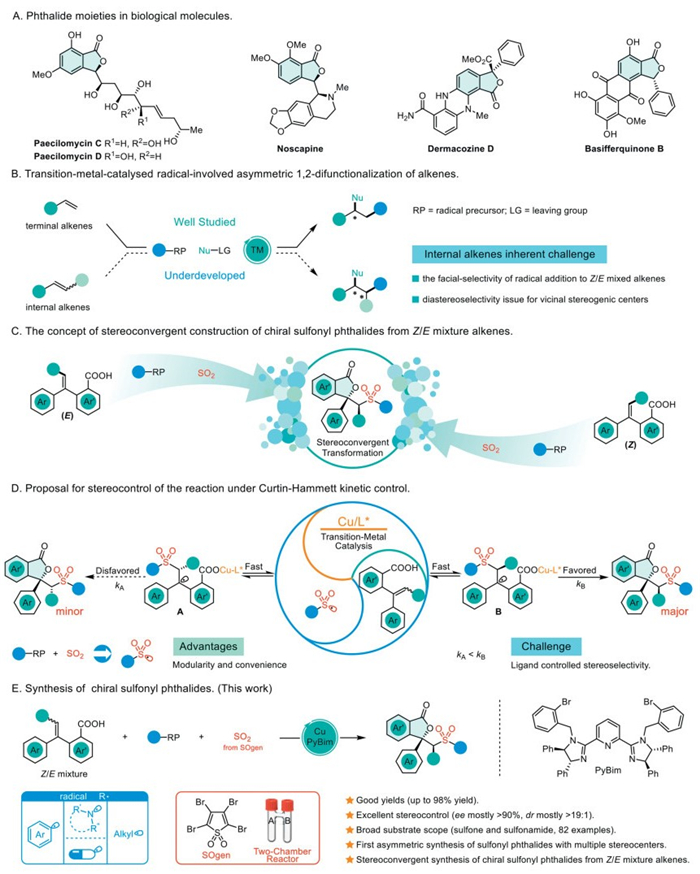
Figure 1.
Copper-catalyzed stereoconvergent transformation synthesis of chiral sulfonyl phthalide.

Chiral phthalides are prevalent as key structural motifs across a broad spectrum of pharmaceuticals, and natural products exhibiting diverse biological properties (Fig. 1A) [1–8]. Additionally, sulfonyl groups are significant pharmacophores capable of enhancing biological activity, modulating acid-base properties, and augmenting solubility [9–11]. Therefore, devising an asymmetric synthesis strategy to amalgamate these two divergent structures is of paramount research significance.
Starting from alkenes is a crucial approach for synthesizing phthalides [12–17], but classical methods for producing internal alkenes primarily yield Z/E mixtures that are challenging to separate [18]. Stereoconvergence strategy, on the other hand, can achieve the conversion of substrates with poor stereoselectivity into products with a single stereo configuration through precise control of reaction conditions and mechanisms, with a theoretical yield of 100% [19–22]. Therefore, the direct stereoconvergent transformation of Z/E alkene mixtures into optically pure products holds substantial practical importance. Recent advances in transition-metal-catalyzed, radical-initiated asymmetric alkene difunctionalization have significantly enhanced the asymmetric formation of carbon-carbon and carbon-heteroatom bonds [23–46]. Predominantly, these reactions have mainly been successful with terminal alkenes, as the radical addition to internal alkenes often encounters issues with facial-selectivity in the addition to Z/E mixed alkenes and diastereoselectivity at vicinal stereogenic centers. Consequently, there are limited studies that simultaneously generate two vicinal stereocenters using internal alkenes (Fig. 1B) [25,29,47].
Drawing inspiration from the research conducted by Liu [25] and Han [48], we propose employing Z/E internal alkene mixtures for the construction of chiral sulfonyl phthalides utilizing a stereoconvergent strategy (Fig. 1C). Recognizing that the addition of sulfonyl radicals to internal alkenes constitutes a rapid and reversible process [49–51], combined with the modularity and convenience advantages of sulfur dioxide insertion for the construction of diverse sulfonyl fragments [52–61], we envisaged that sulfonyl radicals, generating via sulfur dioxide insertion into radical precursors, were added to Z/E alkene mixtures, leveraging Curtin-Hammett kinetic control to yield diastereoisomers A and B (Fig. 1D). The disparity in the rates of subsequent cyclization processes (kA < kB) facilitates the swift conversion of intermediate A to intermediate B, facilitating stereoconvergence in the synthesis of chiral sulfonyl phthalides [62,63]. The key element of this strategy lies in the selection of suitable ligands, which not only enable enantioselective control but also modulate the cyclization rates of intermediates A and B, thereby facilitating diastereoselective control. The successful implementation of this strategy is expected to open new avenues for the stereoconvergence of Z/E mixed alkene substrates.
Recently, our group designed a cost-effective, solid, and bench-stable external SO2 surrogate (SOgen) in combination with two-chamber reactors for sulfur dioxide insertion reactions [43,64–66]. Herein, we present an efficient approach to synthesize chiral sulfonyl phthalides via SO2 insertion enabled by SOgen and the Cu/PyBim catalytic system (Fig. 1). This approach exhibits high yield, exceptional stereoselectivity, and a broad substrate scope, enabling the stereoconvergent synthesis of chiral sulfonyl phthalides from Z/E alkene mixtures. Notably, it represents the first successful synthesis of a series of sulfonyl phthalide compounds with multiple stereocenters. The reaction achieves stereoconvergence of Z/E mixed alkenes through Curtin-Hammett kinetic control. The rapid and reversible addition of sulfonyl radicals to internal alkenes, combined with the strategic selection of appropriate ligands, is crucial for achieving high stereoselectivity in this reaction.
To assess the effectiveness of a stereoconvergent transformation strategy, we selected a 1:2 Z/E ratio mixture of 2-(1-phenylprop-1-en-1-yl)benzoic acid (1a) as the model substrate. This substrate was then reacted with 4-Me-PhN2BF4 (2a) and SO2 (from SOgen), to evaluate the reaction outcomes (Table 1). In our initial experiments, we investigated various chiral ligands including Box (L1), PyBox (L2), and PyBim (L3–L10). The findings demonstrated that the 2-bromobenzyl substituent in ligand L8 substantially enhances both enantioselectivity and diastereoselectivity, making it the most effective for exceptional stereoselectivity. As reported in the literature, we hypothesize that although both PyBox and PyBim ligands share similar chiral environments, they exhibit differences in their electronic properties. In this reaction, the stereoselectivity is significantly influenced by the distinct electronic characteristics of the ligands [61]. Building on these results, we further explored additional parameters of the reaction. Initially, various solvents were evaluated, with dichloromethane (DCM) emerging as the optimal choice (entries 1–5). Regarding the choice of base, 2,6-di-tert-butylpyridine proved more effective than either 2,6-di-Me-pyridine, Na2CO3 or K2CO3 (entries 6–8). This efficacy may be attributed to the steric hindrance provided by the tert-butyl groups in 2,6-di-tert-butylpyridine, which effectively mitigates catalyst deactivation by pyridine. When the substrate 1a, characterized by an 8:1 Z/E ratio, was employed, the reaction consistently yielded product retaining the same configuration, suggesting that the reaction is governed by Curtin-Hammett kinetic control and achieves high stereoselectivity through stereoconvergent transformation. The substitution of sulfur dioxide surrogates or TsCl for SOgen invariably led to diminished yield and stereoselectivity in the reaction (entries 10–13). These findings clearly underscore the indispensable role of SOgen in enabling this efficient transformation.
 DownLoad:
CSV
DownLoad:
CSV
 |
||||
| Entry | Deviation from standard conditions | Yield (%)b | ee (%)c | drd |
| 1 | None | 96 (92) | 96 | >20:1 |
| 2 | MeCN instead of DCM | 67 | 83 | >20:1 |
| 3 | 1,4-Dioxane instead of DCM | trace | – | – |
| 4 | 2-Me-THF instead of DCM | 5 | 96 | >20:1 |
| 5 | DCE instead of DCM | 97 | 92 | 19:1 |
| 6 | 2,6-Di-Me-pyridine instead of 2,6-di-tert-butylpyridine | 21 | 85 | 17:1 |
| 7 | Na2CO3 instead of 2,6-di-tert-butylpyridine | 32 | 94 | 14:1 |
| 8 | K2CO3 instead of 2,6-di-tert-butylpyridine | 74 | 94 | >20:1 |
| 9 | Z/E = 8:1 of 1a as the substrate | 95 | 96 | >20:1 |
| 10e | DABSO as SO2 source | 83 | 92 | 16:1 |
| 11e | Na2S2O5 as SO2 source | 58 | 92 | 14:1 |
| 12e | K2S2O5 as SO2 source | 60 | 90 | 10:1 |
| 13e | TsCl as sulfonyl precursor | 8 | 96 | >20:1 |
| a Reaction conditions: Chamber A (SO2 generation): SOgen (0.41 mmol), 1-methyl-4-vinylbenzene (0.40 mmol), tetradecane (1.0 mL), at 100 ℃ for 10 min. Chamber B: 1a (0.20 mmol), 2a (0.40 mmol, 2 equiv.), Cu(MeCN)4PF6 (0.01 mmol, 5 mol%), L (0.012 mmol, 6 mol%), Base (0.40 mmol, 2 equiv.), DCM (2.0 mL) at room temperature for 12 h under argon atmosphere. b Yields were determined by 1H NMR using 1,3,5-trimethoxybenzene as the internal standard. c The ee value of 3a was determined by HPLC analysis on a chiral stationary phase. d The dr value of 3a was determined by 1H NMR. e The reaction was set up in an 8 mL vial. |
||||
Upon establishing the optimized reaction conditions, we then evaluated their general applicability. We initially explored the scope of the transformation with respect to Z/E mixture alkenes 1 (Scheme 1). These alkenes, characterized by electron-neutral substituents, yielded the corresponding products (3a–3c) with moderate to good yields and high stereoselectivities. Additionally, internal alkenes featuring both electron-donating and electron-withdrawing substituents were found to be compatible with this transformation, producing a series of products (3d–3m). The absolute configuration of 3k was determined using X-ray crystallographic analysis.
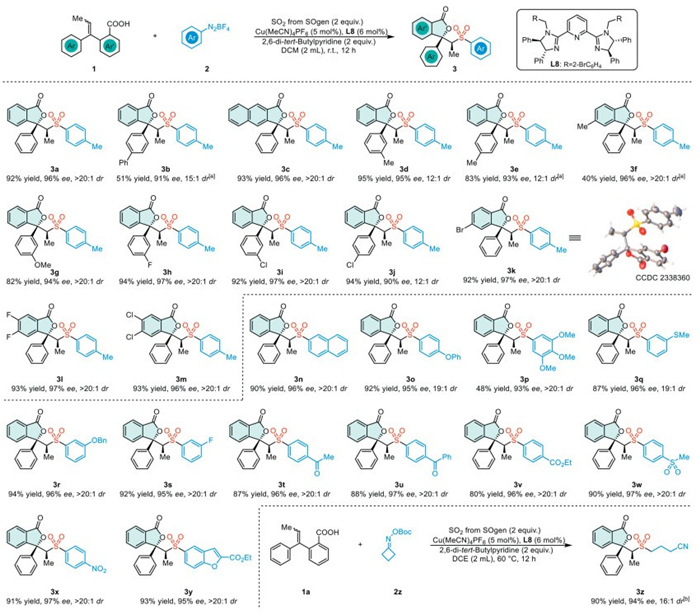
Furthermore, we extended our investigation to a variety of substituted aryldiazonium salts in the transformation (Scheme 1). These salts, featuring electron-neutral, electron-donating, and electron-withdrawing substituents, demonstrated excellent stereoselectivity in this reaction (3n–3x). Additionally, the heterocyclic diazonium salt has demonstrated efficacy as a radical precursor, reliably producing the corresponding product (3y) with excellent stereoselectivity. Moreover, with precise adjustments of the reaction conditions, cyclobutanone oxime ester can be effectively transformed, exhibiting notable control over stereoselectivity (3z).
Encouraged by these promising results, we explored the compatibility of terminal alkenes in our experiments. To our satisfaction, the reaction between terminal alkenes 4 and aryldiazonium salts 2 proceeded smoothly, consistently yielding the anticipated products (Scheme 2). The catalytic system exhibited remarkable tolerance to a variety of functional groups, efficiently converting various substituted terminal alkenes into products with moderate to high yields and excellent enantioselectivities (5a–5l). We further investigated a spectrum of aryl diazonium salts bearing a variety of substituents. These included electron-neutral groups such as naphthalene and biphenyl, electron-donating groups (Me, i-Pr, Bn, and OMe), and electron-withdrawing groups (m-SMe, m-OBn, halogens, acetylene, ketones, ester, and NO2). All the investigated substituents were compatible with the reaction, producing the corresponding products (5m–5af) in 49%–98% yields with 85%–96% ee. The absolute configuration of 5m was determined using X-ray crystallographic analysis. Heterocyclic compounds such as dibenzothiophene and benzofuran diazonium salts can also be utilized in this reaction, yielding products 5ag and 5ah with favorable outcomes, respectively.
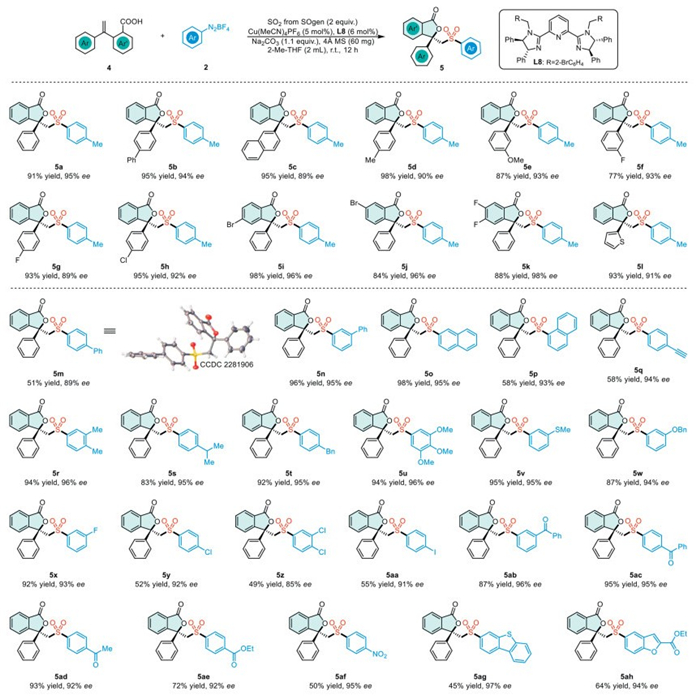
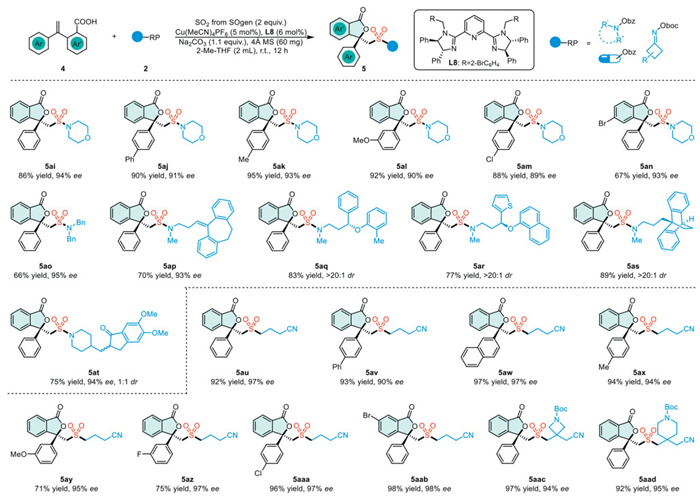
We subsequently investigated the feasibility of synthesizing chiral sulfonyl phthalides using radical species other than aryl diazonium salts (Scheme 3). Carboxylic acids with diverse functional groups on the phenyl ring or O-acyl-N-hydroxylamines consistently yielded the desired chiral sulfonylamide phthalides (5ai−5at). It is noteworthy that sulfur dioxide can act as a pharmaceutical linker, facilitating the one-step synthesis of compounds containing medicinal molecules, sulfonyl groups, and chiral phthalides (5ap−5at). We have established a reaction for vicinal sulfonyl-esterification of alkenes to construct chiral sulfonyl phthalides by using alkyl radicals derived from cycloketone oxime esters. A broad spectrum of carboxylic acids and cycloketone oxime esters was found to be compatible with the established reaction, resulting in the formation of products with moderate to high yields and high enantioselectivities (5au–5aad).
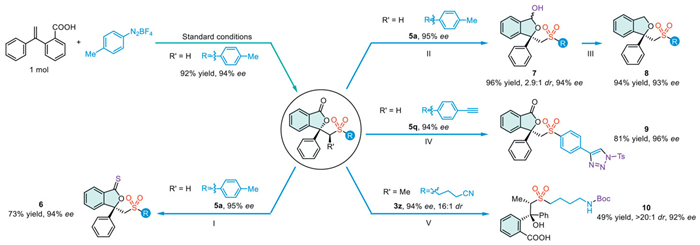
To demonstrate the practical applicability, we conducted a scaled-up reaction (1.0 mmol), which produced the target compound 5a with a 92% yield and 94% ee (Scheme 4). Moreover, the chiral sulfonyl phthalides synthesized via this method can be easily converted into other valuable compounds that while preserving its enantioselectivity. For instance, the reaction of compound 5a with Lawesson reagent resulted in the formation of the thiolated product 6. Furthermore, phthalide in compound 5a can be selectively reduced possess a variety of functional groups, using DIBAl-H to yield hemiacetal 7. This intermediate is then further reduced by Et3SiH to synthesize compound 8. Similarly, compound 5q was subjected to a copper-catalyzed click reaction, which introduced a crucial triazole moiety into the chiral sulfonyl phthalide, resulting in the generation of product 9 with excellent yield. Finally, compound 5au was converted into the Boc-protected amino acid 10 via a nickel-catalyzed reduction process. Compound 10 contains several important functional groups, including an amino group, a carboxyl group, a hydroxyl group, and a sulfonyl group. Additionally, it features two conserved stereocenters, which are commonly found in various pharmaceutical agents and bioactive compounds [25].
To elucidate the mechanism more clearly, a series of experiments was conducted to achieve a comprehensive understanding of the process (Scheme 5). Initially, upon adding 3.0 equiv. of TEMPO under standard conditions, the desired product 5a was not detected. Instead, the formation of TEMPO adducts 11 and 12 was observed (Scheme 5A). Secondly, upon introducing 1,1-diphenylethene into the reaction system, merely trace amount of the sulfonylated products were produced. Conversely, compounds 13 and 14, originating from aryl radicals and sulfonyl radicals, were detected (Scheme 5B). The results suggest that the reaction may involve a radical process. Additionally, a linear correlation was observed between the enantiopurity of product 5a and that of ligand L8 (Scheme 5C). This suggests the involvement of a single chiral ligand and a copper complex in the enantiocontrol step [67]. Subsequently, we pre-synthesized the chiral catalyst Cat1 and employed it to catalyze the reaction under standard conditions, achieving the target product with a 76% yield and 95% ee (Scheme 5D). This outcome suggests that Cat1 possesses the same catalytic active center as the originally synthesized catalyst. Finally, under standard conditions, substrates with E/Z ratios of 2:1 and 1:8 in alkenes consistently yield target product (S,S)-3a, which exhibits a dominant stereoconfiguration (Scheme 5E). This outcome demonstrates the reaction's capability for stereoconvergent synthesis of chiral sulfonyl phthalides from Z/E alkene mixtures. Drawing on previous literature, it is highly probable that the envisioned Curtin-Hammett kinetic control was operative under our experimental conditions [62,63].
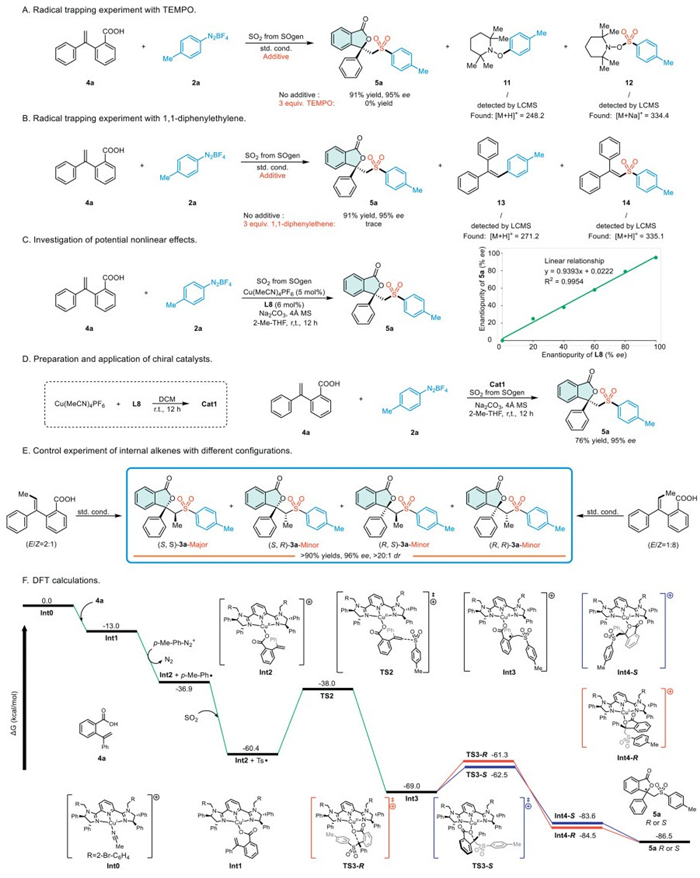
To gain a comprehensive understanding of the underlying mechanisms, we conducted extensive investigations utilizing density functional theory (DFT) calculations in this study. These calculations focused on the reaction pathway of the terminal alkene substrate 4a in the asymmetric construction of the chiral sulfonyl phthalide 5a. The precatalyst [Cu(MeCN)4]⊕ engages in a ligand exchange with L8, leading to the formation of the complex [CuⅠMeCN-L8]⊕ (Int0) (Scheme 5F). First, the deprotonated terminal alkene substrate (4a) complexes with Int0 to form the CuⅠ-L8-aryl carboxylic acid complex (Int1), accompanied by a decrease in Gibbs free energy of 13.0 kcal/mol. Next, through a single electron transfer (SET) process, Int1, in conjunction with the p-methylphenyl diazonium compound, generates the [CuⅡ-L8-aryl carboxylic acid]⊕ complex (Int2) and an aryl radical, resulting in a decrease in Gibbs free energy of 23.9 kcal/mol. Subsequently, the aryl radical captures sulfur dioxide, undergoing a barrierless process to generate an aryl sulfonyl radical (Gibbs free energy decrease by 23.5 kcal/mol). While aryl radicals are theoretically predisposed to engage in direct addition reactions with alkenes, DFT calculations conducted by the Lei group reveal the presence of an energy barrier in this addition process [68]. Therefore, under conditions where sulfur dioxide is present, aryl radicals are more inclined to capture sulfur dioxide to form sulfonyl radicals, rather than undergoing radical addition with alkenes. This calculation result is consistent with the observation that no by-products from the direct addition of aryl radicals to alkenes, in the absence of sulfur dioxide capture, were detected in the experiment. Then, the sulfonyl radical undergoes radical addition to the C = C bond of [CuⅡ-L8-aryl carboxylic acid]⊕ (Int2) via the transition state TS2, leading to the formation of a new divalent copper complex (Int3) that contains a carbon radical. The process releases an energy of 8.6 kcal/mol, which aligns with the well-documented tendency of radicals to react with unsaturated bonds [69]. Following the radical addition, the oxygen atom coordinated to copper in Int3 approaches the carbon radical and transitions through the TS3 transition state to generate the new copper complex Int4. Int3 undergoes distinct reaction intermediates TS3-R and TS3-S to produce the diastereoisomers Int4-R and Int4-S. In this process, an intramolecular radical attack on the carboxylate oxygen results in the generation of a chiral carbon atom, which is influenced by the orientation of the approaching planar phenyl radical. This leads to the formation of Int4-R and Int4-S, which release distinct enantiomers. Computational analysis demonstrates that TS3-S is lower in energy by 1.2 kcal/mol compared to TS3-R, indicating a preference for the formation of the S-product.
To gain a more comprehensive understanding of the enantioselectivity of the reaction, we conducted a comparative analysis of the non-covalent interactions in the transition states TS3-R and TS3-S (Scheme 6A) [70]. The analysis revealed that the phenyl group on the ligand in TS3-S forms stronger π–π interactions with the aryl group on the substrate compared to TS3-R. Furthermore, TS3-S features additional π–π interactions between the phenyl groups on the substrate and both the phenyl group on the ligand and the Ts group on the substrate. These interactions result in a larger RDG surface area in TS3-S relative to TS3-R, thereby providing a more favorable environment for enantioselectivity. Overall, the analysis indicates that TS3-S demonstrates more favorable non-covalent interactions than TS3-R, which likely contributes to its enhanced enantioselectivity.
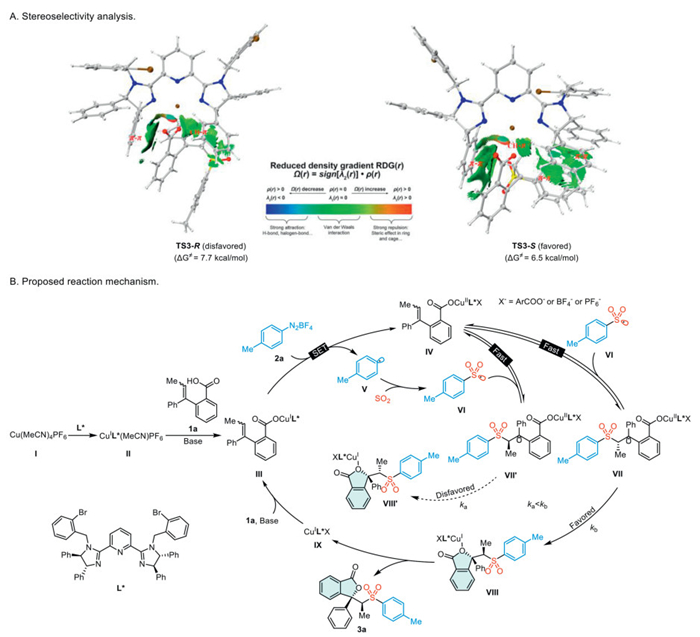
Based on the combined evidence from experimental results and literature reports, we propose a potential mechanism for the reaction (Scheme 6B) [25,62,63]. In the presence of a base, species Ⅱ, CuⅠ-L*(MeCN)PF6, reacts with reactant 1a, ultimately forming species Ⅲ, [CuⅠ-L*O2CAr]. Subsequently, species Ⅲ undergoes a SET process with aryldiazonium salt 2a, resulting in the formation of [CuⅡ-L*O2CArX] species Ⅳ and p-methylphenyl radical Ⅴ. Then, the radical V captures sulfur dioxide to generate a sulfonyl radical Ⅵ. Subsequently, the sulfonyl radical rapidly and reversibly adds to intermediate Ⅳ, resulting in the formation of alkyl radicals Ⅶ and Ⅶ'. Due to the significantly slower rate of intramolecular radical substitution reactions in intermediate Ⅶ' compared to intermediate Ⅶ (ka < kb), intermediate Ⅶ' rapidly and reversibly converts back to Ⅶ. Subsequently, species Ⅶ undergoes an intramolecular radical substitution, leading to the formation of the CuⅠ complex Ⅷ. This is followed by the dissociation of species Ⅷ, resulting in the production of compound 3a and [CuⅠ-L*X] Ⅸ.
In summary, we have developed a method that allows for the stereoconvergent synthesis of chiral sulfonyl phthalides using Z/E alkene mixtures under a Cu/PyBim catalytic system. This method not only enables the successful synthesis of sulfonyl phthalides with multiple stereocenters for the first time, but also exhibits broad applicability across a range of terminal and internal alkene substrates, as well as diverse aryl, alkyl, and nitrogen radical precursors, under exceptionally mild reaction conditions. The Cu/PyBim catalytic system is crucial for controlling the stereoselectivity of this reaction. The experimental results suggest that the reaction utilizes a Curtin-Hammett kinetic control strategy to achieve stereoconvergent construction of compounds with high stereoselectivity. We anticipate that this strategy will have broad implications and practical applications in the field of organic synthesis.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Zhiqian Chang: Writing – original draft, Formal analysis, Data curation, Conceptualization. Xiaochun He: Writing – original draft, Methodology, Formal analysis, Data curation, Conceptualization. Xuemei Zhang: Writing – review & editing, Writing – original draft, Project administration, Investigation, Funding acquisition, Conceptualization. Zhong Lian: Writing – review & editing, Supervision.
This work is supported by National Natural Science Foundation of China (No. 22301192), Sichuan Science and Technology Program (No. 2024NSFSC1124), West China Hospital 135 project (No. ZYYC23017), Postdoctor Research Fund of West China Hospital, Sichuan University (No. 2024HXBH027) and Hebei Province Higher Education Institutions Scientific Research Project (No. QN2025195). The authors would like to thank Shiyanjia Lab (
Supplementary material associated with this article can be found, in the online version, at doi:
L. Xu, Z. He, J. Xue, et al., J. Nat. Prod. 73 (2010) 885–889. doi: 10.1021/np900853n
B. Sung, K.S. Ahn, B.B. Aggarwal, Cancer Res. 70 (2010) 3259–3268.
W.M. Abdel-Mageed, B.F. Milne, M. Wagner, et al., Org. Biomol. Chem. 8 (2010) 2352–2362. doi: 10.1039/c001445a
M. Azuma, M. Yoshida, S. Horinouchi, et al., Agric. Biol. Chem. 54 (1990) 1441–1446.
S.K. Gadakh, A. Sudalai, Tetrahedron Lett. 57 (2016) 25–28. doi: 10.1016/j.tetlet.2015.11.047
H. Mitsuhashi, T. Muramatsu, U. Nagai, et al., Chem. Pharm. Bull. 11 (1963) 1317–1319. doi: 10.1248/cpb.11.1317
W. Wang, X.-X. Cha, J. Reiner, et al., Eur. J. Med. Chem. 45 (2010) 1941–1946. doi: 10.1016/j.ejmech.2010.01.036
D. Xingxing, D. Pan, X. Cen, et al., Drug Metab. Dispos. 41 (2013) 430. doi: 10.1124/dmd.112.049684
P. Ertl, E. Altmann, J.M. McKenna, J. Med. Chem. 63 (2020) 8408–8418. doi: 10.1021/acs.jmedchem.0c00754
D.H. Jornada, G.F. Dos Santos Fernandes, D.E. Chiba, et al., Molecules 21 (2016) 42.
P. Wu, Y. Su, X. Liu, et al., MedChemComm 3 (2012) 659–662. doi: 10.1039/c2md00255h
Y. Ge, Z. Han, Z. Wang, et al., Angew. Chem. Int. Ed. 57 (2018) 13140–13144. doi: 10.1002/anie.201807639
J.M. Cabrera, J. Tauber, M.J. Krische, Angew. Chem. Int. Ed. 57 (2018) 1390–1393. doi: 10.1002/anie.201712015
Y.L. Pan, H.L. Zheng, J. Wang, et al., ACS Catal. 10 (2020) 8069–8076. doi: 10.1021/acscatal.0c02585
D.H. Dethe, N.C. Beeralingappa, S.A. Siddiqui, et al., J. Org. Chem. 87 (2022) 4617–4630. doi: 10.1021/acs.joc.1c02961
A.R. Kizhakkayil Mangadan, J. Liu, A. Aponick, Angew. Chem. Int. Ed. 60 (2021) 22224–22229. doi: 10.1002/anie.202108336
Y. Li, J. Xu, J.C.A. Oliveira, et al., ACS Catal. 14 (2024) 8160–8167. doi: 10.1021/acscatal.4c01886
T. Takeda, Synthesis 2004 (2004) 1532 -1532. doi: 10.1055/s-2004-829036
V. Bhat, E.R. Welin, X. Guo, et al., Chem. Rev. 117 (2017) 4528–4561. doi: 10.1021/acs.chemrev.6b00731
E.L. Eliel, S.H. Wilen, Stereochemistry of Organic Compounds, John Wiley & Sons, 1994.
Z.C. Litman, Y. Wang, H. Zhao, et al., Nature 560 (2018) 355–359. doi: 10.1038/s41586-018-0413-7
R. Mao, D.M. Taylor, D.J. Wackelin, et al., Nat. Synth. 3 (2024) 256–264.
Z.L. Li, G.C. Fang, Q.S. Gu, et al., Chem. Soc. Rev. 49 (2020) 32–48. doi: 10.1039/c9cs00681h
Q.S. Gu, Z.L. Li, X.Y. Liu, Acc. Chem. Res. 53 (2020) 170–181. doi: 10.1021/acs.accounts.9b00381
X.T. Li, L. Lv, T. Wang, et al., Chem 6 (2020) 1692–1706. doi: 10.3390/app10051692
W. Zhang, Y. Tian, X.D. Liu, et al., Angew. Chem. Int. Ed. 63 (2024) e202319850. doi: 10.1002/anie.202319850
L. Fu, X. Chen, W. Fan, et al., J. Am. Chem. Soc. 145 (2023) 13476–13483. doi: 10.1021/jacs.3c04498
F. Wang, P. Chen, G. Liu, Acc. Chem. Res. 51 (2018) 2036–2046. doi: 10.1021/acs.accounts.8b00265
D. Wang, F. Wang, P. Chen, et al., Angew. Chem. Int. Ed. 56 (2017) 2054–2058. doi: 10.1002/anie.201611850
W. Sha, L. Deng, S. Ni, et al., ACS Catal. 8 (2018) 7489–7494. doi: 10.1021/acscatal.8b01863
Z. Nie, M.F. Chiou, J. Cui, et al., Angew. Chem. Int. Ed. 61 (2022) e202202077. doi: 10.1002/anie.202202077
R. Zhu, S.L. Buchwald, Angew. Chem. Int. Ed. 52 (2013) 12655–12658. doi: 10.1002/anie.201307790
R. Zhu, S.L. Buchwald, J. Am. Chem. Soc. 137 (2015) 8069–8077. doi: 10.1021/jacs.5b04821
M. Xue, J. Cui, X. Zhu, et al., Angew. Chem. Int. Ed. 62 (2023) e202304275. doi: 10.1002/anie.202304275
F.D. Lu, L.Q. Lu, G.F. He, et al., J. Am. Chem. Soc. 143 (2021) 4168–4173. doi: 10.1021/jacs.1c01260
J. Chen, Y.J. Liang, P.Z. Wang, et al., J. Am. Chem. Soc. 143 (2021) 13382–13392. doi: 10.1021/jacs.1c06535
P.Z. Wang, X. Wu, Y. Cheng, et al., Angew. Chem. Int. Ed. 60 (2021) 22956–22962. doi: 10.1002/anie.202110084
G. Lei, H. Zhang, B. Chen, et al., Chem. Sci. 11 (2020) 1623–1628. doi: 10.1039/c9sc04029c
X. Ma, G. Zhang, ACS Catal. 11 (2021) 5108–5118. doi: 10.1021/acscatal.0c05576
L.F. Fan, R. Liu, X.Y. Ruan, et al., Nat. Synth. 1 (2022) 946–955. doi: 10.1038/s44160-022-00172-8
Y. Jin, L.F. Fan, E.W.H. Ng, et al., J. Am. Chem. Soc. 145 (2023) 22031–22040. doi: 10.1021/jacs.3c07008
N. Fu, L. Song, J. Liu, et al., J. Am. Chem. Soc. 141 (2019) 14480–14485. doi: 10.1021/jacs.9b03296
L. Chen, X. Zhang, M. Zhou, et al., ACS Catal. 12 (2022) 10764–10770. doi: 10.1021/acscatal.2c02297
L. Ge, H. Zhou, M.F. Chiou, et al., Nat. Catal. 4 (2021) 28–35.
D. Lv, Q. Sun, H. Zhou, et al., Angew. Chem. Int. Ed. 60 (2021) 12455–12460. doi: 10.1002/anie.202017175
H.T. Zhao, J.N. Lin, W. Shu, Chem. Eur. J. 30 (2024) e202402712. doi: 10.1002/chem.202402712
Z. Bai, H. Zhang, H. Wang, et al., J. Am. Chem. Soc. 143 (2021) 1195–1202. doi: 10.1021/jacs.0c12333
Y. Wang, L. Deng, J. Zhou, et al., Adv. Synth. Catal. 360 (2018) 1060–1065. doi: 10.1002/adsc.201701532
A.S. Gozdz, P. Maslak, J. Org. Chem. 56 (1991) 2179–2189. doi: 10.1021/jo00006a040
V.I. Timokhin, S. Gastaldi, M.P. Bertrand, et al., J. Org. Chem. 68 (2003) 3532–3537. doi: 10.1021/jo026870b
M. Tamba, K. Dajka, C. Ferreri, et al., J. Am. Chem. Soc. 129 (2007) 8716–8723. doi: 10.1021/ja070626q
S.M. Hell, C.F. Meyer, G. Laudadio, et al., J. Am. Chem. Soc. 142 (2020) 720–725. doi: 10.1021/jacs.9b13071
M.J. Tilby, D.F. Dewez, L.R.E. Pantaine, et al., ACS Catal. 12 (2022) 6060–6067. doi: 10.1021/acscatal.2c01442
J. Huang, F. Liu, L.H. Zeng, et al., Nat. Commun. 13 (2022) 7081.
J. Zhang, P. Wang, Y. Li, et al., Chem. Commun. 59 (2023) 3821–3826. doi: 10.1039/d2cc06339e
Y. Li, M. Wang, X. Jiang, Chin. J. Chem. 38 (2020) 1521–1525. doi: 10.1002/cjoc.202000198
Y. Li, S. Chen, M. Wang, et al., Angew. Chem. Int. Ed. 59 (2020) 8907–8911. doi: 10.1002/anie.202001589
H. Li, Y. Zhang, X. Yang, et al., Angew. Chem. Int. Ed. 62 (2023) e202300159.
H. Li, Y. Zhang, X. Zou, et al., ACS Catal. 14 (2024) 3664–3674. doi: 10.1021/acscatal.4c00049
S. Cao, W. Hong, Z. Ye, et al., Nat. Commun. 12 (2021) 2377.
Z. Chang, X. Zhang, H. Lv, et al., Adv. Sci. 11 (2024) 2309069.
J.I. Seeman, Chem. Rev. 83 (1983) 83–134. doi: 10.1021/cr00054a001
J.I. Seeman, J. Chem. Educ. 63 (1986) 42. doi: 10.1021/ed063p42
X. Jia, S. Kramer, T. Skrydstrup, et al., Angew. Chem. Int. Ed. 60 (2021) 7353–7359. doi: 10.1002/anie.202014111
L. Luo, X. Zhang, C. Huang, et al., Org. Chem. Front. 11 (2024) 1678–1684. doi: 10.1039/d3qo02004e
C. Huang, X. Zhang, L. Luo, et al., Org. Chem. Front. 11 (2024) 4275–4283. doi: 10.1039/d4qo00671b
T. Satyanarayana, S. Abraham, H.B. Kagan, Angew. Chem. Int. Ed. 48 (2009) 456–494. doi: 10.1002/anie.200705241
W. Yu, S. Wang, M. He, et al., Angew. Chem. Int. Ed. 62 (2023) e202219166.
B. Giese, Angew. Chem. Int. Ed. 22 (1983) 753–764.
J. Contreras-García, E.R. Johnson, S. Keinan, et al., J. Chem. Theory Comput. 7 (2011) 625–632. doi: 10.1021/ct100641a

Figure 1 Copper-catalyzed stereoconvergent transformation synthesis of chiral sulfonyl phthalide.

Scheme 1 Substrate scope. Reaction conditions: Chamber B: 1 (0.20 mmol), 2 (0.40 mmol, 2 equiv.), Cu(MeCN)4PF6 (0.01 mmol, 5 mol%), L8 (0.012 mmol, 6 mol%), 2,6-di-tert-butylpyridine (0.40 mmol, 2 equiv.), DCM (2.0 mL) at room temperature for 12 h under argon atmosphere. a Na2CO3 (0.22 mmol, 1.1 equiv.) as base, 4 Å molecular sieve (60 mg) as additive, 2-Me-THF (2.0 mL) as solvent. b DCE (2.0 mL) as solvent, 60 ℃ for 12 h.

Scheme 2 Substrate scope. Reaction conditions: Chamber B: 4 (0.20 mmol), 2 (0.40 mmol, 2 equiv.), Cu(MeCN)4PF6 (0.01 mmol, 5 mol%), L8 (0.012 mmol, 6 mol%), Na2CO3 (0.22 mmol, 1.1 equiv.), 4 Å molecular sieve (60 mg), 2-Me-THF (2.0 mL) at room temperature for 12 h under argon atmosphere.

Scheme 3 Substrate scope. Reaction conditions: Chamber B: 4 (0.20 mmol), 2 (0.40 mmol, 2 equiv.), Cu(MeCN)4PF6 (0.01 mmol, 5 mol%), L8 (0.012 mmol, 6 mol%), Na2CO3 (0.22 mmol, 1.1 equiv.), 4 Å molecular sieve (60 mg), 2-Me-THF (2.0 mL) at room temperature for 12 h under argon atmosphere.

Scheme 4 Scale-up reaction and transformations of chiral sulfonyl phthalides. Reaction conditions: (Ⅰ) Lawesson reagent (2.25 equiv.), toluene, reflux; (Ⅱ) DIBAl-H (1.1 equiv.), CH2Cl2, −78 ℃, 1 h; (Ⅳ) Et3SiH (5 equiv.), BF3·Et2O (4 equiv.), CH2Cl2, r.t., 3 h; (Ⅳ) TsN3 (1.1 equiv.), CuTc (10 mol%), toluene, r.t., 12 h; (Ⅴ) NiCl2·6H2O (3.0 equiv.), (Boc)2O (3.0 equiv.), NaBH4 (10 equiv.), MeOH, r.t., 12 h.
Table 1. Optimization of reaction conditions.a
|
||||
| Entry | Deviation from standard conditions | Yield (%)b | ee (%)c | drd |
| 1 | None | 96 (92) | 96 | >20:1 |
| 2 | MeCN instead of DCM | 67 | 83 | >20:1 |
| 3 | 1,4-Dioxane instead of DCM | trace | – | – |
| 4 | 2-Me-THF instead of DCM | 5 | 96 | >20:1 |
| 5 | DCE instead of DCM | 97 | 92 | 19:1 |
| 6 | 2,6-Di-Me-pyridine instead of 2,6-di-tert-butylpyridine | 21 | 85 | 17:1 |
| 7 | Na2CO3 instead of 2,6-di-tert-butylpyridine | 32 | 94 | 14:1 |
| 8 | K2CO3 instead of 2,6-di-tert-butylpyridine | 74 | 94 | >20:1 |
| 9 | Z/E = 8:1 of 1a as the substrate | 95 | 96 | >20:1 |
| 10e | DABSO as SO2 source | 83 | 92 | 16:1 |
| 11e | Na2S2O5 as SO2 source | 58 | 92 | 14:1 |
| 12e | K2S2O5 as SO2 source | 60 | 90 | 10:1 |
| 13e | TsCl as sulfonyl precursor | 8 | 96 | >20:1 |
| a Reaction conditions: Chamber A (SO2 generation): SOgen (0.41 mmol), 1-methyl-4-vinylbenzene (0.40 mmol), tetradecane (1.0 mL), at 100 ℃ for 10 min. Chamber B: 1a (0.20 mmol), 2a (0.40 mmol, 2 equiv.), Cu(MeCN)4PF6 (0.01 mmol, 5 mol%), L (0.012 mmol, 6 mol%), Base (0.40 mmol, 2 equiv.), DCM (2.0 mL) at room temperature for 12 h under argon atmosphere. b Yields were determined by 1H NMR using 1,3,5-trimethoxybenzene as the internal standard. c The ee value of 3a was determined by HPLC analysis on a chiral stationary phase. d The dr value of 3a was determined by 1H NMR. e The reaction was set up in an 8 mL vial. |
||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们


 下载:
下载: