Received Date:
26 February 2025 Accepted Date:
19 May 2025 Revised Date:
17 May 2025 Available Online:
15 May 2026
Abstract:
The advent of the most representative commercially available formulations of paclitaxel, Taxol and Abraxane®, resolved the intravenous challenge of paclitaxel by increasing the water solubility. However, the severe excipient-related toxicity and poor stability of Taxol, along with the low drug loading (10%), complex preparation processes, and poor tumor selectivity of Abraxane®, present significant clinical dilemma. To overcome the challenges, 16-methylheptadecanoic acid (16-MH), with excellent biocompatibility was selected as the assembly module. The paclitaxel-16-MH prodrug nanoassemblies (PSSMH NPs) were constructed by conjugating 16-MH with redox-sensitive disulfide bonds and paclitaxel through an ethylene glycol. PSSMH NPs featured the advantages of easy preparation, high drug loading (> 50%) and superior stability (stable storage for 60 days at 25 ℃). Notably, the area under the concentration−time curve (AUC0–24 h) of PSSMH NPs was 14.95-fold compared with Taxol, indicating a significant improvement in the in vivo fate of paclitaxel. Moreover, the existence of redox-sensitive disulfide bonds endowed PSSMH NPs with increased tumor selectivity, resulting in exceptional tolerance and antitumor efficacy. Overall, the redox-triggered prodrug nano-system with high tumor selectivity and biocompatibility exhibits substantial potential for clinical translation.
Paclitaxel, the first U.S. Food and Drug Administration-approved taxane chemotherapeutic agent derived from a natural plant, is known as the "ageless legend" of antitumor therapeutics [1]. However, the intravenous administration of paclitaxel is constrained by its poor water solubility (< 0.4 mg/L) [2]. The commercially available injection, Taxol, utilizes Cremophor EL and ethanol to enhance the water solubility of paclitaxel, leading to serious side effects [2–4]. Furthermore, the poor diluent stability (stable storage for 27 h at 25 ℃) and severe excipient-related toxicity of Taxol pose considerable challenges for clinical application [5,6]. The emergence of nano-delivery technology has revitalized chemotherapeutic agents. A prominent example of the commercially available nano-formulations is Abraxane®, an albumin-bound nanoparticle that significantly enhances the solubility of paclitaxel by the encapsulation of human serum albumin (HSA) [7]. The application of nanoparticle albumin-bound technology mitigates the toxic side effects associated with traditional solubilizers [7]. Nevertheless, the clinical application of Abraxane® is limited by several factors, including low drug loading (10%), a complex preparation process, non-selective toxicity, and the poor solution stability (stable storage for 4 days at 25 ℃) [7–10]. Therefore, there is an urgent need to develop a novel drug delivery system to break through the antitumor dilemma of paclitaxel.
Recently, nano-delivery platforms with long blood circulation time and high tumor selectivity have been extensively studied for cancer treatment [11–16]. The rational selection and precise design of nano-delivery platforms are crucial for enhancing the physicochemical properties of chemotherapeutic agents and improving therapeutic efficacy [11,13,14]. Prodrug nanoassemblies, which utilize prodrug strategies improving the physicochemical properties of the parent drug and forming nanoparticles by self-assembly of the prodrugs, are simple to prepare and possess ultra-high drug loading [17–22]. In comparison to traditional nano-delivery systems, prodrug nanoassemblies have the advantages of high drug loading, excellent stability, low toxicity, and facile manufacturing processes, which provide an effective strategy to overcome the delivery deficiency of paclitaxel [17,23]. It is noteworthy that the structure of prodrugs is one of the key factors influencing the physiological functions of prodrug nanoassemblies. Prodrugs are typically composed of drug modules, response modules, and assembly modules [24]. Among them, the response modules trigger the selective activation of the prodrugs at tumor sites [25,26]. The abnormal metabolism of tumor cells results in redox imbalance compared to normal tissues, characterized by the overexpression of reactive oxygen species (ROS) and glutathione (GSH) [26–28]. Disulfide bonds with dual-redox sensitivity are promising response modules with potential for development [25,29,30].
For the assembly modules, fatty acids or fatty alcohols are frequently selected to enhance the self-assembly capacity of prodrugs [31–35]. Notably, recent studies have demonstrated that fatty acids offer unique advantages in enhancing the self-assembly capabilities of chemotherapeutic agents by introducing "structural defects" [36]. Among the fatty acids, monomethyl branched-long-chain fatty acids (MBLCFA) are present in various human tissues and organs, such as the liver and the adipose tissue [37,38]. As a representative MBLCFA, 16-methylheptadecanoic acid (16-MH) exhibits excellent biocompatibility and metabolic safety [39]. Therefore, 16-MH is a promising option for conjugation with paclitaxel to improve its self-assembly performance and safety.
In this study, 16-MH was selected as the assembly module, while the prodrug (PSSMH) was synthesized by conjugating 16-MH with a redox-sensitive disulfide bond and paclitaxel through an ethylene glycol. The redox-triggered prodrug nanoassemblies (PSSMH NPs) were prepared using a facile manufacturing process with ultra-high drug loading (> 50%) and superior stability. Through an analysis of the structure-activity relationship and a comprehensive exploration of the biological effects, the following conclusions could be drawn (Scheme S1 in Supporting information): (ⅰ) In comparison to Taxol and Abraxane®, PSSMH NPs demonstrated significantly enhanced stability, remaining stably stored at 25 ℃ for 60 days. (ⅱ) The area under the concentration−time curve (AUC0–24 h) of PSSMH NPs was 14.95-fold compared with Taxol, indicating a significant improvement in the in vivo fate of paclitaxel. (ⅲ) In contrast to Taxol and Abraxane®, the presence of disulfide bonds imparted increased tumor selectivity to PSSMH NPs, contributing to the excellent tolerance and antitumor efficacy of PSSMH NPs. The findings demonstrated that redox-triggered PSSMH NPs with high tumor selectivity and biocompatibility could help to break through the antitumor dilemma of paclitaxel.
The synthesis procedure of PSSMH was shown in Fig. S1 (Supporting information). The high-resolution mass spectrometry (HRMS) and proton nuclear magnetic resonance (1H NMR) spectra verified the structure and relative molecular mass of PSSMH (Fig. S2 in Supporting information). The purity of PSSMH was over 99% (determined by the high-performance liquid chromatographic determination (HPLC), Fig. S2), which satisfied the requirements for subsequent experiments.
To explore the differences in the assembly behaviors of paclitaxel and PSSMH, different concentrations of paclitaxel and PSSMH were dropped into deionized water. Paclitaxel with poor water solubility precipitated immediately upon dropping into deionized water and failed to self-assemble to form nanoassemblies even at a low concentration of 0.1 mg/mL (Table S1 and Fig. S3A in Supporting information). After the modification of 16-MH, PSSMH could self-assemble into non-PEGylated nanoassemblies with uniform particle sizes at concentrations of 0.1 and 0.2 mg/mL, which might be attributed to the strong hydrophobic interactions between PSSMH (Figs. S3B–D in Supporting information). In addition, the particle size of non-PEGylated PSSMH NPs at a concentration of 0.1 mg/mL was 95.56 nm, which was approximately 30 nm smaller than the particle size of Abraxane® at the same concentration (Table S1 and Fig. S3D). The optimal particle size for enhanced permeability and retention (EPR) is generally considered to be approximately 100 nm, whereas smaller particle sizes are more effective for deeper penetration into tumors [40–42].
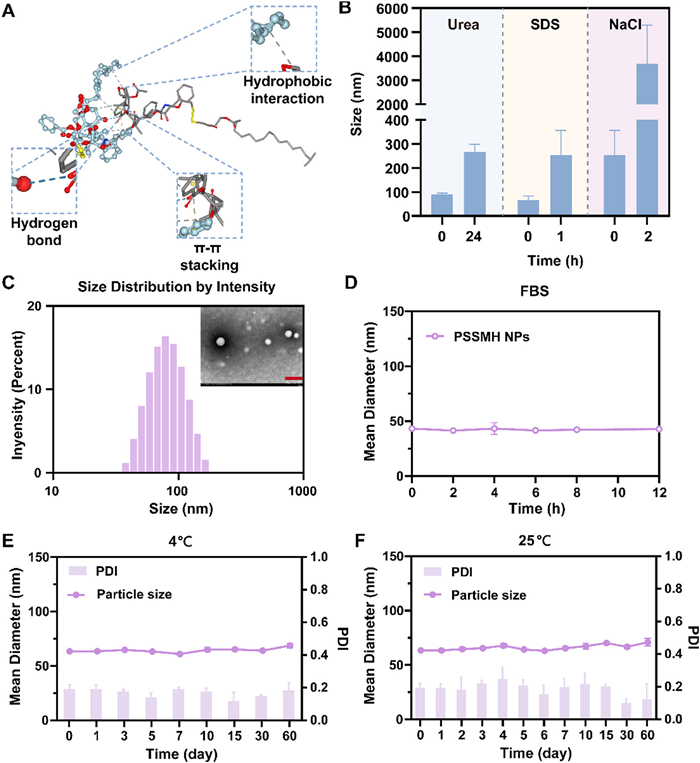
To further explore the self-assembly mechanism of PSSMH, the molecular simulation was conducted. As shown in Fig. 1A, hydrophobic interactions, hydrogen bonding forces, and π-π stacking forces were involved in the self-assembly process of PSSMH. The particle sizes of non-PEGylated PSSMH NPs increased by 2 times after incubation in 0.1 mol/L urea (hydrogen bonding forces blocker) for 24 h or 0.1 mol/L sodium dodecyl sulfate (SDS, hydrophobic interactions disruptor) for 1 h, which confirmed the existence of hydrogen bonding forces and hydrophobic interactions (Fig. 1B, Figs. S4A and B in Supporting information). Moreover, electrostatic interactions also participated in the self-assembly of PSSMH, which was manifested by a significant increase in the particle sizes of non-PEGylated PSSMH NPs after incubation with 0.1 mol/L NaCl (ionic competitor) aqueous solution for 2 h (Fig. 1B and Fig. S4C in Supporting information). Thus, hydrophobic interactions, hydrogen bonding forces, π-π stacking forces, and electrostatic interactions promoted the stable assembly of PSSMH.
Figure 1
Figure 1.
Characterization and stability of PSSMH NPs. (A) Molecular simulation of PSSMH. (B) The variation of the particle sizes of non-PEGylated PSSMH NPs after co-incubated with urea, SDS or NaCl. (C) Particle size distribution and TEM image of PEGylated PSSMH NPs. Scale bar: 200 nm. (D) The colloidal stability. (E, F) Storage stability of PEGylated PSSMH NPs. Data are presented as mean ± standard deviation (SD) (n = 3).
To further improve the stability and prolong the blood circulation time of PSSMH NPs, N-(carbonyl‑methoxy polyethylene glycol 2000)-1,2-distearoyl-sn-glycerol-3-phosphoryl ethanolamine (DSPE-PEG2K) was introduced as the surface modifier (Fig. S4D in Supporting information). As displayed in Table S2 (Supporting information), the drug loading of PSSMH NPs exceeded 50%, which was significantly higher than other paclitaxel prodrug nano-delivery systems (< 40%) [43,44]. Moreover, PSSMH NPs had a polydispersity index (PDI) of < 0.2 and a particle size of 70 nm, which was smaller than 130 nm of Abraxane® at the same concentration. Abraxane® had a zeta potential of less than −10 mV, while PSSMH NPs possessed a zeta potential close to −20 mV. This increased negative charge may enhance the thermodynamic stability of the nano-structures by promoting greater repulsion between the negatively charged particles (Table S2). The transmission electron microscope (TEM) images verified the spherical structures of PEGylated PSSMH NPs (Fig. 1C).
The stability of PEGylated PSSMH NPs in phosphate buffer saline (PBS) containing 10% (v/v) fetal bovine serum (FBS), storage stability at 4 and 25 ℃ were investigated. After 12 h of culture in PBS with 10% FBS, the particle sizes remained relatively unchanged (Fig. 1D), indicating that the DSPE-PEG2K modification enhanced the colloidal stability of PSSMH NPs. In comparison to Taxol and Abraxane® solutions, which could only be stably stored at 25 ℃ for 27 h and 4 days [5,9], PEGylated PSSMH NPs exhibited excellent storage stability, as evidenced by the negligible changes in particle size when stored at 4 or 25 ℃ for 60 days (Figs. 1E and F). The above results demonstrated that DSPE-PEG2K modification enhanced the stability of PSSMH NPs, which may help to improve the pharmacokinetic of paclitaxel. Given the excellent stability of PEGylated PSSMH NPs, subsequent experiments were conducted using PEGylated PSSMH NPs.
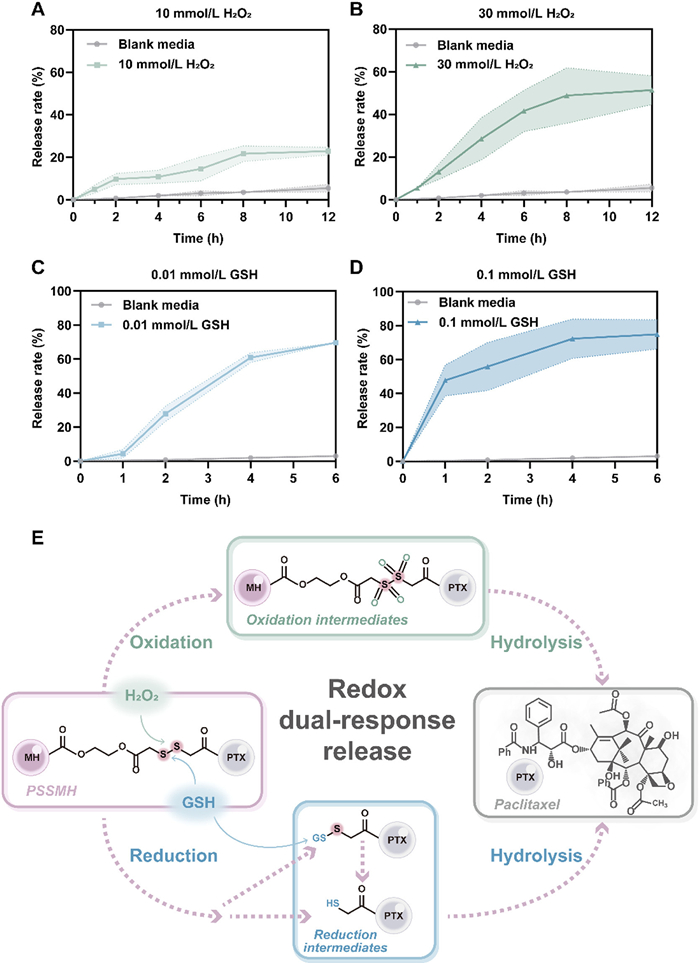
The concentration of ROS and GSH in tumor cells is 100 times and 7–10 times higher than that in normal cells. To enhance the tumor selectivity of paclitaxel, redox-sensitive disulfide bonds were selected as response modules to be introduced into the prodrug structural design. To investigate the redox release behavior and mechanism, PSSMH NPs were incubated with media without or with different concentrations of H2O2 (10 and 30 mmol/L) or GSH (0.01 and 0.1 mmol/L). In the blank release media, < 10% of paclitaxel was released by PSSMH NPs within 12 h (Figs. 2A and B). On the contrary, the release of paclitaxel from PSSMH NPs was significantly increased by the addition of H2O2 or GSH. The oxidative release of PSSMH NPs was concentration-dependent. When the concentration of H2O2 was 10 mmol/L, approximately 20% of paclitaxel was released from PSSMH NPs after incubation for 12 h (Fig. 2A). When the concentration of H2O2 was increased to 30 mmol/L, the release rate of paclitaxel from PSSMH NPs rose to 50% within 12 h (Fig. 2B). In the media containing H2O2, the disulfide bonds were oxidized to form hydrophilic sulfones or sulfoxides, facilitating the hydrolysis of the ester bonds and the release of paclitaxel (Fig. 2E, Figs. S5A and C in Supporting information).
Figure 2
Figure 2.
Redox dual-response release. Paclitaxel release profiles under (A) 10 mmol/L H2O2, (B) 30 mmol/L H2O2, (C) 0.01 mmol/L GSH, and (D) 0.1 mmol/L GSH. (E) The redox dual-response mechanism of PSSMH NPs. Data are presented as mean ± SD (n = 3).
The reductive release of PSSMH NPs also exhibited a concentration dependence. In the release medium containing 0.01 mmol/L GSH, the paclitaxel release rate within 2 h was 30% (Fig. 2C). In contrast, when the concentration of GSH was increased to 0.1 mmol/L, the paclitaxel release was increased to 50% within 2 h (Fig. 2D). In the existence of GSH, the disulfide bonds were reduced to thiol intermediates (Fig. 2E, Figs. S5B and C in Supporting information). The generation of hydrophilic intermediates in the above process promoted the release of paclitaxel. Thus, the introduction of disulfide bonds ensured selective activation of PSSMH NPs in tumor cells with high redox levels.
The effective internalization of nanoassemblies by tumor cells is a prerequisite for potent antitumor efficacy. Thus, the cell internalization of PSSMH NPs was assessed using coumarin-6 fluorescent labelling (Fig. S6A in Supporting information). The internalization efficiency was positively correlated with the green fluorescence of coumarin-6. As shown in Fig. S6A, 4T1 cells treated with coumarin-6 labeled PSSMH NPs exhibited stronger green fluorescence compared to coumarin-6 solution, indicating that PSSMH NPs could be efficiently uptake by tumor cells. In addition, the cell internalization of PSSMH NPs exhibited a time-dependent pattern, as evidenced by the increased intracellular green fluorescence with the extension of incubation time (Fig. S6A). Moreover, the confocal laser scanning microscope (CLSM) images was quantitatively analyzed, and the results were consistent with the above results (Figs. S6B and C in Supporting information).
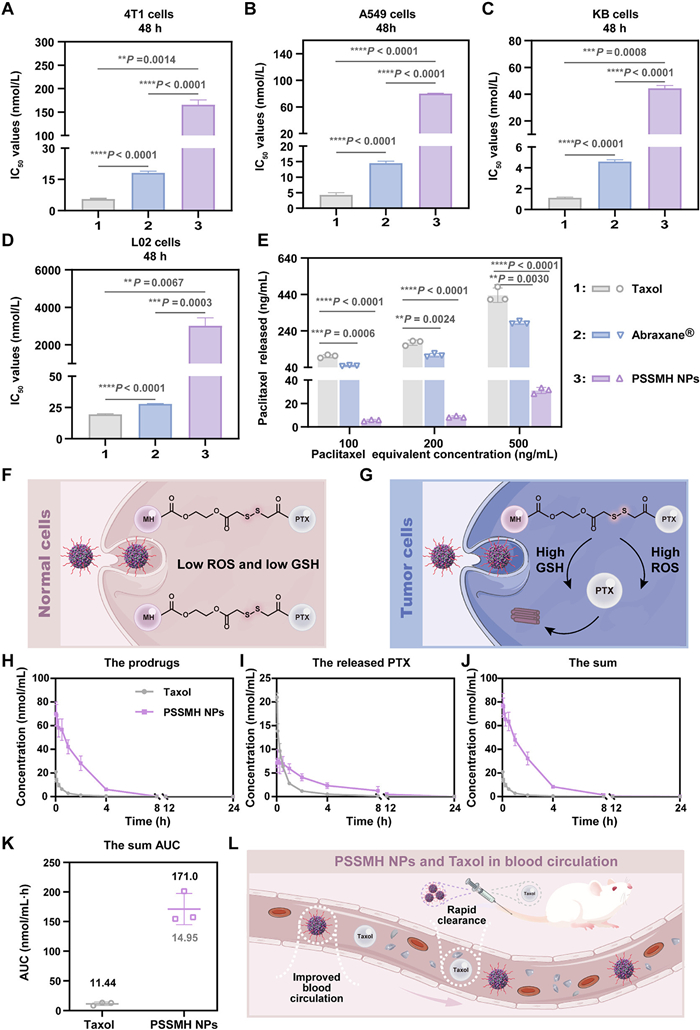
After confirming that PSSMH NPs could be effectively internalized by tumor cells, the cytotoxicity of Taxol, Abraxane®, and PSSMH NPs was evaluated in 4T1, A549, KB, and L02 cells. In three tumor cells (4T1, A549, and KB cells), the order of the cytotoxicity was Taxol > Abraxane® > PSSMH NPs (Figs. 3A–C, Fig. S7 and Table S3 in Supporting information). The cytotoxicity of PSSMH NPs in tumor cells was weaker than that of Taxol and Abraxane®, which was attributed to the delayed intracellular release of paclitaxel in PSSMH NPs (Fig. 3E). PSSMH could only exert the tumor cell killing effects when the disulfide bonds were cleaved and paclitaxel was released, resulting in inferior killing ability against tumor cells compared to Taxol and Abraxane®.
Figure 3
Figure 3.
Cytotoxicity and pharmacokinetics. (A–D) The IC50 of Taxol, Abraxane® and PSSMH NPs in 4T1, A549, KB, and L02 cells. (E) Paclitaxel released by different concentrations of PSSMH NPs in 4T1 cells. (F, G) Tumor selective activation mechanism of PSSMH NPs. (H–J) Plasma drug concentration-time curves (PTX equivalent). (K) The sum AUC0–24 h (prodrug and paclitaxel, paclitaxel equivalent). (L) The diagram of PSSMH NPs and Taxol in blood circulation. **P < 0.01, ***P < 0.001, ****P < 0.0001 by two-tailed Student's t-test. Data are presented as mean ± SD (n = 3).
In normal cells (L02 cells), Taxol and Abraxane® still exhibited strong cytotoxicity, while the cytotoxicity of PSSMH NPs was significantly reduced (Fig. 3D and Fig. S7). Compared to Taxol and Abraxane®, PSSMH NPs demonstrated excellent tumor selectivity due to the introduction of disulfide bonds (Table S4 in Supporting information). The disulfide bonds acted as a switch and were specifically triggered under high concentrations of ROS or GSH in the tumor microenvironment (Fig. 3G). The sulfur atoms in the disulfide bonds could be oxidized by ROS to form hydrophilic sulfones or sulfoxides, which enhanced the hydrophilicity of the system. The increase in hydrophilicity facilitated the hydrolysis of the ester bonds, promoting the release of paclitaxel (Fig. S8A in Supporting information). In the presence of GSH, the sulfur atoms of the disulfide bonds are attacked by -SH groups to form hydrophilic thiol intermediates. As in the oxidation process, hydrophilic thiol intermediates also promoted the hydrolysis of ester bonds and the release of paclitaxel (Fig. S8B in Supporting information). On the contrary, the disulfide bonds were less likely to be triggered in normal cells, indicating that PSSMH NPs exhibited good safety for normal cells (Fig. 3F).
In addition, the apoptosis of tumor cells induced by Taxol, Abraxane®, and PSSMH NPs was further investigated. As shown in Fig. S9 and Table S5 (Supporting information), the apoptosis rate (Q2 (%) + Q3 (%)) of tumor cells was positively correlated with the cytotoxicity of the formulations. The apoptosis rates induced by Taxol and Abraxane® were 54.5% and 51.2%. However, the proportion of apoptotic cells was 36.52% after treatment with PSSMH NPs, which was significantly lower than Taxol and Abraxane®. This difference could be attributed to the delayed release of paclitaxel in PSSMH NPs. The result was consistent with the cytotoxicity assay.
Paclitaxel promoted microtubule polymerization by binding to microtubule proteins, thereby disrupting cell mitosis and leading to apoptosis. The microtubule depolymerization inhibition of PSSMH NPs was investigated to verify the antitumor mechanism. As shown in Fig. S10 (Supporting information), the intensity of red fluorescence was directly proportional to the inhibition degree of microtubule depolymerization. The inhibition capacity was Taxol > Abraxane® > PSSMH NPs. This finding was consistent with the results of cytotoxicity and apoptosis assay. The delayed release of paclitaxel led to the weaker inhibition of microtubule depolymerization of PSSMH NPs compared to Taxol and Abraxane®.
The pharmacokinetic behavior of antitumor agents constitutes a key factor influencing the efficiency of drug delivery in vivo. Therefore, the pharmacokinetic characteristics of Taxol and PSSMH NPs were explored. The plasma drug concentration-time curves were shown in Figs. 3H–J, and the pharmacokinetic parameters were listed in Table S6 (Supporting information). The mean residence time (MRT) of PSSMH NPs was 2.27 times higher than Taxol. This finding suggested that Taxol was rapidly cleared in plasma, whereas PSSMH NPs exhibited prolonged blood retention time. Moreover, the sum area under the plasma drug concentration-time curves of PSSMH NPs increased 14.95-fold compared with Taxol (Fig. 3K). The remarkable improvement in area under the concentration-time curve (AUC0–24 h) and blood retention time of PSSMH NPs was attributed to the excellent colloidal stability (Fig. 3L). In addition, only a small amount (< 8%) of paclitaxel was released from PSSMH NPs after a 24 h incubation with rat plasma (Fig. S11 in Supporting information). This result further demonstrated that PSSMH NPs exhibited excellent stability in plasma, which was beneficial for enhancing the pharmacokinetic behavior.
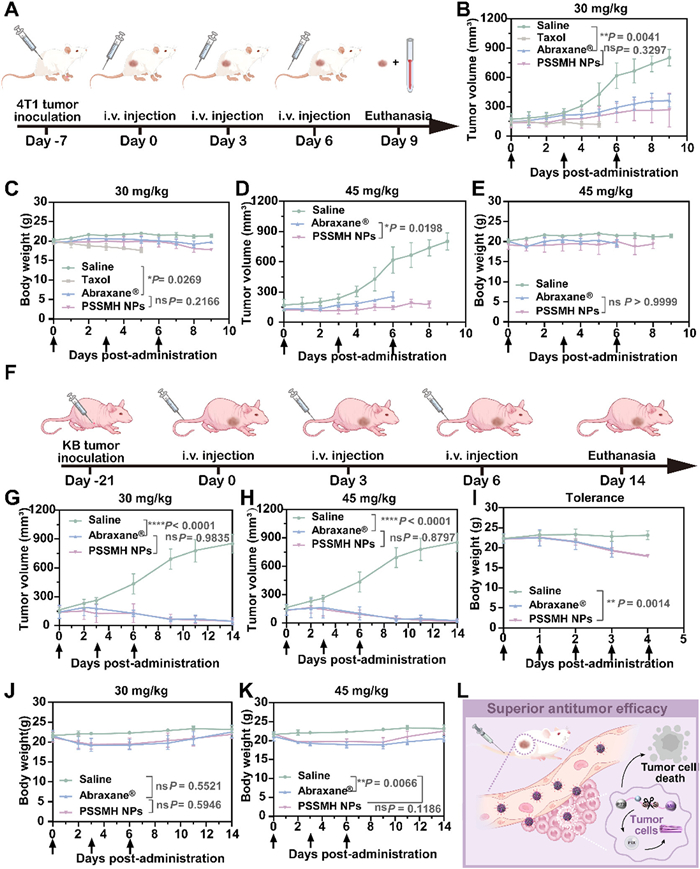
The antitumor efficacy of PSSMH NPs at different doses (30 and 45 mg/kg, paclitaxel equivalent) was evaluated on BALB/c mice subjected 4T1 tumors (Fig. 4A). As shown in Fig. 4B, all treatment groups demonstrated effective tumor growth inhibition at low doses (30 mg/kg, paclitaxel equivalent). Among them, Taxol exhibited the strongest antitumor efficacy. Although the tumor inhibitory capacity of PSSMH NPs was weaker than Taxol, it was comparable to Abraxane® (Fig. 4B and Fig. S12 in Supporting information). However, the severe toxicity of Taxol was reflected in the significant decrease in body weight and the death of all mice on the 5th day (Fig. 4C). Compared to Taxol, the treatment of PSSMH NPs and Abraxane® did not result in dramatic weight loss or the death of the mice (Fig. 4C). Since Taxol exhibited excessive toxicity when administered at low doses, higher doses were not set. At high doses (45 mg/kg, paclitaxel equivalent), PSSMH NPs demonstrated a stronger tumor inhibition capability than Abraxane® (Fig. 4D). The superior antitumor efficacy of PSSMH NPs could be attributed to the small particle size (73.48 nm). The size of the nano-delivery systems is a critical factor influencing the tumor penetration depth. The small particle size of < 100 nm enabled PSSMH NPs to penetrate deeply into the tumor, thus improving the antitumor efficacy. In contrast, Abraxane®, with a relatively larger particle size of approximately 130 nm, may exhibit limited tumor penetration depth, resulting in a weaker antitumor efficacy compared to PSSMH NPs [45]. In addition, Abraxane® presented poor safety, as evidenced by the death of all mice on the 6th day. In contrast, no significant weight loss in mice was observed after the treatment of PSSMH NPs (Figs. 4D and E, Fig. S13 in Supporting information). To further evaluate the safety of PSSMH NPs and Abraxane®, the blood of mice was taken for blood routine assessment and hepatorenal toxicity analysis. As shown in Figs. S14 and S15 (Supporting information), the treatment of PSSMH NPs did not lead to abnormal changes in the amount of major blood cells. Besides, the continuous treatment of PSSMH NPs did not cause abnormal changes in liver and kidney function (Fig. S16 in Supporting information). These results indicated that PSSMH NPs exhibited good safety.
Figure 4
Figure 4.
Antitumor efficacy and tolerance of PSSMH NPs. (A) The administration schedule of the BALB/c mice. (B–E) Tumor volume and body weight of BALB/c mice. (F) The administration schedule of the BALB/c nude mice. (G‒K) Tumor volume and body weight of BALB/c nude mice. (L) The antitumor mechanism of PSSMH NPs. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. ns, not significant by two-tailed Student's t-test. Data are presented as mean ± SD (n = 3).
Encouraged by the positive results of the above antitumor assay, BALB/c nude mice models inoculated with KB cells were established to eliminate the influence of immune rejection of HSA on the antitumor efficacy (Fig. 4F). At a low dosage of 30 mg/kg, Abraxane® and PSSMH NPs significantly inhibited the growth of tumors and did not cause weight loss in mice (Figs. 4G and J). When the dosage increased to 45 mg/kg, the antitumor efficacy of PSSMH NPs was similar to Abraxane® (Fig. 4H). However, although the antitumor efficacy of Abraxane® was enhanced, a slight weight loss in mice was observed, indicating that Abraxane® might be mild toxicity. In contrast, the mice treated with PSSMH NPs did not experience weight loss at the end of the assay, indicating that PSSMH NPs was safer than Abraxane® (Fig. 4K). The superior antitumor efficacy and safety of PSSMH NPs could be attributed to two key factors: (ⅰ) High drug loading and superior stability increased the delivery efficiency of PSSMH NPs. (ⅱ) Disulfide bonds enhanced the tumor selectivity of PSSMH NPs, improving the antitumor efficacy and safety (Fig. 4L).
Finally, the tolerance of Abraxane® and PSSMH NPs was evaluated in healthy female BALB/c nude mice. Since Taxol exhibited notable toxicity at the dose of 30 mg/kg, its tolerance was not investigated. The administration dosage was set at 75 mg/kg, which was higher than the dosage given in the antitumor assay. After the third administration, the mice treated with Abraxane® and PSSMH NPs showed a continuous decrease in body weight (Fig. 4I). At the end of the tolerance assay, the mortality rate of mice treated with Abraxane® was 33.33%, while no mice death was observed after the treatment of PSSMH NPs (Table S7 in Supporting information). This result demonstrated that the tolerance of PSSMH NPs was superior to Abraxane®. The animal assays conducted in this study followed the Guidelines for the Management and Use of Laboratory Animals. The study received ethical support from the Institutional Animal Ethical Care Committee (IAEC) of Shenyang Pharmaceutical University.
In this study, the redox-triggered PSSMH NPs were prepared to overcome the deficiencies of paclitaxel. For the preparation of PSSMH NPs, 16-MH with excellent biocompatibility was selected as the assembly module and was linked to redox-sensitive disulfide bonds and paclitaxel by ethylene glycol. PSSMH NPs had the advantages of higher drug loading, superior stability and easy preparation. Comprehensive studies were conducted to evaluate the storage stability, drug release efficiency, cytotoxicity, pharmacokinetic behavior, antitumor efficacy and safety of PSSMH NPs. The conclusions were as follows: (ⅰ) PSSMH NPs were stable as a liquid for 60 days at 25 ℃ compared to Taxol and Abraxane®. (ⅱ) PSSMH NPs improved the in vivo fate of paclitaxel, as evidenced by the AUC0–24 h was 14.95-fold compared with Taxol. (ⅲ) The existence of redox-sensitive disulfide bonds allowed PSSMH NPs to be redox-triggered at the tumor sites, which enhanced the tumor selectivity, thus resulting in excellent tolerance and antitumor efficacy. Overall, the redox-triggered prodrug nano-system with high tumor selectivity and biocompatibility provided valuable insights for breaking through the antitumor dilemma of paclitaxel.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by Key research and development program of Liaoning Province (No. 2024JH2/102500061), Youth innovation team of Liaoning Province Department of Education (No. LJ222410163049), Liaoning Revitalization Talents Program (No. XLYC2203083), the Open Fund of High-level Key Discipline of Chemistry of Chinese Medicine of the State Administration of Traditional Chinese Medicine, Anhui University of Chinese Medicine (No. HKDCCM2024007), Postdoctoral Fellowship Program of CPSF (No. GZC20231732), China Postdoctoral Science Foundation (Nos. 2023TQ0222, 2023MD744229), General Program of Department of Education of Liaoning Province (No. JYTMS20231372), Doctoral Scientific Research Staring Foundation of Liaoning Province (No. 2024-BS-073).
Supplementary materials
Supplementary material associated with this article can be
found, in the online version, at doi:10.1016/j.cclet.2025.111350.
[1]
M.C. Wani, H.L. Taylor, M.E. Wall, P. Coggon, A.T. McPhail, J. Am. Chem. Soc. 93 (1971) 2325–2327. doi: 10.1021/ja00738a045
[2]
T. Konno, J. Watanabe, K. Ishihara, J. Biomed. Mater. Res. 65A (2003) 209–214. doi: 10.1002/jbm.a.10481
H. Liang, X. Ren, J. Qian, et al., ACS Appl. Mater. Interfaces 8 (2016) 10136–10146. doi: 10.1021/acsami.6b00668
Figure 1
Characterization and stability of PSSMH NPs. (A) Molecular simulation of PSSMH. (B) The variation of the particle sizes of non-PEGylated PSSMH NPs after co-incubated with urea, SDS or NaCl. (C) Particle size distribution and TEM image of PEGylated PSSMH NPs. Scale bar: 200 nm. (D) The colloidal stability. (E, F) Storage stability of PEGylated PSSMH NPs. Data are presented as mean ± standard deviation (SD) (n = 3).
Figure 3
Cytotoxicity and pharmacokinetics. (A–D) The IC50 of Taxol, Abraxane® and PSSMH NPs in 4T1, A549, KB, and L02 cells. (E) Paclitaxel released by different concentrations of PSSMH NPs in 4T1 cells. (F, G) Tumor selective activation mechanism of PSSMH NPs. (H–J) Plasma drug concentration-time curves (PTX equivalent). (K) The sum AUC0–24 h (prodrug and paclitaxel, paclitaxel equivalent). (L) The diagram of PSSMH NPs and Taxol in blood circulation. **P < 0.01, ***P < 0.001, ****P < 0.0001 by two-tailed Student's t-test. Data are presented as mean ± SD (n = 3).
Figure 4
Antitumor efficacy and tolerance of PSSMH NPs. (A) The administration schedule of the BALB/c mice. (B–E) Tumor volume and body weight of BALB/c mice. (F) The administration schedule of the BALB/c nude mice. (G‒K) Tumor volume and body weight of BALB/c nude mice. (L) The antitumor mechanism of PSSMH NPs. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. ns, not significant by two-tailed Student's t-test. Data are presented as mean ± SD (n = 3).

 DownLoad:
DownLoad:

 下载:
下载:





 下载:
下载:
