Key Laboratory of Cluster Science of Ministry of Education, School of Chemistry and Chemical Engineering, Liangxiang Campus, Beijing Institute of Technology, Beijing 102488, China
b.
College of Physical Science and Technology, Yangzhou University, Yangzhou 225009, China
c.
Analysis & Testing Center, School of Chemistry and Chemical Engineering, Liangxiang Campus, Beijing Institute of Technology, Beijing 102488, China
Received Date:
27 March 2025 Accepted Date:
28 May 2025 Revised Date:
20 May 2025 Available Online:
15 June 2026
Abstract:
Tuning molecular chirality through external stimuli stands at the forefront of chemical science. Here, we systematically examine the stereodynamic properties of a series of Cu(Ⅱ)-based perovskite single crystals, CystaH2[CuCl(4-x)Brx] (CystaH2 = protonated cystamine cation, x = 0.74, 0.80, 1.05, 1.11, 1.18), which undergo a polar-to-nonpolar ferroelectric phase transition. In these compounds, the symmetry breaking structural change is governed by an unusually thermally driven P ↔ M helical inversion of the helical CystaH22+ organic cations. The conformational inversion can be substantially modulated by bromide doping. As the bromide content increases, the phase-transition temperature progressively shifts closer to room temperature, decreasing by up to 49 K. Detailed structural analyses reveal that the chiral inversion of CystaH22+ is highly sensitive to local chemical environments, and this doping strategy offers an effective route for precisely governing temperature-mediated chiral transformations, thereby expanding the design space for advanced chiral materials.
Chirality, the property of being non-superimposable on its mirror image, is extensively observed in nature and living organisms and profoundly influences fundamental biological processes, including asymmetric synthesis, protein engineering, chiral sensing, and information science. Owing to its diverse applications in chiral recognition, asymmetric catalysis, and optical modulation, numerous studies have explored the underlying mechanisms of chirality [1–8]. Manipulating molecular chirality via external stimuli represents a cutting-edge frontier in chemical science [9–11]. Several strategies have already been reported in soft materials such as liquids, polymers, and gel systems. In these reports, the molecular chirality can be tuned by photoirradiation [12–14], pH variation [15,16], solvents [17,18], and redox reactions [19,20]. For example, Huang et al. describe a unimolecular chirality switch based on a pillar[5]azacrown pseudo [1]catenane, wherein acid/base triggers can reversibly invert the chirality through protonation- or deprotonation-induced conformational changes between self-inclusion and self-exclusion arrangements [15]. Katsonis et al. illustrate a light-driven dynamic helix inversion of liquid crystals by incorporating photoreactive molecular motors capable of trans–cis photoisomerization into a nematic liquid crystal. In contrast, reversing molecular chirality in solid-state materials remains a considerable challenge [14].
In this context, protonated cystamine (CystaH22+) has attracted substantial attention due to its propensity for adopting either M- or P- helicity upon crystallization, and the disulfide moiety exhibits remarkable structural flexibility arising from the low-energy barrier for C–S–S–C flipping [21–26]. Consequently, the molecular chirality can be interconverted between the M- and P- helical conformations under external stimuli. This stereodynamic property was first reported by Prof. Mercier in a single crystal of CystaH2[PbI5]·H3O, where half of the homochiral CystaH22+ cations are converted into their enantiomers upon heating to 348 K [26]. More recently, it has been demonstrated that the chiral inversion of CystaH22+ in the two-dimensional perovskite ferroelectric CystaH2[CuCl4] can reverse the spontaneous polarization, enabling the simultaneous switching of molecular chirality and polarization under an electric field [27]. Such chiral inversion in solid-state materials may find widespread applications in advanced enantioselective sensors, spintronics, and other chiroptical electronic devices. Nonetheless, a detailed investigation of the stereodynamic nature of CystaH22+ in crystalline materials is still lacking.
In perovskite ferroelectrics, halogen substitution is commonly employed to tailor phase-transition temperatures [28–33]. Introducing Br⁻ dopants into the structure of CystaH2[CuCl4] could potentially regulate the dynamic behavior of CystaH22+. In this study, we prepared a series of bromine-substituted Cu(Ⅱ)-based organic–inorganic hybrid materials, CystaH2[CuCl(4-x)Brx] (x = 0.74, 0.80, 1.05, 1.11, 1.18) via halogen doping strategy, briefly denoting as 1Cl0.81Br0.19, 1Cl0.80Br0.20, 1Cl0.74Br0.26, 1Cl0.72Br0.28, and 1Cl0.70Br0.30. Analogous to the parent compound, the CystaH22+ units in these crystals adopt a homochiral M- or P-helicity in a single crystal at room temperature. Upon heating, the linear organic cations undergo an order-disorder phase transition that induces chiral inversion. As Br doping alters the octahedral configuration and enlarges the lattice cavity, the chiral-inversion temperature progressively shifts to room temperature with increasing Br⁻ content.
Plate-like crystals of CystaH2[CuCl(4-x)Brx] (CystaH2 = [(NH3(CH2)2SS(CH2)2NH3)2+]) (x ≠ 0) were synthesized by evaporating aqueous HCl solutions containing CuBr2 and CystaH2Cl2. By varying the molar ratio of chloride to bromide ions in the precursor solution, a series of small brownish-yellow crystals distinct from the light-yellow CystaH2[CuCl4] were obtained, presumably due to bromide incorporation. Notably, when the CuBr2 exceeds five equivalents relative to CystaH22+, a distinct pale-purple crystal containing Cu(Ⅰ) is obtained (Fig. S1 in Supporting information).
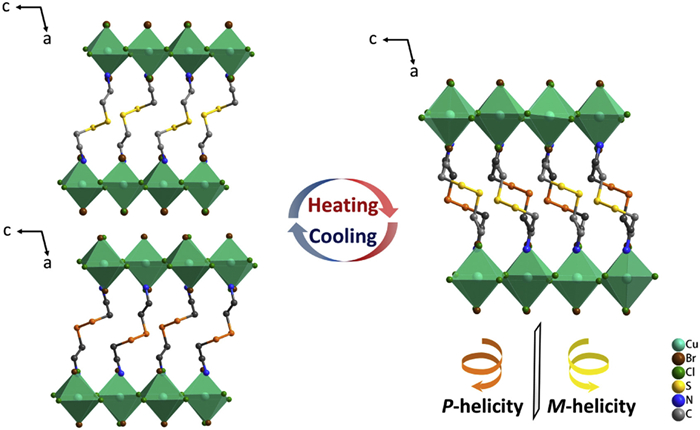
Single-crystal X-ray diffraction (SC-XRD) analyses confirmed that these crystals are isostructural and crystallize in the chiral polar space group P21, retaining the same packing arrangement as CystaH2[CuCl4]. Each doped compound contains one CystaH22+ cation and a bromide-substituted copper halide anion in the asymmetric unit. The corner-sharing octahedra of the hybrid copper halides form two-dimensional layers, with CystaH22+ cations situated in the interlayer space. In the LTP, the CystaH22+ cations adopt a homochiral P- or M- conformation anchored to the inorganic framework by N–H···X hydrogen bonds (Fig. 1 and Fig. S2 in Supporting information).
Figure 1
Figure 1.
Crystal structures at the low-temperature and high-temperature phase (LTP, HTP) of CystaH2[CuCl(4-x)Brx]. The organic cations in an individual single crystal adopt either P- or M- helicity at the LTP. At the HTP, both P- and M- helical conformations exist.
To determine the extent of bromide incorporation in crystals grown from solutions with varying chloride-to-bromide ratios, combined scanning electron microscopy-energy dispersive X-ray spectroscopy (SEM-EDS) analyses were performed. Elemental mapping revealed a uniform halogen distribution across the crystal surface, which retains its layered morphology as observed in the SEM images (Fig. S3 in Supporting information). Quantitative EDS measurements indicated that the bromide content in these crystals is x = 0.74, 0.80, 1.05, 1.11, and 1.18, increasing in parallel with the bromide ratio in the precursor solution (Table 1).
Table 1
Table 1.
Summary of elemental composition determined from EDS.
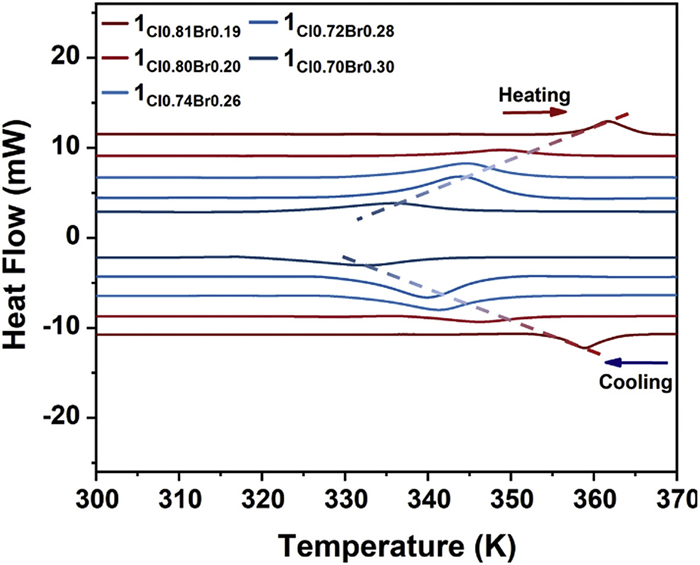
Differential scanning calorimetry (DSC) analyses were conducted to investigate the thermal behavior of the bromide-substituted crystals. As shown in Fig. 2 and Fig. S4 (Supporting information), exothermic and endothermic events were observed upon cooling and heating, respectively. Consistent with prior studies, the phase transition temperature of CystaH2[CuCl4] is 384 K. In contrast, the transition points of 1Cl0.81Br0.19, 1Cl0.80Br0.20, 1Cl0.74Br0.26, 1Cl0.72Br0.28, and 1Cl0.70Br0.30 occur at 362, 355, 345, 343, and 335 K, respectively. Hence, increasing the bromide content progressively lowers the phase transition temperature, culminating in a reduction of approximately 49 K relative to CystaH2[CuCl4] (Fig. S5 in Supporting information).
Figure 2
Figure 2.
DSC curves of the crystals CystaH2[CuCl(4-x)Brx] at a scanning rate of 10 K/min.
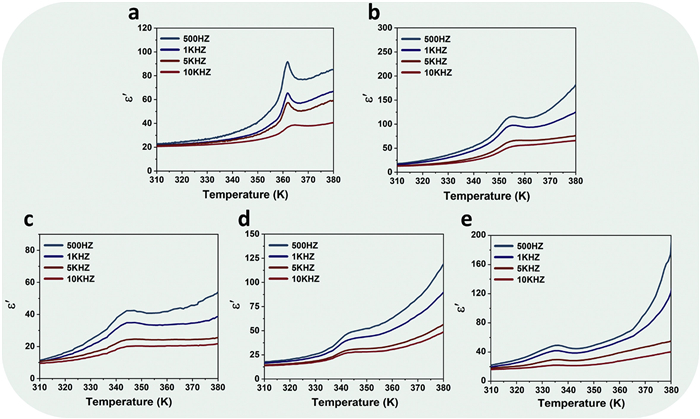
The occurrence of molecular phase transitions is closely connected with their dielectric properties. Accordingly, temperature-dependent dielectric constants were measured for this series of crystals (Fig. 3). Upon heating, the dielectric constant curves exhibit pronounced peaks at the phase transition temperature, with amplitudes several times higher than those in the LTP. Moreover, these peaks shift to lower temperatures with increasing bromine content, consistent with the phase transition temperatures derived from DSC. Concurrently, the peak profiles evolve from sharp to broadened, likely due to local inhomogeneities introduced by doping. According to the literature, halogen substitution can distort the lattice, generate internal stress and point defects, and create an inhomogeneous local polar environment. These factors broaden the distribution of local phase-transition temperatures, leading to the observed widening of temperature-dependent dielectric peak [34,35]. In addition, the dielectric switching behavior is frequency-dependent: At lower frequencies, the dielectric constants undergo significant variation that gradually diminishes in magnitude at higher frequencies. Meanwhile, this change also displays a bromine inclusion-related feature which may facilitate their application in piezoelectric and electrocaloric materials [36–38].
Figure 3
Figure 3.
Temperature-dependent dielectric constant curves at different frequencies. The dielectric constant for (a) 1Cl0.81Br0.19, (b) 1Cl0.80Br0.20, (c) 1Cl0.74Br0.26, (d) 1Cl0.72Br0.28, and (e) 1Cl0.70Br0.30.
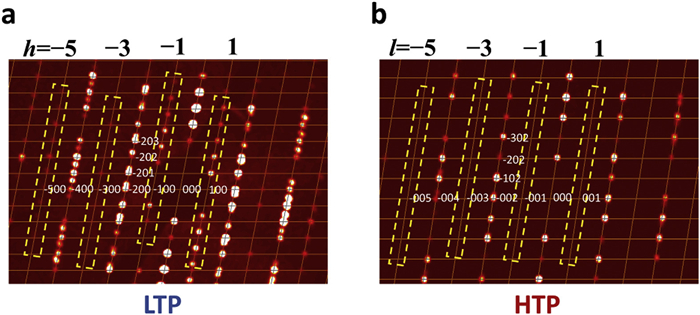
The phase transition is related to the conformational change of the protonated CystaH22+ cation. As these compounds are isostructural, 1Cl0.81Br0.19, 1Cl0.74Br0.26, and 1Cl0.70Br0.30 were selected to investigate the phase transition mechanisms. This transformation entails a pronounced symmetry shift from the polar chiral space group P21 in the LTP to the centrosymmetric space group P21/c in the HTP (Fig. 4 and Fig. S6 in Supporting information). The two symmetry elements (E, C2) in the LTP increase to four (E, C2, i, and σh) in the HTP (Fig. S7 in Supporting information). The emergence of these new symmetry elements reflects a change in molecular chirality. In the LTP, each single crystal adopts a homochiral P- or M-conformation. Upon heating, the sulfur atoms' displacement ellipsoids become considerably more pronounced, indicating two mirror orientations emerge at specific positions in the HTP: The disorder states can be deconvoluted into two components in both P- and M- conformations, confirming the chiral inversion of the organic cation during the phase transition (Fig. 1). The corresponding noncentrosymmetric-to-centrosymmetric transition was substantiated by reciprocal space analysis: In the LTP, diffraction spots h0l: l = 2n + 1 are observed (due to the exchange of the a and c axes, h = 2n + 1 in LTP) but are absent in the HTP. Detailed crystallographic data are listed in Tables S2–S4 (Supporting information). The acentric-to-centrosymmetric transition of single crystal induced by the chiral inversion of CystaH22+ was verified by temperature-dependent second-harmonic generation (SHG) measurements. For 1Cl0.81Br0.19, a strong SHG response appears below 362 K and disappears above this temperature, indicating that bromide incorporation allows fine control over the critical temperature of switchable SHG halogenides (Fig. S8 in Supporting information).
Figure 4
Figure 4.
Reciprocal spaces reconstructed from the frames of SCXRD. Presented are the diffraction patterns of h0l in (a) LTP and (b) HTP. During the phase transition, the crystallographic a and c axes undergo interconversion.
Notably, the N–H···X hydrogen bond interactions of 1Cl0.81Br0.19, 1Cl0.74Br0.26, and 1Cl0.70Br0.30 also undergo adjustments during this process, with the shortest N···X distances changing from 3.271, 3.288, 3.357 Å in LTP to 3.301, 3.268, 3.336 Å in HTP respectively (Fig. S9 in Supporting information).
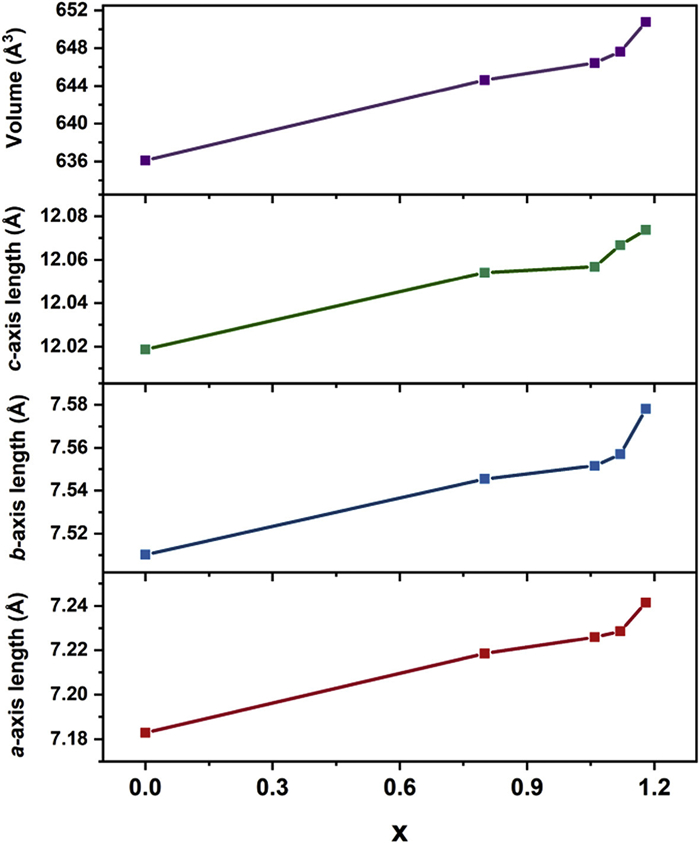
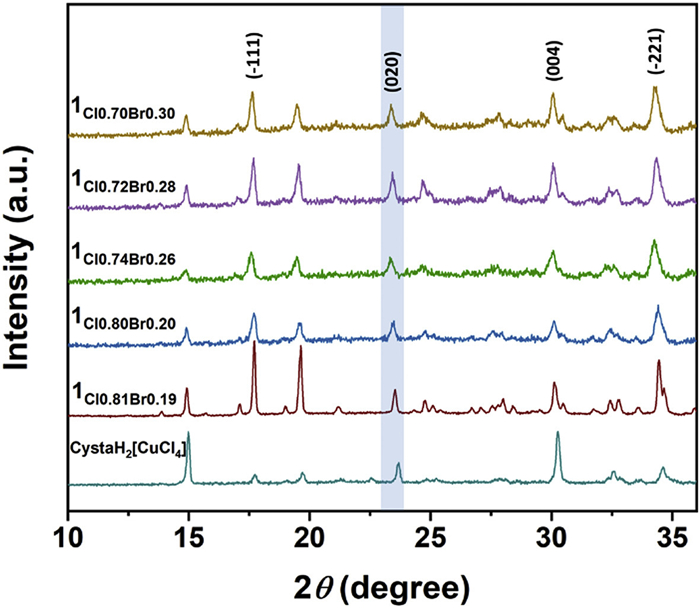
The shift in the critical temperature for molecular-chirality inversion is closely connected to the bromine incorporation. The increasing bromine content results in an enlargement in the unit cell parameters. The length of a-axis increases from 7.183 Å to 7.241 Å, the b-axis changes from 7.510 Å to 7.578 Å, and the c-axis increases from 12.019 Å to 12.074 Å. The elongation of cell length leads to the notable expansion of the unit cell volume from 636.09 Å3 to 650.76 Å3 (Fig. 5). The powder X-ray diffraction (PXRD) patterns of CystaH2[CuCl(4-x)Brx] crystals display the same trend. The diffraction peaks exhibit similar positions and shapes, suggesting the same space group, and as the value of x increases, the diffraction peaks shift to lower angle (Fig. 6 and Fig. S10 in Supporting information). Taking the (020) plane as an example, the diffraction peak (2θ) is located at 23.69° without bromine substitution (x = 0) and it shifts to 23.35° when x = 1.18. According to Bragg's law nλ = 2dsinθ (n = 1, 2, 3···), such shifts denote continuous expansion of the interplanar distances as the bromide ratio rises [39–41].
Figure 5
Figure 5.
Influence of bromine ratio x on the crystallographic a-, b-, c-axis lengths and unit cell volume.
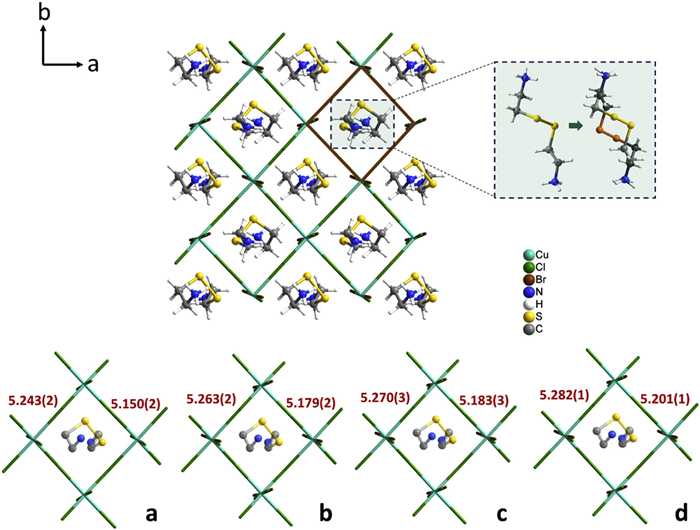
Furthermore, bromine incorporation enlarges halogen-ion size and weakens electrostatic interactions between the cations and the framework, given that smaller, more electronegative chlorine atoms (radius 99 pm, electronegativity 3.16) are replaced by larger, less electronegative bromine atoms (radius 114 pm, electronegativity 2.96). As for the specific substitution sites, the crystallographic investigation was carefully conducted through structural refinement by a full matrix least-squares against F2 method within SHELX [42,43]. Initial refinement attempts considering all halogen sites as fully occupied chlorine atoms were proved unreasonable as the unusually large displacement parameters and the elongated thermal ellipsoids at the interlayer halogen position near the apex of the cation. Considering the site selectivity of the halogen substitution, these apical halogen sites were assigned as mixed Cl/Br occupancy with free variable refinement in the subsequent process, and the disappearance of strange residual electron peaks in differential Fourier electron density maps of the three refinement schemes confirms the reasonability of the statistically disordered structure at the apical position (Fig. S11 in Supporting information). Actually, the substituted sites may be influenced by the steric hindrance or the coordination environment. As shown in Fig. S12 (Supporting information), the difference of Cu–Cl bond lengths between apical and equatorial positions of CystaH2[CuCl(4-x)Brx] progressively increases from 0.018 Å to 0.102 Å with higher bromine doping concentration, demonstrating bromine ions' preferential occupation of the less sterically hindered apical sites. The incorporation of bromine leads to an increase in the cavity where the CystaH22+ cation is located. As shown in Fig. 7 and Fig. S13 (Supporting information), the lengths of the anionic octahedral framework (measured by Cu–Cu distance) elongate from 5.197 Å to 5.242 Å as the bromine ratio x increases which endows the cations with greater freedom for conformational inversion. The reduced intermolecular interactions and larger free volume confer the CystaH22+ cation with greater molecular degree of freedom, resulting in a decrease in the chiral inversion temperature of this compound toward room temperature.
Figure 7
Figure 7.
Octahedral framework of anion observed along the c-axis. The Cu–Cu distance (Å) is used to represent the size of the anion framework for (a) CystaH2[CuCl4], (b) 1Cl0.81Br0.19, (c) 1Cl0.74Br0.26, and (d) 1Cl0.70Br0.30.
In conclusion, a series of Cu-based organic-inorganic hybrid perovskites, CystaH2[CuCl(4-x)Brx], with varying degrees of bromine doping, has been synthesized. These compounds undergo a structural transformation from a homochiral P-/M- conformation to a dynamically racemic arrangement as the stereo-flexible cations rotate in response to thermal stimuli. Bromine incorporation, which modifies both the unit cell parameters and the chemical environment of copper-halide framework surrounding the cystamine cation, influences the molecular mobility within the crystalline lattice. Consequently, the chiral transformation temperature decreases by about 49 K, shifting from 384 K to 335 K, and leads to systematic changes in the dielectric properties. Future investigations may explore alternative chemical or physical stimuli for tuning the chiral transition temperature, advancing the broader application potential of chiral chemistry and informing the design of new functional materials.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by the National Natural Science Foundation of China (Nos. 22071009 (Z.-S. Yao), 22371015 and 92061106 (J. Tao)) and Xiaomi Young Scholar Program (Z.-S. Yao). The technical support from the staff at the Analysis & Testing Center, Beijing Institute of Technology is also appreciated.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.111386.
Figure 1
Crystal structures at the low-temperature and high-temperature phase (LTP, HTP) of CystaH2[CuCl(4-x)Brx]. The organic cations in an individual single crystal adopt either P- or M- helicity at the LTP. At the HTP, both P- and M- helical conformations exist.
Figure 3
Temperature-dependent dielectric constant curves at different frequencies. The dielectric constant for (a) 1Cl0.81Br0.19, (b) 1Cl0.80Br0.20, (c) 1Cl0.74Br0.26, (d) 1Cl0.72Br0.28, and (e) 1Cl0.70Br0.30.
Figure 4
Reciprocal spaces reconstructed from the frames of SCXRD. Presented are the diffraction patterns of h0l in (a) LTP and (b) HTP. During the phase transition, the crystallographic a and c axes undergo interconversion.
Figure 7
Octahedral framework of anion observed along the c-axis. The Cu–Cu distance (Å) is used to represent the size of the anion framework for (a) CystaH2[CuCl4], (b) 1Cl0.81Br0.19, (c) 1Cl0.74Br0.26, and (d) 1Cl0.70Br0.30.

 DownLoad:
DownLoad:

 下载:
下载:








 下载:
下载:
