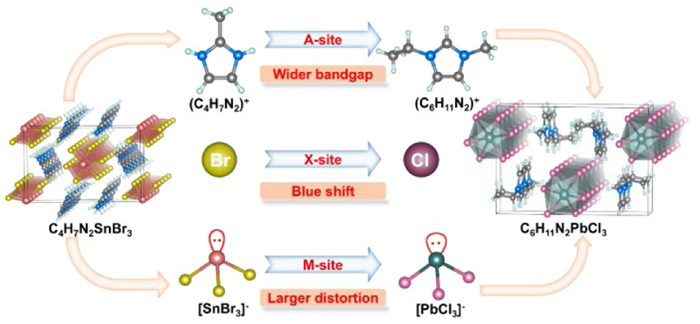
Figure 1.
Design ideas.
Synergistic triple-site engineering in ABX3-type hybrid halides for high-performance nonlinear optical crystals
Yuwei Kang , Can Yang , Jun Zhang , Qi Wu

ABX3-type organic-inorganic hybrid halides have garnered significant attention as a promising class of materials within the optoelectronics domain, characterized by their remarkable optical attributes and the capacity for structural customization [1–3]. Their nonlinear optical (NLO) characteristics are vital for their applications in photodetection, light-emitting diodes (LEDs), and solar energy conversion [4–11]. To fully exploit their potential, the development of non-centrosymmetric (NCS) structures in NLO optical crystals is imperative for optimal performance. However, achieving this is a complex task, as it requires a delicate balance between maximizing the second-harmonic generation (SHG) response and broadening the optical bandgap, suitable birefringence, all of which are critical for high-performance NLO materials [12–15].
The ABX3 framework offers a versatile platform for constructing NCS structures and enhancing NLO performance through modifications at the A-site, B-site, and X-site. The introduction of stereochemically active lone-pair cations at the B-site, such as Pb2+, Sn2+, Bi3+, and Sb3+, is a strategic move to impart high NLO properties to anionic groups like [PbCl3]–, [PbBr3]– and [SnBr3]– [16–18]. Although the geometric arrangement of these anionic groups plays a significant role in determining the macroscopic NLO coefficients, it is not sufficient to ensure non-centrosymmetric crystallization, which is essential for the development of NLO-active materials with desired properties. Therefore, circumventing the centrosymmetric crystallization of anionic groups is a fundamental challenge. Moreover, for high-performance NLO crystals, the anionic groups must be aligned parallelly with large second-order micropolarizability to enable synergistic action, presenting another challenge in achieving the optimal arrangement [19–23].
Structural analysis of ABX3-type organic-inorganic hybrid halides has revealed the crucial role of cations in modulating the arrangement of ABX3 anions, which is intricately linked to the cations' variable coordination environments [24,25]. Consequently, the strategic design of A-site cations has become an effective approach to achieving ideal performance, attracting extensive research attention [26,27]. For instance, Prof. Mao reported that the judicious tuning of A-site cations can significantly enhance the SHG response in the perovskite structure of AGeBr3 [28]. Our previous studies also confirmed that incorporating larger planar π-conjugated organic cations can lead to an augmentation of the bandgap [3]. In the realm of halide anion design for the X-site, it is widely acknowledged that the contribution of these anions to SHG decreases in the order of I > Br > Cl, while the opposite trend is observed for their impact on the optical bandgap [29–32]. These strategies have led to notable advancements.
Despite advances in A/B/X-site modifications, most studies focus on single/dual-site engineering, which fails to simultaneously address symmetry breaking, bandgap widening, and polarization alignment. A holistic triple-site modulation strategy is urgently needed. The tri-site modulation approach has proven to be a highly efficient strategy for synthesizing target compounds, enhancing the precision of crystal engineering and promising new avenues for material property optimization. Building on this foundation, our research aims to orchestrate modulation across the three critical sites to achieve the optimal performance attributes essential for NLO crystals. By meticulously fine-tuning the interactions at these sites, we strive to unlock the full potential of NLO materials.
In our quest to push the boundaries of NLO crystals, we have strategically incorporated planar π-conjugated organic cations, namely 2-methylimidazole cation (C4H7N2)+ and 1-ethyl-3-methylimidazole cation (C6H11N2)+, into the ABX3 (B = Pb, Sn; X = Cl, Br) template (Fig. 1). This innovative approach has led to the synthesis of nine distinct organic-inorganic hybrid NLO crystals, effectively disrupting the conventional centrosymmetric structure, a crucial step towards enhancing NLO properties. As we systematically varied the composition from C4H7N2SnBr3 to C6H11N2PbCl3, we observed a gradual improvement in performance. Specifically, the SHG efficiency increased from 1.6 times that of KDP to an impressive 3.8 times, highlighting the effectiveness of our synthetic strategy. Concurrently, the bandgap energy expanded significantly, ranging from 2.75 eV to 3.87 eV. The UV cut-off edge, a key parameter for NLO crystals' utility in the UV region, exhibited a substantial blue shift from 383 nm to 300 nm, aligning with the growing demand for materials with deeper UV capabilities. Furthermore, the birefringence, crucial for phase-matching in NLO processes, increased notably from 0.1 to 0.14, enhancing the potential for efficient frequency conversion. Lastly, the thermal stability of these crystals, a testament to their robustness for practical applications, saw a substantial enhancement, with the temperature rising from 190 ℃ to a remarkable 294 ℃. This comprehensive enhancement across various performance parameters underscores the efficacy of our multisite modulation strategy in crafting NLO crystals.
Nine novel crystalline compounds were successfully synthesized through hydrothermal and low-temperature solid-phase methods. Single-crystal X-ray diffraction analysis (CCDC deposition numbers: 2426108–2426116) revealed distinct structural features: (C4H7N2)2SnCl6 (1), (C4H7N2)2SnBr6 (2), and C6H11N2SnCl3 (4) adopt centrosymmetric space groups (Pnnm, P1 and P21/n, respectively), while the remaining six compounds C4H7N2SnBr3 (3), C6H11N2SnBr3 (5), C6H11N2SnBr1.4Cl1.6 (6), C6H11N2PbBr3 (7), C6H11N2PbBr1.8Cl1.2 (8) and C6H11N2PbCl3 (9) crystallize in the non-centrosymmetric space group P212121. The bond valence sums (BVS) of Sn and Pb were calculated, revealing oxidation states of +2 for both elements, which is consistent with their expected valence states in the crystal structure. Detailed synthetic procedures, crystallographic data, and structural characterization are provided in Supporting information (Tables S1-S6 in Supporting information). The phase purity and structural integrity of the synthesized compounds were confirmed through powder X-ray diffraction (PXRD) analysis, with the experimental patterns demonstrating excellent agreement with the simulated patterns generated from single-crystal X-ray diffraction data (Fig. S1 in Supporting information). Energy-dispersive X-ray spectroscopy (EDS) analysis unequivocally demonstrated the existence of C, N, Sn, Pb, Cl, and Br elements (Fig. S2 in Supporting information).
The single-crystal structural analysis reveals that the asymmetric unit of compound 1 comprises a discrete [SnCl2]2+ group and one (C4H7N2)+ organic cation (Fig. S3a in Supporting information). The Sn(Ⅳ) center exhibits nearly ideal octahedral coordination geometry, with six Sn-Cl bond distances ranging from 2.416 Å to 2.446 Å and Cl-Sn-Cl bond angles varying between 89.975° and 90.026°, demonstrating minimal distortion from perfect octahedral symmetry. The organic cations adopt an ordered antiparallel arrangement around the inorganic framework, with their molecular dipoles effectively canceling each other, thereby generating a centrosymmetric crystal structure in the Pnnm space group (Fig. 2a).
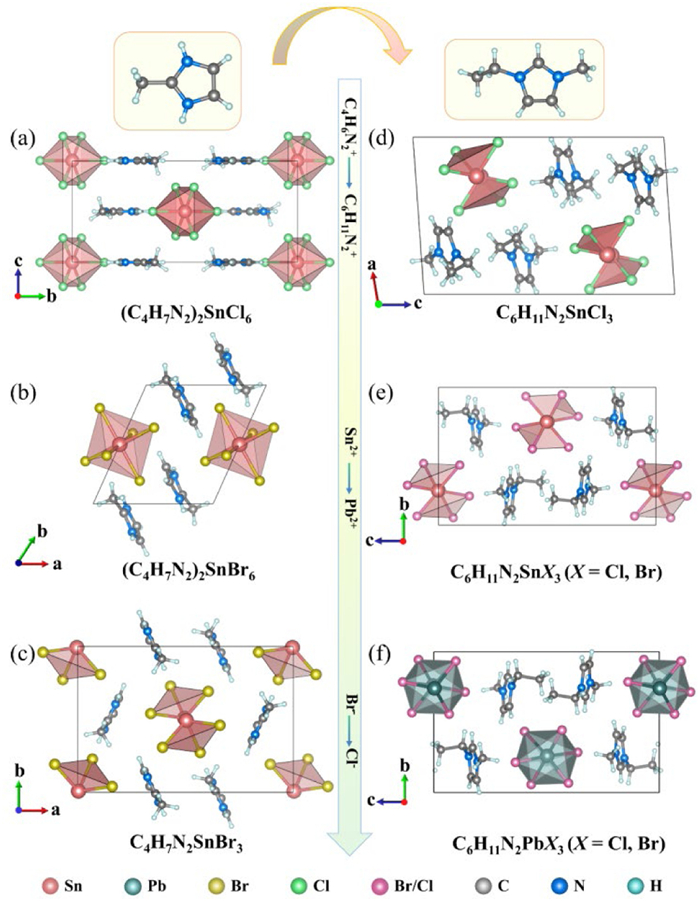
Structural analysis of compound 2 reveals an asymmetric unit containing [SnBr3]+ group and a (C4H7N2)+ cations (Fig. S3b in Supporting information). The substitution of bromide anions induces distinct structural modifications compared to the chloride analogue, with Sn-Br bond distances ranging from 2.5845 Å to 2.6051 Å and Br-Sn-Br bond angles varying between 89.493° and 90.507°. Despite these geometric perturbations, the [SnBr6]2- octahedra maintain high structural symmetry through mutual cancellation of bond dipoles within the coordination sphere. The planar (C4H7N2)+ cations adopt an antiparallel arrangement similar to compound 1, with their molecular planes oriented parallel to the inorganic layers (Fig. 2b). This structural configuration results in an overall centrosymmetric packing arrangement in the P1 space group.
Crystal 3 exhibits a distinct structural configuration compared to its analogues, despite sharing the same asymmetric unit composition of Sn(Ⅱ), three bromide ligands, and (C4H7N2)+ cation. The organic cations adopt two distinct orientations, forming dihedral angles of 49.54° between their molecular planes (Fig. S4a in Supporting information). Notably, the Sn(Ⅱ) center coordinates with only three bromide anions, forming a significantly distorted [SnBr3]- trigonal pyramidal unit. The Sn-Br bond distances range from 2.674 Å to 2.711 Å, with Br-Sn-Br bond angles varying between 89.99° and 93.30°. The [SnBr3]- units adopt a mirror-symmetric arrangement within the crystal lattice, resulting in partial cancellation of local dipole moments; however, the overall structure maintains a non-centrosymmetric configuration, crystallizing in the polar Pna21 space group (Fig. 2c).
Whereas the structure of crystal 4 has the same highly distorted triangular cone unit [SnCl3]- as crystal 3, where the Sn-Cl bond length ranges from 2.491 Å to 2.534 Å and the Cl-Sn-Cl bond angle ranges from 90.34° to 92.54°, unfortunately, the [SnCl3]- triangular cones are arranged in an opposite manner, which leads to complete cancellation of polarization and ultimately to a centrosymmetric structure. In addition, the planar organic cation (C6H11N2)+ intersperses the [SnCl3]- anion antiparallelly with a dihedral angle of 53.28° (Fig. 2e and Fig. S4b in Supporting information).
Crystals 5–9 are isomorphic and discussed in detail in terms of crystal 9, with an asymmetric unit consisting of one Pb atom, three Cl atoms, and a (C6H11N2)+ cation (Fig. S3d in Supporting information). The Pb is surrounded by six Cl atoms to form the highly distorted octahedral unit [PbCl3]-, in which the Pb-Cl bond lengths range from 2.618 Å to 3.567 Å, and each [PbCl3]- octahedral units share planes with neighboring [PbCl3]- octahedra, and [PbCl3]∞- anionic chains are grown along the [100] direction, where the planar π-conjugation and the (C6H11N2)+ cations are interspersed in two different orientations, with mutually non-parallel organic cations with dihedral angles of about 29.86° (Fig. S4c in Supporting information) interspersed among the anions in an orderly manner.
The crystal packing is stabilized by an extensive network of weak hydrogen-bonding interactions between the anionic metal halide frameworks and organic cations, as quantitatively analyzed in Table S7 (Supporting information). These supramolecular interactions play a crucial role in maintaining the structural integrity and influencing the overall crystal packing arrangement.
The infrared spectra of the compounds were collected using an FT-IR spectrometer in the wavenumber range of 4000–400 cm-1, and the functional groups of them were analyzed (Fig. S5 in Supporting information). The peaks at 568 and 1420 cm-1 are the C═N stretching vibrations on the imidazole ring, and the peaks observed at 2112 and 2067 cm-1 correspond to the C—N vibrations. The absorption peaks in the range of 3364–3090 cm-1 can be ascribed to the N—H stretching vibrations, while the peaks observed at 616–840 cm-1 and 1391 cm-1 correspond to the C—H bending vibrations. All of the above proved the presence of imidazole ring and the attribution of all the absorption peaks matches the literature reports [3,13].
Optical properties of compounds 1–9 were systematically investigated through UV–vis diffuse reflectance spectroscopy (200–800 nm). The UV absorption cut-off edges of compounds 1–9 were 280, 342, 383, 300, 310, 338, 346, 332, and 300 nm, respectively. By using Kubelka-Munk function [33], the band gaps of compounds 1–9 were 3.97, 2.83, 2.75, 3.68, 3.46, 3.05, 3.39, 3.47, and 3.87 eV, respectively (Fig. S6 in Supporting information). The analysis of the bandgap variation law confirms that larger sizes of planar π-conjugated organic cations are favorable for bandgap enhancement [34,35].
Meanwhile, compounds 1–9 were tested for thermal stability (Fig. S7 in Supporting information). Different hydrogen bonding profiles in the structures through the introduction of different planar π-conjugated organic cations, and the complex hydrogen bonding network is favorable for thermal stability, so they were stabilized at 160, 220, 190, 260, 270, 280, 290, 290, and 294 ℃, respectively. We analyzed in detail the weight loss of the compounds in the different temperature intervals and compounds 1, 2, 3, 5, and 6 all decomposed completely in one step, and the remaining compounds underwent a two-step decomposition (Fig. S5). The weight loss patterns of compounds 4 and 8 are similar, with the initial weight loss occurring in the temperature range of 260/290 ℃ to 400 ℃. This corresponds to the loss of the organic component (C6H11N2)+. Subsequently, the second stage of weight loss is observed beyond 400 ℃, which is attributed to the decomposition of the inorganic component, namely [SnCl3]- and [PbBr1.8Cl1.2]-. Compounds 7 and 9 exhibit a comparable thermal decomposition process. The first step of their decomposition reaction takes place within the temperature interval of 290 ℃/294 ℃ to 400 ℃, during which both compounds lose one molecule of the organic component (C6H11N2)+ and one molecule of either Br or Cl atoms. Upon further heating, the residual [PbCl2] and [PbBr2] begins to decompose [36]. The experimental findings reveal that compounds 4, 8, and 9 did not reach 100% weight loss at the end of the reaction, and it is presumed that the high temperature at 800 ℃ was not sufficient to cause the compounds to achieve complete weight loss in the second step.
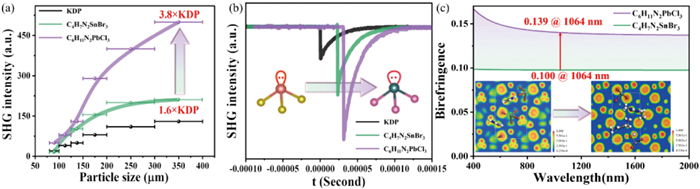
The inorganic anions of compounds 1 and 2, which are highly symmetric octahedra, both crystallize in centrosymmetric space groups. Despite the inorganic anion [SnCl3]- in compound 4 being a highly distorted triangular cone, the completely antiparallel arrangement of the molecules results in the cancellation of polarizations, leading to a centrosymmetric structure. Compounds 3, 5, 6, 7, 8 and 9, however, exhibit non-centrosymmetric structures. We evaluated the SHG strength of their sieved powder samples using the Kurtz-Perry method [37]. As depicted in Fig. 2, these compounds demonstrate superior frequency-doubling performance under 1064 nm laser irradiation, with enhancements of 1.6, 1.5, 1.3, 6, 4 and 3.8 times that of KDP, respectively. The SHG intensities increase and approach saturation with increasing particle size, suggesting that these compounds have the potential to be effectively utilized in the 1064 nm wavelength band (Fig. S8 in Supporting information). Through an in-depth investigation of the structure-property relationship, we elucidate that the superior SHG response originates from the enhanced hyperpolarizability (β) induced by pronounced electron cloud distortion. The stereochemically active 6s² lone pair of Pb²⁺ exhibits more pronounced lone pair stereoactivity compared to Sn²⁺, resulting in greater electronic distortion and hyperpolarizability. Furthermore, the larger ionic radius of Br⁻ and the more polarizable 4p orbitals (versus Cl⁻ 3p orbitals) significantly enhance the electron cloud deformability of anionic frameworks (e.g., [SnBr₃]⁻ or [PbBr₃]⁻). This facilitates stronger nonlinear polarization under an external electric field. C4H7N2⁺, featuring a smaller organic cation, demonstrates restricted intramolecular charge transfer magnitude compared to C6H11N2⁺ counterparts, consequently exhibiting a diminished NLO response. The experimental findings demonstrate a pronounced enhancement in the SHG effect, achieved through precise modulation of the three-point positioning. This result underscores the efficacy of the proposed approach in optimizing the desired outcome (Figs. 3a and b).
We employed the first-principles methods, utilizing density functional theory (DFT) [38–40], to calculate the energy band structures, densities of states (DOS), and optical properties of compounds 1–9. The ELF of compounds 1–9 is shown in Fig. S11 (Supporting information), clearly demonstrating the localization behavior of the electrons. Given that compounds 5–9 are isostructural, the ELF analysis of compound 5, as depicted in Fig. 3c, is presented as a representative case for the compounds 5–9. The calculated band gaps, which are 3.64, 2.24, 3.51, 3.63, 3.57, 3.50, 3.81, 3.66, and 4.19 eV, closely match the experimentally determined band gaps (Fig. S9 in Supporting information). Consequently, we applied scissor corrections of 0.33, 0.59, 0.76, 0.05, 0.11, 0.45, 0.42, 0.19, and 0.32 eV to refine the calculation of their optical properties. Upon analyzing the density of states diagrams, it is evident that the optical properties of compound 1 are primarily influenced by the Cl-4p and C-2p orbitals at the top of the valence band, and the H-1s and N-2p orbitals at the bottom of the conduction band (Fig. S9a). In compound 2, the valence band top is predominantly composed of Br-4p and C-2p orbitals, while the conduction band bottom is mainly constituted by Sn-5s and Br-4p orbitals (Fig. S9b). For compound 3, the electronic activity near the band gap is governed by the C, N, Sn, and Br atoms, with the Br-4p and Sn-5s orbitals at the top of the valence band and the C-2p, N-2p, Sn-5p, and Br-4s orbitals at the bottom of the conduction band playing a significant role (Fig. S9c). The energy band structures of compounds 4–9 are similar to that of compound 6 (Fig. S9f), and thus the DOS diagram of compound 9 was selected for detailed analysis. The top of the valence band is occupied by Cl-4p, Pb-5s, and C-2p orbitals, while the bottom of the conduction band consists of C-2p, N-2p, H-1s orbitals, and the Pb-5p orbitals within the (C6H11N2)+ moiety. Consequently, the electronic activity in the vicinity of the band gap for compounds 1–9 is determined by the interplay between the anionic group and the π-conjugated organic group.
Birefringence is a pivotal property of optically functional crystals, playing an indispensable role in nonlinear optical crystals for phase matching and regulating the polarization of light. Crystals with large birefringence can meet the application demands across specific wavelength bands, including the visible, ultraviolet, and deep ultraviolet regions, which is essential for expanding the application spectrum of birefringent crystals. Compounds 1–9 are classified as biaxial crystals, and their birefringence at λ = 1064 nm is calculated to be 0.17, 0.07, 0.10, 0.09, 0.12, 0.12, 0.14, 0.15, and 0.14, respectively (Fig. S10 in Supporting information). Notably, the birefringence of the six non-centrosymmetric compounds exhibits a clear increasing trend, suggesting that a three-point modulation strategy can effectively enhance birefringence.
Through a systematic three-point modulation strategy, we have successfully achieved structural transformation from highly symmetric polyhedral units to significantly distorted trigonal pyramidal configurations. This structural engineering approach has enabled simultaneous optimization of multiple functional properties, including (1) a substantial increase in optical band gap, (2) a pronounced blue-shift in absorption edges, (3) enhanced birefringence, (4) improved thermal stability. These property enhancements, as illustrated in Fig. 4, demonstrate the effectiveness of our synergistic modulation approach in tailoring the structure-property relationships of hybrid materials.
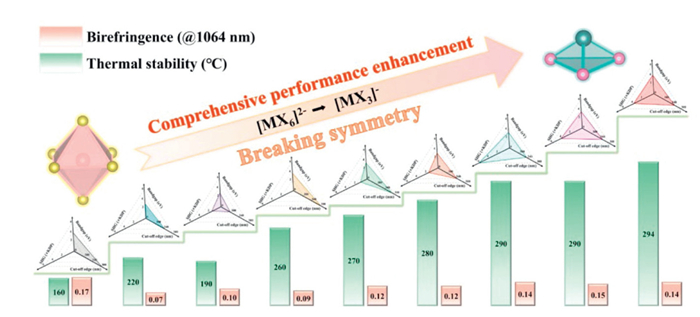
In conclusion, we have successfully synthesized nine ABX3-type organic-inorganic hybrid halides through strategic modulation of the three pivotal components: cations (A), metals (B), and halogens (X). Notably, the synthesis of C6H11N2PbCl3 using (C4H7N2)2SnCl6 as a template effectively disrupts the crystal structure symmetry and achieves a delicate balance between a strong SHG response, an expanded optical bandgap, and increased birefringence. Our triple-site modulation strategy establishes a universal paradigm for designing NCS hybrid crystals, decoupling the traditional constraints between optical nonlinearity and transparency. The exceptional transparency (cut-off edge: 300 nm) and thermal stability (294 ℃) position these materials as promising candidates for frequency conversion and high-power laser applications. Future work will focus on further optimizing these materials and exploring their potential in other advanced optical applications.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Yuwei Kang: Writing – review & editing, Writing – original draft, Data curation, Conceptualization. Can Yang: Writing – original draft, Investigation, Data curation. Jun Zhang: Writing – review & editing, Investigation, Data curation. Qi Wu: Writing – review & editing, Writing – original draft, Data curation, Conceptualization.
This work was supported by the National Natural Science Foundation of China (No. 22275052) and Department of Science and Technology of Hubei Province (Nos. 2025AFA111 and 2024CSA076).
Supplementary material associated with this article can be found, in the online version, at doi:
K. Zhou, B. Qi, Z. Liu, et al., Adv. Funct. Mater. 34 (2024) 2411671. doi: 10.1002/adfm.202411671
R. Tang, D. Yang, L. Ma, et al., Adv. Opt. Mater. 13 (2025) 2403044. doi: 10.1002/adom.202403044
Y. Kang, C. Yang, J. Gou, et al., Inorg. Chem. 63 (2024) 2725–2731. doi: 10.1021/acs.inorgchem.3c04148
W. Zeng, Y. Tian, H. Zeng, et al., Angew. Chem. Int. Ed. 64 (2025) e202422818. doi: 10.1002/anie.202422818
J.J. Zhao, S.F. Li, M.H. Lv, et al., Inorg. Chem. 64 (2025) 807–812. doi: 10.1021/acs.inorgchem.4c05135
H.M. Ngo, Y. Kuk, J. Lee, et al., Adv. Opt. Mater. 13 (2025) 2403241. doi: 10.1002/adom.202403241
K. Liu, J. Zhao, Q. Liu, Laser Photonics Rev. 18 (2024) 2400345. doi: 10.1002/lpor.202400345
Q. Zhang, R. An, X. Long, et al., Angew. Chem. Int. Ed. 64 (2025) e202415066. doi: 10.1002/anie.202415066
Y. Kang, C. Yang, J. Gou, et al., Inorg. Chem. 63 (2024) 2725–2731. doi: 10.1021/acs.inorgchem.3c04148
S. Cui, S. Yang, H. Wu, et al., Adv. Funct. Mater. 35 (2025) 2424059. doi: 10.1002/adfm.202424059
B. Ding, X. Cheng, H. Mi, et al., Inorg. Chem. 64 (2025) 1153–1163. doi: 10.1021/acs.inorgchem.4c04869
X. Kong, J. Chai, H. Zhao, et al., Inorg. Chem. Front. 12 (2025) 630–636. doi: 10.1039/d4qi02469a
Y. Kang, C. Yang, J. Gou, et al., Angew. Chem. Int. Ed. 63 (2024) e202402086. doi: 10.1002/anie.202402086
S. Choi, Y. Li, Y. Kuk, et al., Adv. Sci. 12 (2025) 2414503. doi: 10.1002/advs.202414503
P. Li, C. Hu, F. Kong, et al., Angew. Chem. Int. Ed. 62 (2023) e202301420. doi: 10.1002/anie.202301420
J.H. Wu, C.L. Hu, Y.F. Li, et al., Chem. Sci. 15 (2024) 8071–8079. doi: 10.1039/d4sc01716a
R. Tang, C. Hu, B. Wu, et al., Angew. Chem. Int. Ed. 58 (2019) 15358–15361. doi: 10.1002/anie.201909735
S. Liu, L. He, Y. Wang, et al., Chin. Chem. Lett. 33 (2022) 1032–1036. doi: 10.1016/j.cclet.2021.07.039
Y. Hu, X. Jiang, C. Wu, et al., Chem. Mater. 33 (2021) 5700–5708. doi: 10.1021/acs.chemmater.1c01434
K. Liu, J. Zhao, Q. Liu, Laser Photonics Rev. 18 (2024) 2400345. doi: 10.1002/lpor.202400345
S. Yang, H. Wu, Z. Hu, et al., Inorg. Chem. 63 (2024) 1404–1413. doi: 10.1021/acs.inorgchem.3c03928
Y. Li, J. Luo, S. Zhao, Acc. Chem. Res. 55 (2022) 3460–3469. doi: 10.1021/acs.accounts.2c00542
F. Chen, H. Wu, Z. Hu, et al., Chem. Sci. 16 (2025) 2015–2023. doi: 10.1039/d4sc05747c
W. Han, P. Cheng, J. Guan, et al., Angew. Chem. Int. Ed. 16 (2025) e202500786.
J.W. Lee, S. Tan, S.I. Seok, et al., Science 375 (2022) eabj1186. doi: 10.1126/science.abj1186
L. Zhou, P.P. Shi, X.M. Liu, et al., NPG Asia Mater. 11 (2019) 1–9. doi: 10.1038/s41427-018-0100-z
L. He, Y. Liu, P. Shi, et al., ACS Appl. Mater. Interfaces 12 (2020) 53799–53806. doi: 10.1021/acsami.0c16180
Y. Liu, Y. Gong, S. Geng, et al., Angew. Chem. Int. Ed. 61 (2022) e202208875. doi: 10.1002/anie.202208875
Y. Zheng, J. Xu, X. Bu, Adv. Opt. Mater. 10 (2022) 2101545. doi: 10.1002/adom.202101545
D. Chen, S. Hao, L. Fan, et al., Chem. Mater. 33 (2021) 8106–8111. doi: 10.1021/acs.chemmater.1c02896
Q. Shui, H. Tang, R. Fu, et al., Angew. Chem. Int. Ed. 60 (2021) 2116–2119. doi: 10.1002/anie.202013088
J. Guan, Y. Zheng, P. Cheng, et al., J. Am. Chem. Soc. 145 (2023) 26833–26842. doi: 10.1021/jacs.3c09395
P. Kubelka, F. Munk, Z. Tech. Phys. 12 (1931) 259–274.
C.C. Stoumpos, L. Frazer, D.J. Clark, et al., J. Am. Chem. Soc. 137 (2015) 6804–6819. doi: 10.1021/jacs.5b01025
M. Mittal, A. Jana, S. Sarkar, et al., J. Phys. Chem. Lett. 7 (2016) 3270–3277. doi: 10.1021/acs.jpclett.6b01406
Y. Deng, X. Dong, M. Yang, et al., Dalton Trans. 48 (2019) 17451–17455. doi: 10.1039/c9dt04102h
S. Kurtz, T. Perry, J. Appl. Phys. 39 (1968) 3798–3813. doi: 10.1063/1.1656857
M.D. Segall, P.J.D. Lindan, M.J. Probert, et al., J. Phys. Condens Mat. 14 (2002) 2717–2744. doi: 10.1088/0953-8984/14/11/301
V. Milman, B. Winkler, J.A. White, et al., Int. J. Quantum Chem. 77 (2000) 895–910. doi: 10.1002/(SICI)1097-461X(2000)77:5<895::AID-QUA10>3.0.CO;2-C
J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77 (1996) 3865–3868. doi: 10.1103/PhysRevLett.77.3865

Figure 2 The crystal structure of compound (a) 1, (b) 2, (c) 3, (d) 4, (e) 5–6 and (f) 7–9.

Figure 3 (a) SHG intensity vs. particle size of compounds under 1064 nm laser irradiation. (b) The oscillo scope traces of the SHG signals for powders of compounds. (c) Calculated birefringence of compounds 3 and 9, the illustrations are electronic localization function (ELF) analysis.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:

 下载:
下载: