Discovery of α-mangostin derivatives as novel PDE4 inhibitors for the treatment of Alzheimer's disease: An artificial intelligence-driven synergized strategy
Citation:
Zhi-Pei Sang, Teng Xue, Qian-Ru Xing, Qi-Yao Zhang, Hong-Song Chen, Xue Wang, Fu-Rong Zhang, Wen-Ling Fu, Wu Dong, Shu-Heng Huang, Yi-You Huang, Hai-Bin Luo. Discovery of α-mangostin derivatives as novel PDE4 inhibitors for the treatment of Alzheimer's disease: An artificial intelligence-driven synergized strategy[J]. Chinese Chemical Letters,
2026, 37(5): 111318.
doi:
10.1016/j.cclet.2025.111318
Discovery of α-mangostin derivatives as novel PDE4 inhibitors for the treatment of Alzheimer's disease: An artificial intelligence-driven synergized strategy
English
Discovery of α-mangostin derivatives as novel PDE4 inhibitors for the treatment of Alzheimer's disease: An artificial intelligence-driven synergized strategy
Key Laboratory of Tropical Biological Resources of Ministry of Education and Hainan Engineering Research Center for Drug Screening and Evaluation, School of Pharmaceutical Sciences, Hainan University, Haikou 570228, China
b.
College of Animal Science and Technology. Inner Mongolia Minzu University, Tongliao 028000, China
c.
Song Li's Academician Workstation of Hainan University (School of Pharmaceutical Sciences), Yazhou Bay, Sanya 572000, China
hbluo@hainanu.edu.cn (H.-B. Luo). 1 These authors contributed equally to this work.
Received Date:
06 February 2025 Accepted Date:
14 May 2025 Revised Date:
13 May 2025 Available Online:
15 May 2026
Abstract:
Alzheimer's disease (AD) is a chronic, progressive neurodegenerative disorder with no effective therapeutic agents currently available. Inhibiting phosphodiesterase 4 (PDE4) has emerged as a promising strategy for AD treatment. In this study, we employed a synergistic approach combining generative recurrent neural network (RNN)-driven combinatorial compound design, virtual screening, and structure-activity relationship (SAR) analysis to discover novel PDE4 inhibitors. Utilizing α-mangostin as a hit compound (half maximal inhibitory concentration (IC50) = 1.31 µmol/L), we identified a novel PDE4 inhibitor, 13d (IC50 = 72.8 nmol/L) with moderate liver microsomal stability (rat liver microsomes (RLM), t1/2 = 32.4 min). In vitro activity results indicated that 13d exhibited favorable anti-inflammatory effects and promising neuroprotective activity. In vivo experiments demonstrated that 13d significantly improved AlCl3-induced zebrafish AD model by inhibiting PDE4 and reducing inflammatory cytokine. Further, 13d significantly alleviated AlCl3/d-galactose-induced AD mouse model. These findings highlight the potent PDE4 inhibitor 13d with promising anti-AD activity, underscoring the potential of artificial intelligence-driven drug discovery for novel therapeutic agents for AD.
Alzheimer's disease (AD) is a progressive neurodegenerative disorder characterized by deteriorating memory, declining language skills, and a spectrum of cognitive impairments in older adults [1]. Currently, over 55 million individuals worldwide are diagnosed with AD, and this number is projected to reach 139 million by 2050 [2]. The disease poses significant social and economic challenges. Currently available Food and Drug Administration (FDA)-approved treatments include cholinesterase inhibitors (such as rivastigmine, donepezil, and galantamine), N-methyl-d-aspartic acid (NMDA) receptor antagonists (like memantine), and monoclonal antibodies (including aducanumab and lecanemab). Although these therapies offer limited symptomatic relief, they do not stop, delay, or reverse the progression of the disease [3]. Additionally, many patients experience substantial adverse effects, including gastrointestinal disturbances, dizziness, heart failure, seizures, and infusion-related reactions associated with monoclonal antibodies [4-7]. Therefore, the search for effective treatments has become an urgent priority.
Phosphodiesterase IV (PDE4) which specifically hydrolyzes cAMP (cyclic adenosine monophosphate), has four isoforms (PDE4A, PDE4B, PDE4C, and PDE4D), and is widely expressed in the brain of demented patients [8-10]. Since the 1990s, many reports have confirmed that PDE4 inhibitors (such as roflumilast [11], rolipram [12], HT-0712 [8], and FFPM [13]) could significantly improve learning and memory through cAMP/cAMP-response element binding protein (CREB)/brain derived neurotrophic factor (BDNF) and anti-inflammatory effects. By increasing intracellular cAMP levels, PDE4 inhibitors enhance CREB phosphorylation, which in turn upregulates the expression of BDNF, a crucial neurotrophic factor involved in synaptic plasticity, neuronal survival, and memory formation. This neuroprotective mechanism contributes to reduced neuronal apoptosis, mitigates neuroinflammation, and counteracts oxidative stress [8,9]. More importantly, PDE4 inhibitors have had positive results in clinical studies, such as, roflumilast has completed Phase Ⅱ clinical trials (NCT04658654, NCT01433666) investigating cognitive-related outcomes [14,15], BPN14770 is currently undergoing Phase Ⅱ clinical trials for AD (NCT03817684), and HT-0712 has completed Phase Ⅱ clinical trials (NCT02013310) targeting age-related memory impairment [8,9]. Therefore, PDE4 as a target for AD has received widespread attention.
α-Mangostin (α-M, Fig. 1A) is a major polyphenolic xanthone extracted from the pericarps, bark, and dried sap of the mangosteen fruit (Garcinia mangostana L.), first isolated in 1855. Traditionally employed in Southeast Asian medicine, α-M has gained substantial research interest for its multifaceted biological activities [16,17]. Notably, emerging evidence indicates its potential in mitigating cognitive decline and providing neuroprotection in AD, attributed to its powerful antioxidant, anti-inflammatory, and neuroprotective properties [18]. In our group's preliminary work, we discovered α-M as a natural PDE4 inhibitor (half maximal inhibitory concentration (IC50) = 1.31 µmol/L) [19], warranting further development into a potent PDE4 inhibitor for the treatment of AD.
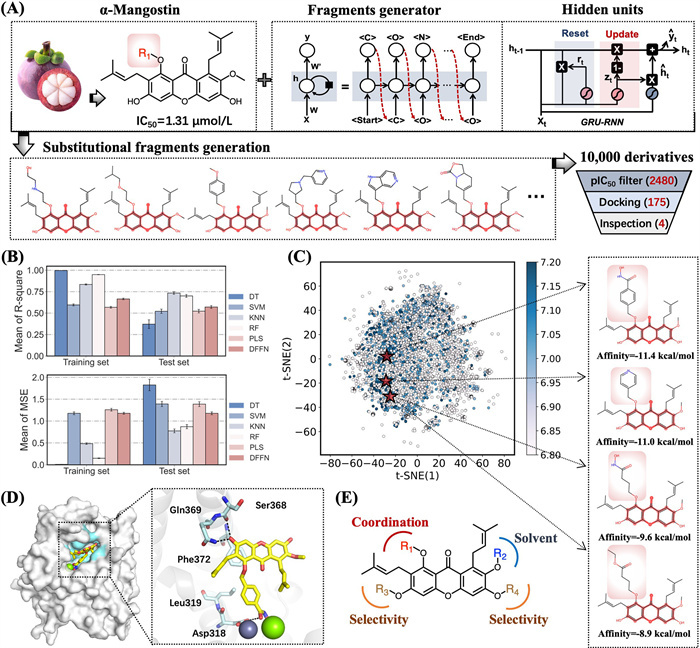
Figure 1
Figure 1.
Discovery of novel α-M derivatives as lead compounds. (A) Schematic illustration of the molecular design and development of noval α-M derivatives. (B) The performance of the 5 ML models and the DFNN model on the training and test dataset. Default parameters were used for the ML models if not specified. (C) The distribution in the chemical space of the 10,000 generated α-M derivatives. (D) Binding modes of representative compound, where key residues were represented as stick models. The H-bond interactions were indicated by black dashed lines. (E) General modification methods of structure-based molecular design.
Traditional drug discovery processes, particularly in medicinal chemistry, are often characterized by a degree of blindness and unpredictability. Due to the vastness of chemical space and the inherent complexity of molecular interactions, it is difficult to comprehensively explore all possible compounds and their corresponding biological activities [20,21]. This limitation leads to inefficiencies and high costs in the drug development pipeline. However, with the rapid advancement of artificial intelligence (AI) and machine learning (ML) technologies, new opportunities have emerged to overcome these challenges. Integrating generative models with virtual screening techniques enables a more systematic and efficient exploration of the chemical space [22,23]. This combination of AI-driven approaches significantly enhances the efficiency of drug discovery, reducing both the time and resources required to identify viable drug candidates.
Herein, we present a novel approach that combines generative recurrent neural network (RNN)-driven combinatorial compound design, virtual screening, and structure-activity relationship (SAR) analysis for the de novo design of PDE4 inhibitors and the evaluation of their potential anti-AD activity. This integrated strategy synergistically combines the strengths of AI-powered molecular generation with traditional medicinal chemistry principles, enabling systematic exploration of chemical space. The synergistic strategy consists of the following steps: The synergized strategy for the intelligent drug design of PDE4 inhibitors is as follow: (1) Generating 10,000 unique substitutional fragments by RNN-based "Fragment Generator"; (2) Constructing a virtual library of α-M derivatives by substituent modification; (3) Virtual screening of PDE4 inhibitors by ML modeling and molecular docking; (4) Optimizing the lead compounds by SAR studies; (5) Evaluating the anti-AD of 13din vitro and in vivo experiments (Fig. 1A). By using this strategy, we discovered a novel PDE4 inhibitor, designated as compound 13d (IC50 = 72.8 ± 9.6 nmol/L). The significant efficacy of 13d warranted comprehensive investigations into its drug-likeness profile. In vivo studies were conducted using AlCl3-induced zebrafish AD model and AlCl3/galactose-induced mice. These findings represent a substantial advancement in the design and development of potent PDE4 inhibitors, positioning compound 13d as a promising lead for the treatment of AD.
To fully explore the chemical space of α-M derivatives, we first utilized a self-built RNN-based "Fragment Generator" to generate 10,000 substituent fragments [24]. To optimize the generation of small molecular substituents, we curated a specialized training set from ChEMBL consisting of 1179,477 unique drug-like molecules (MW < 200 Da). After 30,000 training steps, the loss value converged and plateaued at 23.1, while the validity rate of the generated SMILES strings reached 93.4% (Fig. S1 in Supporting information), demonstrating the model's capability to consistently produce structurally valid molecular fragments. Then, 10,000 fragments were systematically substituted onto the α-M scaffold at the R1 position constructing a virtual library of α-M derivatives, thereby maximizing structural diversity. Subsequently, for developing a reliable ML model for predicting PDE4 inhibitory activities, we evaluated the performance of five ML algorithms, i.e., decision tree (DT), support vector machine (SVM), k-nearest neighbor (K-NN), random forest (RF), partial least squares (PLS), along with the deep learning model deep feedforward neural network (DFNN). Based on prediction accuracy and balanced performance on the test set, K-NN was selected as the optimal model, with R2 of 0.83 and 0.73 on the training and test set, respectively (Fig. 1B). Then, the virtual library was subjected to activity prediction using the optimal activity predictor model, which identified 2480 compounds with predicted pIC50 values greater than 7, implying strong activity in the nanomolar range (Fig. 1C).
A high-throughput virtual screening method was used to perform molecular docking on the screened compounds. Based on the docking results, 175 compounds with affinities lower than −8 kcal/mol were selected as high-affinity candidates. A synthetic feasibility assessment was then conducted to ensure the practicality of these compounds. Finally, four lead structures with favorable binding modes and high synthetic feasibility were selected for subsequent chemical synthesis and SAR studies. It is noteworthy that molecular docking studies revealed a binding mode similar to that observed in the previously reported crystal structure of PDE4 in complex with α-M derivative (PDB: 6KJZ) [19]. It can be observed the α-M's scaffold embedded well into the catalytic pocket of PDE4, with its benzene fragment forming a π-π stacking interaction with Phe372. In addition, the catalytic site contained a metal binding pocket composed of residues such as Asp318 and Leu319, which formed hydrogen bonds and metal coordination interactions with the R1 side chain. The R2 substituent was mainly involved in the solvation region. The R3 and R4 substituents respectively extend into the Q1 and Q2 pockets, which are unique subpockets present among PDE4 isoforms, thereby contributing to enhanced selectivity (Figs. 1D and E) [19].
To further optimize the hit structure, we performed structure-based drug design based on the docking results. As shown in Fig. 1E, the R1 position was suitable for introducing long-chain charged groups, while the R2 position was modified with CH3O group. The R3 and R4 positions were tailored to enhance the selectivity of PDE4 inhibitors. Subsequently, we designed a series of α-M derivatives by modifying the R1 position with four selected high-scoring fragments (i.e., pyridine, carboxylic ester, carboxylic acid and hydroximic acid) (Fig. 1C). In consideration of the synthetic accessibility and potential for systematic structural diversification, the second compound was selected as our initial optimization starting point. A total of 22 compounds were synthesized and evaluated for their anti-PDE4 activity.
The synthetic routes for the novel target derivatives 6a, 6b, 8a–8f, 11a–11c, 13a–13d, 19a–19c and 20a–20d were illustrated in Schemes S1–S4 (Supporting information).
In order to evaluate the PDE4 inhibitory potency of the target derivatives, liquid scintillation cocktail assay was used, and rolipram was also tested as a positive control [24,25]. Considering cost and efficiency, the PDE4 inhibitory activity of all target compounds was assessed using single-point inhibition tests at concentrations of 1 µmol/L and 100 nmol/L, respectively. As shown in Table 1, both the two alkoxy side chains R1/R2 and the targeting metal region fragment significantly influenced the PDE4 inhibitory activity. When the targeting metal region fragment was pyridine, compounds 6a and 6b were synthesized by introducing different R1/R2 fragments based on our previous work, the results demonstrated that compounds 6a and 6b did not exhibit a significant improvement in PDE4 inhibitory activity compared to α-M. When R1 and R2 were substituted by the same fragments, compounds 8a–8f were obtained, the PDE4 inhibition data displayed that smaller R1/R2 fragments showed better PDE4 inhibitory activity, such as 8b (methyl fragment), 8d (ethyl fragment), 8e (isobutyl), and 8f (cyclopentyl). Notably, compound 8b, which contained two methoxy groups, exhibited good PDE4 inhibitory activity at 100 nmol/L (52.76%).
Table 1
Table 1.
The PDE4 inhibitory activities of target compounds 6a, 6b, 8a–8f, 11a–11c and 13a–13d.
Furthermore, when both alkoxy groups R1/R2 were methoxy, we synthesized target compounds 11a–11c and 13a–13d (Table 1), and investigated the influence of carboxylic acid and hydroxamic acid fragments on PDE4 inhibitory activity. The test results showed that derivatives containing carboxylic acid fragments, such as 11a–11c, significantly reduced PDE4 inhibitory activity, with no obvious effect of CH2 chain length on PDE4 inhibition. After substituting the carboxylic acid fragment of compounds 11a–11c with hydroxamic acid, derivatives 13a–13c were obtained, the PDE4 inhibitory activity remarkably improved, with no evident influence of CH2 chain length on inhibition. When the aliphatic chain of compounds 13a–13c was replaced by a benzene ring, derivative 13d was obtained. Compound 13d exhibited significantly enhanced PDE4 inhibitory activity, showing 81.75% inhibition at 100 nmol/L with an IC50 of 72.8 ± 9.6 nmol/L (Table S1 in Supporting information), outperforming the positive control rolipram (55.74% inhibition at 800 nmol/L).
To further discuss the SAR, we removed the isopentene group fragments from 11a–11c and 13a–13d, yielding derivatives 19a–19c and 20a–20d (Table S2 in Supporting information), which exhibited potent PDE4 inhibitory activity. And among derivatives 19a–19c and 20a–20d, derivatives containing hydroxamic acids demonstrated better PDE4 inhibitory activity. Thus, 13d gave better PDE4 inhibitory potency. The selectivity of compound 13d across other PDEs was also tested and listed in Table S1. 13d indicated very weak inhibitory potencies against PDE1C2, PDE3A and PDE8A1, PDE9A, and PDE10A with IC50 values higher than 10 µmol/L and selectivity folds greater than 136. Besides, the IC50 values of 13d against PDE2A, PDE7A1 and PDE9A2 were tested to be 1727,300 and 3442 nmol/L with 23-, 4- and 47-fold selectivity, respectively. Therefore, 13d was a highly effective and selective PDE4 inhibitor, deserving for further investigations.
To investigate the binding mechanism of 13d with PDE4, 50-ns molecular dynamics (MD) simulations were performed using the 13d-PDE4 complex, followed by binding free energy calculations to assess binding affinity. The system remained stable over 50 ns, with the RMSD below 2 Å in the final 20 ns, indicating stable binding. 13d interacted with active pocket of PDE4 and fitted into a hydrophobic pocket. The total binding free energy was −11.27 ± 14.12 kcal/mol, with van der Waals interactions playing a particularly significant role (−46.15 ± 15.41 kcal/mol). The benzamide formed hydrogen bonds with Thr271 and Asp272, and had π-π conjugation with Phe372, which was the key catalytic residue of PDE4 (−3.48 ± 0.20 kcal/mol). Moreover, van der Walls interactions could be observed between the 13d and Met273, Ile336, Ser368 and Tyr375 (Fig. S2 in Supporting information).
Before subjected to in vitro and in vivo pharmacodynamics (PD) studies, pharmacokinetic profile, drug-like property, and acute safety of 13d were also evaluated. Liquid chromatograph/mass spectrometer (LC-MS/MS) analysis results demonstrated that 13d indicated moderate metabolic stability (rat liver microsomes (RLM), t1/2 = 32.4 min) [26]. The pharmacokinetic properties of compound 13d were listed in Table S3 (Supporting information). Compound 13d displayed significant half-lives of 6.5 h (intravenous (i.v.)) and 3.7 h (per oral (p.o.)) and exhibited potent systemic exposure with area under the curve (AUC) values of 1439.0 h ng mL−1 (i.v., 2.5 mg/kg) and 68.9 h ng mL−1 (p.o., 5.0 mg/kg). The oral bioavailability of 13d was 2.77%. To establish the safety profile of 13d, twenty C57BL/6T (18–22 g) mice (half males and half females) were divided into control and 13d (500 mg/kg) group, which were administered orally and intragastric for 14 days. Animal studies were conducted in accordance with the approved protocol by the Animal Ethics Committee of Hainan University (approval No. HPIACUC2024090). The results showed that there were not significant changes in movement behavior, mental state, and body weight. Therefore, compound 13d indicated a favorable safety profile.
The AlCl3-induced zebrafish AD model has been widely applied for the high-throughput screening of novel anti-Alzheimer's agents [27-29]. Herein, we first assessed the safety profile of 13d through five different concentrations (10.0, 20.0, 40.0, 80.0 and 160.0 µg/mL) in a zebrafish model (Fig. S3 in Supporting information). Notably, no mortality was observed at 160.0 µg/mL over a 120 h exposure period (Fig. S3A). Following this, Fig. S3B showed that 13d showed significant changes in pericardial edema/body length at 160.0 µg/mL. Moreover, Figs. S3C and D exhibited that 160.0 µg/mL produced obvious influence in swim bladder area. Thus, the maximum tolerable concentration (MTC) of 13d was 80.0 µg/mL.
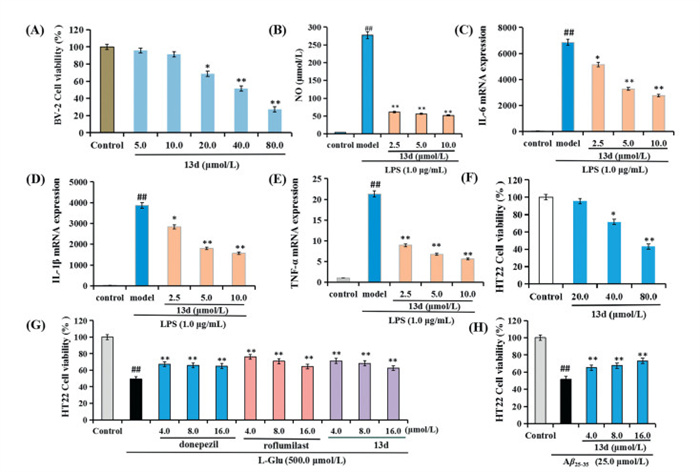
Compound 13d was selected to evaluate its anti-inflammatory activity by assessing the production of nitric oxide (NO), interleukin 6 (IL-6), IL-1β, and tumor necrosis factor-α (TNF-α) in lipopolysaccharide (LPS)-induced BV-2 microglial cells [30]. As shown in Fig. 2A, 13d did not show obvious cytotoxicity at 10.0 µmol/L, suggesting a favorable safety profile. As illustrated in Figs. 2B–E, 13d significantly reduced the production of NO, IL-6, IL-1β, and TNF-α in a dose-dependent manner, suggesting that 13d demonstrated significant anti-inflammatory property.
Figure 2
Figure 2.
Anti-inflammatory activities and neuroprotective effects of compound 13d. (A) The cell viability of 13d on BV-2 cells was assessed using the CCK-8 assay. *P < 0.05, **P < 0.01 vs. control group. Anti-inflammation effects of 13d in LPS-stimulated BV-2 cells. (B) NO release. (C) IL-6 mRNA expression. (D) IL-1β mRNA expression. (E) TNF-α mRNA expression. ##P < 0.01 vs. control group. **P < 0.01 vs. LPS-induced group. (F) Cell viability (%) of 13d in HT22 cells. *P < 0.05, **P < 0.01 vs. control group. (G) Neuroprotective effects of 13d, donepezil and roflumilast on Glu-induced HT22 cells injury. (H) Neuroprotective effects of 13d on Aβ25–35-induced HT22 cells injury. Values were expressed as mean ± SD from three independent experiments. ##P < 0.01 vs. control group. *P < 0.05, **P < 0.01 vs. Glu/Aβ25–35-induced group.
The neuroprotective effects of 13d were evaluated in glutamic (Glu)-/Aβ25–35-induced HT22 cell injury using the cell counting kit-8 (CCK-8) assay, with donepezil and roflumilast as positive control [30,31]. As shown in Fig. 2F, 13d did not produce obvious cytotoxicity in HT22 cells at 20 µmol/L. Fig. 2G illustrated that exposure to 500.0 µmol/l-glutamic acid (l-Glu) significantly decreased cell viability to 49.4% (P < 0.01). In contrast, treatment with various concentrations of 13d (4.0, 8.0, and 16.0 µmol/L) resulted in a significant dose-dependent increase in cell viability, indicating that the neuroprotective potential of 13d was similar with donepezil and roflumilast. Furthermore, as demonstrated in Fig. 2H, compound 13d provided significant neuroprotection against Aβ25–35-induced HT22 cells injury in a dose-dependent manner. Thus, these data demonstrated that 13d presented significant neuroprotective effects against Glu- and Aβ25–35-induced cellular injury.
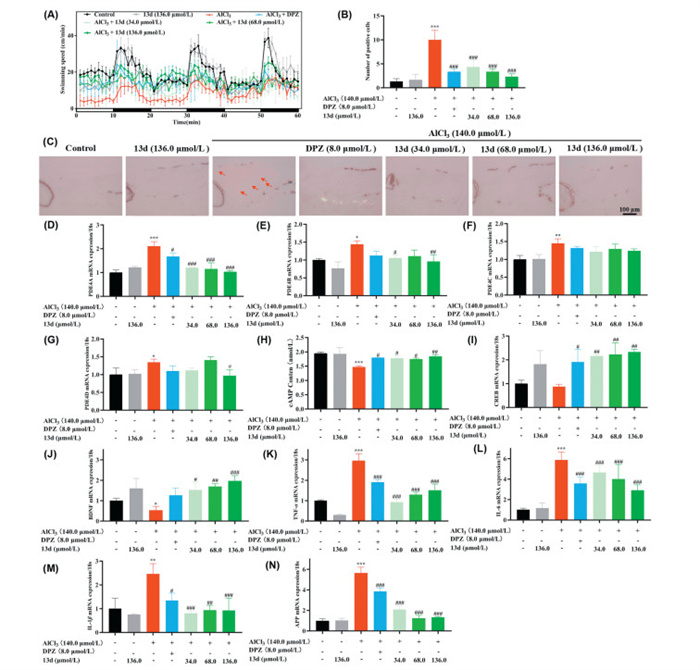
Based on the MTC of 80.0 µg/mL (136 µmol/L) for 13d, we subsequently selected three lower concentrations (34.0, 68.0, and 136 µmol/L) for further evaluation following preliminary screening [30,31]. To assess the therapeutic effects of these treatments, a series of behavioral tests were conducted under alternating light and dark conditions over a 60-min period (Fig. 3A). The experimental design included seven groups: control group, 13d treatment group (136.0 µmol/L), AlCl3-induced model group (140.0 µmol/L), AlCl3 + donepezil (8.0 µmol/L) group, and AlCl3 + 13d (34.0, 68.0, and 136.0 µmol/L) treatment groups. As indicated in Fig. S4 (Supporting information), zebrafish exposed to AlCl3 demonstrated a statistically significant reduction in swimming speed compared to the control group under light and dark conditions (Figs. S4A–C). In contrast, when treatment with donepezil (8.0 µmol/L), the swimming speed significantly increased. And when treating with 13d (34.0, 68.0, and 136.0 µmol/L), the swimming speed presented remarkably improved in a dose-dependent manner. Furthermore, Figs. S4D–F showed that 13d (34.0, 68.0, and 136.0 µmol/L) significantly decreased swimming distance in a dose-dependent manner under the light condition. However, in a dark environment, 13d exhibited an increasing trend, although no significant differences were observed. Further, when the dark environment was transitioned to a light environment, both donepezil and 13d significantly improved reaction capacity. When the light environment was switched back to a dark environment, the reaction capacity of 13d (136.0 µmol/L) was significantly enhanced (Figs. S4G–H). Moreover, the terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) staining results in Figs. 3B and C displayed that donepezil (8.0 µmol/L) and 13d (34.0, 68.0, and 136.0 µmol/L) significantly reduced the number of apoptotic cells, compared to model group.
Figure 3
Figure 3.
(A) Behavioral changes in the zebrafish larvae per minute. (B) The apoptotic body/normal cells in the various zebrafish models was statistically analyzed after TUNEL staining. (C) Apoptotic body changes in the brain of zebrafish were performed using TUNEL staining. Scale bar: 10 µm. (D) PDE4A mRNA expression change. (E) PDE4B mRNA expression changes. (F) PDE4C mRNA expression changes. (G) PDE4D mRNA expression changes. (H) cAMP level changes. (I) CREB mRNA expression changes. (J) BDNF mRNA expression changes. (K) TNF-α mRNA expression changes. (L) IL-6 mRNA expression changes. (M) IL-1β mRNA expression changes. (N) APP mRNA expression changes. Values are expressed as mean ± SD from three independent experiments. *P < 0.05, ***P < 0.01, ***P < 0.001 vs. control group. #P < 0.05, ##P < 0.01, ###P < 0.001 vs. AlCl3-induced zebrafish model.
The anti-AD mechanism of compound 13d was investigated in Figs. 3D–N. Figs. 3D–J indicated that 13d elevated cAMP levels by inhibiting PDE4A, PDE4B and PDE4D, and further improved the levels of CREB and BDNF. Moreover, 13d demonstrated significant anti-inflammatory properties by decreasing levels of TNF-α (Fig. 3K), IL-6 (Fig. 3L) and IL-1β (Fig. 3M). Additionally, the APP levels significantly increased in AlCl3 induced neurotoxicity mouse model [32,33]. As displayed in Fig. 3N, 13d was found to lower amyloid precursor protein (APP) levels in a dose-dependent manner, the possible reason was that 13d indirectly affected APP expression and metabolism by increasing cAMP levels and anti-inflammatory effects. These results demonstrated that 13d significantly ameliorated the AlCl3-induced zebrafish AD model via PDE4A/4B-mediated cAMP/CREB/BDNF signaling. This signaling pathway facilitated the neuroprotective, anti-apoptotic, and anti-inflammatory actions of 13d, which collectively decreased the levels of APP and enhanced its behavioral outcomes.
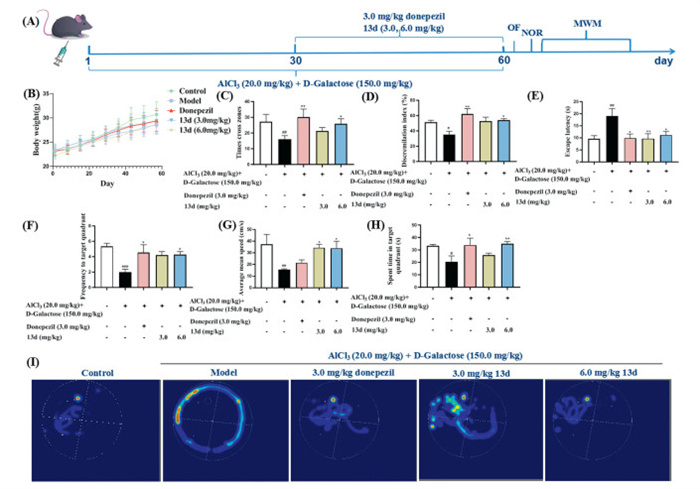
To further evaluate the in vivo effects of compound 13d, the AlCl3 and d-galactose-induced AD mouse model was applied [34,35]. The AD model was established using AlCl3 (20.0 mg/kg) in combination with d-galactose (120.0 mg/kg). AlCl3 was administered via oral gavage at a dose of 0.1 mL/10.0 g daily, and d-galactose was injected intraperitoneally at a dose of 0.1 mL/10.0 g. Mice in the normal control group received an equal volume of carboxymethylcellulose sodium (CMC–Na) (0.1 mL/10.0 g). The modeling was conducted over a continuous period of 60 days. C57BL/6T mice were randomly divided into five groups based on body weight, with each group consisting of 11 mice. The groups included normal control group, model group, positive drug group (3.0 mg/kg donepezil), and 13d groups (3.0 and 6.0 mg/kg). On the 31st day of modeling, each group was administered orally and intragastric the corresponding treatment at a volume of 0.1 mL/10.0 g, with the normal control and model groups receiving an equal volume of CMC–Na (0.1 mL/10.0 g). Treatment was administered once daily for a total of 30 days. Behavioral tests were subsequently conducted, including the open field test (OFT), novel object recognition test (NOR), and Morris water maze (MWM) (Fig. 4A). As shown in Fig. 4B, there were no significant differences in body weight among the different groups of mice. Fig. 4C demonstrated that 13d (6.0 mg/kg) and donepezil significantly improved the times cross zones in the open field test. Fig. 4D indicated 13d (6.0 mg/kg) and donepezil could increase the discrimination index in the NOR test. Fig. 4E exhibited that donepezil and 13d could decrease the locational navigation escape latency compared with the model group in the MWM test. Moreover, Fig. 4F suggested that 13d, especially the dose of 6.0 mg/kg, significantly reduced the spatial exploration escape latency (P < 0.05). Furthermore, Figs. 4G and H demonstrated that 6.0 mg/kg 13d remarkably improved spatial exploration shuttle platform (P < 0.05), average speed (P < 0.05), and the quadrant dwell time in spatial exploration (P < 0.01), respectively. And the localized cruise heat map (Fig. 4I) was consistent with the results. Additionally, Figs. S5A–F (Supporting information) exhibited that there were no significant differences observed in the weights of the heart, liver, and kidneys, nor in the levels of CREA (creatinine) and UREA. Therefore, these observations and data indicated that compound 13d (6.0 mg/kg) significantly enhanced spatial memory and cognitive function. Further, to confirm whether compound 13d could cross the blood-brain barrier, we measured the concentrations of 13d in the cerebral cortex and plasma of the high (6.0 mg/kg) dose group. LC-MS analysis indicated that the concentrations of compound 13d in the cerebral cortex and plasma for the high dose group was 2.2 ng/g and 45.6 ng/mL, respectively, and B/P ratio was 0.05, demonstrating that compound 13d presented weak blood-brain barrier permeability.
Figure 4
Figure 4.
Effects of 13d in the AlCl3/d-galactose mice model. Data are presented as mean ± standard deviation (SD) (n = 9). (A) The in vivo evaluation of 13d in AlCl3/d-galactose mice. (B) Weight changes were recorded in various experimental groups. (C) The times cross zones were evaluated using the field test. (D) The discrimination index was evaluated using the novel object recognition test. (E) The spatial exploration escape latency was recorded in various groups using the MWM. (F) The spatial exploration shuttle platform was recorded in various groups using the MWM. (G) Average speeds were recorded in various groups. (H) The quadrant dwell time in spatial exploration were recorded in various groups. (I) Localized cruise heat map were recorded in various groups using the MWM. #P < 0.05, ##P < 0.01 vs. control group; *P < 0.05, **P < 0.01 vs. model group.
AD is a chronic, progressive neurodegenerative disorder primarily characterized by memory and cognitive impairments. Currently available clinical medications can only provide temporary symptomatic relief without effectively halting or reversing the progression of AD, highlighting the urgent need for the development of novel anti-AD drugs. Inhibiting PDE4 is considered a promising strategy for the treatment of AD. Herein, we present a novel approach that combines RNN-based fragment generator, virtual screening, and SAR analysis for the de novo design of PDE4 inhibitors and the evaluation of their potential anti-AD activity. Our fragment replacement strategy outperforms traditional methods by efficiently exploring chemical space while preserving the α-M scaffold, generating diverse derivatives (> 10,000 compounds) with precise modifications—overcoming RNN randomness and scaffold-constrained yield limitations. Based on the natural PDE4 inhibitor α-M, we utilized it as a hit structure to discover a novel PDE4 inhibitor, compound 13d (IC50 = 72.8 nmol/L). In vitro activity results indicated that compound 13d exhibited significant anti-inflammatory effects by reducing the levels of NO, IL-6, IL-1β and TNF-α. 13d also indicated encouraging neuroprotective effects against Glu- and Aβ25–35-induced HT22 cellular injury. Moreover, compound 13d displayed moderate liver microsome stability (t1/2 = 32.4 min). In vivo experiments demonstrated that 13d significantly improved the AlCl3-induced zebrafish AD model via enhancing cAMP levels by PDE4 inhibition and reducing the levels of inflammatory factors. Additionally, 13d significantly alleviated AlCl3/d-galactose-induced AD mouse model. Collectively, this study demonstrates the discovery of the PDE4 inhibitor 13d with anti-AD activity via AI, warranting further investigation.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was financially supported by the National Key Research and Development Program of China (No. 2023YFF1205102); the National Natural Science Foundation of China (Nos. 22367007, 22377023, and 82304384); the Science Foundation of Hainan Province (Nos. 825MS072, KJRC2023B10 and 824RC500); the China Postdoctoral Science Foundation (No. 2022M712153) and the Fundamental Research Funds for Hainan University (Nos. XTCX2022JKA01, KYQD(ZR)23002).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.111318.
A. Tanas, Ö.Ö. Tozlu, T. Gezmiş, et al., Biomed Res. Int. 2022 (2022) 5467498.
[34]
Y. Hu, X. Fang, J. Wang, et al., Neurotoxicology 91 (2022) 60–68.
[35]
T. Vishnumukkala, P.K. Gopalakrishna, B. Karikalan, et al., Bioinformation 20 (2024) 508–514. doi: 10.6026/973206300200508
Figure 1
Discovery of novel α-M derivatives as lead compounds. (A) Schematic illustration of the molecular design and development of noval α-M derivatives. (B) The performance of the 5 ML models and the DFNN model on the training and test dataset. Default parameters were used for the ML models if not specified. (C) The distribution in the chemical space of the 10,000 generated α-M derivatives. (D) Binding modes of representative compound, where key residues were represented as stick models. The H-bond interactions were indicated by black dashed lines. (E) General modification methods of structure-based molecular design.
Figure 2
Anti-inflammatory activities and neuroprotective effects of compound 13d. (A) The cell viability of 13d on BV-2 cells was assessed using the CCK-8 assay. *P < 0.05, **P < 0.01 vs. control group. Anti-inflammation effects of 13d in LPS-stimulated BV-2 cells. (B) NO release. (C) IL-6 mRNA expression. (D) IL-1β mRNA expression. (E) TNF-α mRNA expression. ##P < 0.01 vs. control group. **P < 0.01 vs. LPS-induced group. (F) Cell viability (%) of 13d in HT22 cells. *P < 0.05, **P < 0.01 vs. control group. (G) Neuroprotective effects of 13d, donepezil and roflumilast on Glu-induced HT22 cells injury. (H) Neuroprotective effects of 13d on Aβ25–35-induced HT22 cells injury. Values were expressed as mean ± SD from three independent experiments. ##P < 0.01 vs. control group. *P < 0.05, **P < 0.01 vs. Glu/Aβ25–35-induced group.
Figure 4
Effects of 13d in the AlCl3/d-galactose mice model. Data are presented as mean ± standard deviation (SD) (n = 9). (A) The in vivo evaluation of 13d in AlCl3/d-galactose mice. (B) Weight changes were recorded in various experimental groups. (C) The times cross zones were evaluated using the field test. (D) The discrimination index was evaluated using the novel object recognition test. (E) The spatial exploration escape latency was recorded in various groups using the MWM. (F) The spatial exploration shuttle platform was recorded in various groups using the MWM. (G) Average speeds were recorded in various groups. (H) The quadrant dwell time in spatial exploration were recorded in various groups. (I) Localized cruise heat map were recorded in various groups using the MWM. #P < 0.05, ##P < 0.01 vs. control group; *P < 0.05, **P < 0.01 vs. model group.

 DownLoad:
DownLoad:















 下载:
下载:





 下载:
下载:
