkenes 4 (0.15 mmol, 1.0 equiv.), fluoroiodane 1 (0.225 mmol, 1.5 equiv.), AgBF4 (0.015 mmol, 10 mol%), 4 Å molecular sieves (37 mg) were stirred in dry DCM (0.1 mol/L). Yield after purification by column chromatography. a The products have a low boiling point and evaporation temperature under reduced pressure is less than 15 ℃.

Scheme 4 (a) Study on the difference in reaction rate between the two reactions. (b) Study on the reactivity of the substrates exerted by the dimethyl substitution. (c) The experiments with isotopically labelled [D2]-2a and [D2]-4l.

Figure 2 DFT-computed potential energy profile for the reaction between 2a and reagent 1 (standard state: 25 ℃, 1 mol/L).

Figure 3 DFT-computed potential energy profile for the reaction between 4a and reagent 1 (standard state: 25 ℃, 1 mol/L).
Table 1. Optimization of reaction conditionsa.
|
|||||
| Entry | Solvent | 1 (equiv.) | Lewis acid (equiv.) | Time (h) | 3a yield (%)b |
| 1 | DCM | 1.5 | - | 12 | 27 |
| 2 | DCM | 1.5 | AgBF4 (1) | 4 | 74 |
| 3 | MeCN | 1.5 | AgBF4 (1) | 4 | 58 |
| 4 | DCM | 1.5 | HBF4 (1) | 2.5 | 23 |
| 5 | DCM | 1.5 | AgBF4 (10%) | 4 | 79 |
| 6 | DCM | 1.5 | Zn(BF4)2 (10%) | 4 | 65 |
| 7 | DCM | 1.5 | BF3·OEt2 (10%) | 4 | 50 |
| 8 | DCM | 1.2 | AgBF4 (10%) | 4 | 61 |
| 9c | DCM | 1.5 | AgBF4 (10%) | 6 | 73 |
| a Reaction conditions: allylic gem-disubstituted alkene 2a (0.1 mmol, 1.0 equiv.), fluoroiodane 1 (1.2−1.5 equiv.), a Lewis acid, and 4 Å molecular sieves (25 mg) were stirred in 1 mL of solvent at room temperature. b Yield after purification by column chromatography. c Under air atmosphere. |
|||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们




 下载:
下载: