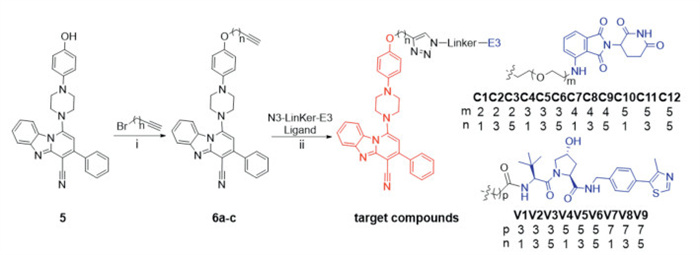
Scheme 1.
The synthesis of the target compounds. Reagents and conditions: (ⅰ) K2CO3, DMF, 90 ℃, 12 h; (ⅱ) sodium ascorbate, CuSO4, DMF/H2O, r.t., 24 h.
PROTAC degraders of FSP1 act as potent GPX4 sensitizers to induce ferroptosis for hepatoma treatment
Jiangmin Zhu , Qimei Tan , Shiying Fan , Yalin Li , Ling Zhu , Lihong Hong , Yuxia Wang , Chuzhen Zhang , Chen Chen , Lingyi Kong , Jianguang Luo

The development of innovative therapy strategies remains crucial, as hepatoma represents a significant global health challenge, characterized by high incidence and mortality rates [1]. Ferroptosis, a distinctive form of regulated cell death dependent on iron and characterized by the excessive accumulation of lipid peroxides (LPO) on cellular membranes, has gained significant attention in recent years [2–4]. One of the most effective strategies to induce ferroptosis is to destroy the defense mechanism of ferroptosis [5,6]. The cystine/glutamate antiporter-glutathione-glutathione peroxidase 4 (system XC−-GSH-GPX4) axis, a glutathione-dependent protective pathway, plays a key role in guarding against ferroptosis and is considered a crucial antioxidant mechanism [7,8]. GPX4, in particular, has become a prominent target in tumor research [9–11]. However, inhibiting GPX4 alone was insufficient to induce ferroptosis in certain cell types or cell lines [12,13]. Hence, it is imperative to investigate innovative approaches to boost the therapeutic efficacy of GPX4 in cancer treatment.
Ferroptosis suppressor protein 1 (FSP1) is a glutathione-independent ferroptosis suppressor and disassociate from the GPX4 [14,15]. Mechanistically, both GPX4 and FSP1 inhibit ferroptosis by reducing LPO production. Notably, knockdown of GPX4 in tumor cells would lead to the up-regulation of FSP1, likely through feedback regulation [16]. In fact, FSP1 inhibition selectively sensitizes tumor cells to ferroptosis inducers, as iFSP1 (FSP1 inhibitor) treatment sensitizes hepatoma cells to GPX4 inhibitor-induced ferroptosis. Furthermore, genetic deletion of FSP1 is more efficient than small-molecule inhibition in sensitizing tumor cells to GPX4 inhibitors [13]. These findings indicate that merely inhibiting the enzymatic activity of FSP1 may not be sufficient to achieve complete functional loss. Instead, agents that degrade FSP1 may be more effective in enhancing the sensitivity of hepatoma cells to GPX4 inhibitors.
Presently, small molecules targeting FSP1 primarily act to directly or indirectly inhibit its biological activity, without affecting the expression level of FSP1 [17,18]. However, recent studies have revealed that the expression of FSP1 protein is modulated through the ubiquitin (Ub)-proteasome system [19], suggesting that the level of FSP1 could potentially be reduced by enhancing its ubiquitination process. As a key regulatory target of ferroptosis, the biological effects of decreased FSP1 protein level are still unclear. Therefore, it is urgent to develop FSP1 degradation agents to elucidate the biological functions of FSP1. Proteolysis targeting chimeras (PROTACs) represent a cutting-edge technology specifically designed to degrade pathogenic proteins via the Ub-proteasome pathway. This innovative PROTACs strategy has garnered significant attention and is poised as a highly promising approach in the exploration and development of novel therapeutics for the treatment of various diseases [20–22]. More significantly, the entry of PROTAC molecules into clinical trials indicates that PROTAC degraders hold practical application prospects [23,24]. iFSP1s were the first identified class of FSP1 inhibitors. Subsequent studies have shown that iFSP1s targeted the quinone-binding pocket of FSP1 [15]. Based on structure-activity relationship study, we speculated that structural modification of the chlorine atom on the benzene ring in iFSP1–8 would not significantly affect its biological activity [13]. The docking model of iFSP1–8 and FSP1 showed that the chlorine atom was oriented towards the solvent region (Fig. S1 in Supporting information), which further supported the feasibility of the modification. Therefore, a series of PROTAC degraders of FSP1 were designed and synthesized using iFSP1–8 as the FSP1 ligand. Among them, C7 showed the most potent FSP1 degradation potency. More importantly, we also found that C7 was able to increase the susceptibility of hepatoma cells to GPX4 inhibitors, with a more pronounced effect than iFSP1. In combination therapy studies, C7 and ML162 (a GPX4 inhibitor) showed significantly enhanced in vivo antitumor efficacy against HepG2 xenograft tumors compared to iFSP1 and ML162. These findings underscored the potential of PROTAC degraders of FSP1, as potent GPX4 sensitizers to induce ferroptosis, thus representing a promising strategy for hepatoma treatment.
The PROTAC molecule consists of three distinct parts, a warhead that targets the protein of interest, a suitable linker and an E3 Ub ligase-recruiting ligand. Compound iFSP1–8 was selected for modification to serve as an FSP1 ligand in the design of PROTACs. Initially, the iFSP1–8 derivative 5 was obtained through a de novo synthesis approach. Compound 5 exhibited comparable biological activity to iFSP1–8 (Fig. S2 in Supporting information), with its hydroxyl group serving as a conjugation site for the further attachment of target molecules. To identify the most effective E3 ligase ligand, we selected pomalidomide (cereblon, CRBN) and VH032 (von Hippel-Lindau, VHL), which are well-established ligands for E3 ligases, for PROTACs construction. Additionally, the efficiency of protein degradation mediated by PROTACs is influenced not only by the ligands' affinity and selectivity for their respective targets but also by the length of the chemical linker [25,26]. Consequently, we chose a variety of linkers with different lengths to connect the FSP1 ligand with the E3 ligase ligand. The structures of target compounds C1–12 and V1–9 were depicted in Scheme 1.
To evaluate the FSP1 degradation efficiency of the designed PROTACs, FSP1 protein levels in HepG2 cells were analyzed after treatment with 21 distinct target compounds. Our observations revealed that the majority of VHL-series compounds exhibited insignificant degradation efficiency for FSP1, with only V7–9 showing a slight effect on FSP1 degradation (Fig. S3A in Supporting information). In contrast, the majority of CRBN-series compounds displayed substantial degradation activity (Fig. S3B in Supporting information). Notably, compounds C6, C7, C9, C10, and C11 achieved degradation rates of exceeding 75%. These results suggested that CRBN outperformed VHL in degrading FSP1, and that the length of linker chain was a critical determinant of the biological activity of PROTAC molecules. Given its demonstration of the most significant FSP1 degradation potential, compound C7 was selected for further investigation.
To assess the FSP1 degradation efficiency of C7, HepG2 cells were exposed to escalating concentrations of C7, after which the intracellular levels of FSP1 protein were quantified. The results indicated that C7 significantly reduced FSP1 expression in a concentration-dependent manner, with a half-maximal degradation concentration (DC50) value of 0.66 µmol/L (Fig. 1A). Reassuringly, the maximum effective degradation rate (Dmax) of C7 reached 86%. Moreover, to determine the time required for C7 to exert its biological activity, HepG2 cells were incubated with C7 at various time points. The results showed that C7 initiates FSP1 degradation after 6 h and achieved Dmax at 24 h (Fig. 1B).
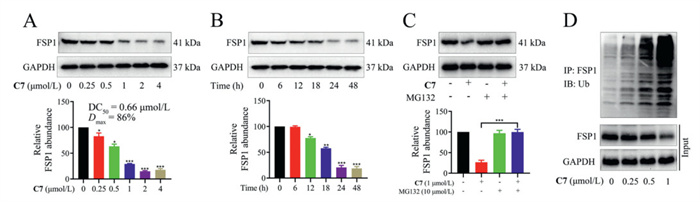
To further investigate the degradation mechanism of FSP1 by C7 in HepG2 cells, a series of rescue experiments were conducted. Initially, HepG2 cells were pretreated with iFSP1, which competed with C7 for binding to FSP1 (Fig. S4 in Supporting information). The results showed that iFSP1 attenuated C7-induced degradation of FSP1 (Fig. S4A). Next, pretreatment of HepG2 cells with pomalidomide substantially impaired the potency of C7 to degrade FSP1 (Fig. S4B). The proteasome inhibitor MG132 also rescued the decrease of FSP1 level in HepG2 cells (Fig. 1C). We further assessed the impact of C7 on the protein levels of ikaros family zinc finger 1 (IKZF1), ikaros family zinc finger 3 (IKZF3) and G1 to S phase transition 1 (GSPT1). Our data revealed no significant alteration in the abundance of these proteins upon C7 treatment (Fig. S4C), thereby confirming that FSP1 degradation by C7 occurs in a target-selective manner. Additionally, C7N (a negative control of C7), containing a methyl group in glutarimide NH, lost the ability to target CRBN and had no significant effect on FSP1 level (Figs. S5A and B in Supporting information). Furthermore, co-immunoprecipitation results demonstrated that C7 significantly increased the ubiquitination of FSP1 in HepG2 cells (Fig. 1D). Surface plasmon resonance (SPR) assay showed that C7 has an obvious binding signal to the FSP1 protein, high affinity and strong concentration dependence, with a Kd of 0.87 µmol/L (Fig. S6 in Supporting information). Molecular dynamics (MD) simulation indicated that C7 could recruit CRBN to FSP1 and form a ternary complex (Fig. S7 in Supporting information). These findings confirmed that C7 degrades FSP1 through the Ub-proteasome pathway.
Next, we evaluated the potential of C7 to increase the sensitivity of HepG2 cells to GPX4 inhibitor. Attentionally, ML162 (GPX4 inhibitor), at a concentration of 100 nmol/L, did not substantially inhibit the growth of HepG2 cells. However, ML162 demonstrated potent effect of growth inhibition in combination with C7 (Fig. 2A, Figs. S8A and B in Supporting information). Notably, C7 exhibited stronger effect than iFSP1 at the same concentration, suggesting that reducing FSP1 expression is more advantageous than merely inhibiting its activity in tumor suppression. However, the sensitizing effect of C7N (a negative control of C7) on GPX4 inhibitor was greatly diminished (Fig. S8C in Supporting information). Additionally, cell viability was evaluated over a temporal gradient following drug treatment. Co-treatment with ML162 and C7 for 6 h did not affect HepG2 cell viability. However, prolonged exposure to ML162 and C7 led to a marked reduction in cell viability in a time-dependent manner (Fig. 2B). The time required for the combination of ML162 and C7 to produce cytotoxicity was consistent with the time required for C7 to start degrading FSP1, indicating that the antiproliferative effects of ML162 and C7 are mediated primarily by inhibiting GPX4 activity and reducing FSP1 level. These results suggested that FSP1 degrader has greater potential to increase the sensitivity of HepG2 cells to GPX4 inhibitor.
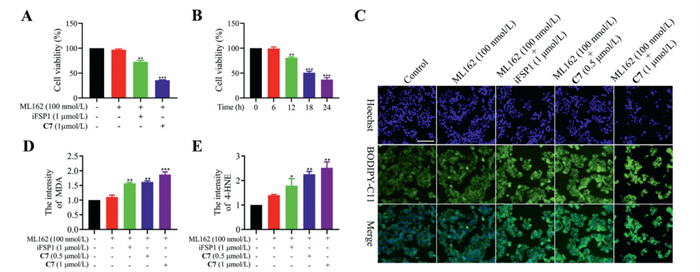
To investigate the mechanism underlying the activity of C7 and ML162, the ferroptosis inhibitor Fer-1 was employed. The results demonstrated that the cytotoxic effect induced by the combination of C7 and ML162 in HepG2 cells could be reversed by Fer-1 (Fig. S8D in Supporting information), highlighting the involvement of ferroptosis in their mechanism of action. The LPO accumulation was a key characteristic of ferroptosis [27]. In HepG2 cells, the LPO level did not change after administration of ML162 or C7 alone. While the intracellular LPO level was significantly increased after co-treatment with C7 and ML162 (Fig. 2C, Figs. S9A and B in Supporting information). Significantly, at equivalent concentration, C7 proved to be more effective than iFSP1 in increasing LPO. Furthermore, LPO breakdown leads to generate reactive toxic aldehydes, such as malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE). These aldehydes inactivate proteins that are critical for cellular processes by crosslinking, thereby promoting ferroptosis [28]. Upon co-treatment with C7 and ML162, the intracellular levels of MDA and 4-HNE elevated in a dose-dependent manner (Figs. 2D and E, Figs. S9C and D in Supporting information). Additionally, after treatment with C7 and ML162 for 24 h, the Fe2+ and ROS content in HepG2 cells was obviously increased, accompanied by a significant downregulation of GSH level (Figs. S10A–D in Supporting information). These results indicated that the combination of C7 and ML162 inhibited HepG2 cells viability by inducing ferroptosis. Moreover, C7 demonstrated superior activity compared to iFSP1, suggesting that FSP1 degraders hold the much greater potential than FSP1 inhibitors in hepatoma therapeutics.
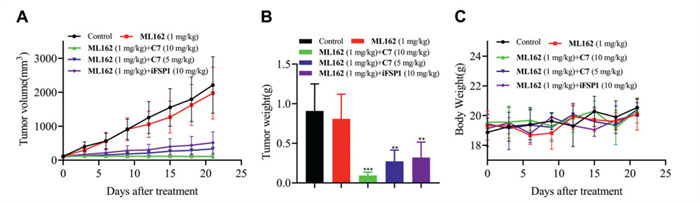
To evaluate the antitumor potency of combination of C7 and ML162 in vivo, the nude mouse HepG2 xenograft models were established. All protocols were approved by the Animal Experimentation Committee of China Pharmaceutical University (IACUC Number: 2024–05–067). As shown in Fig. 3 and Fig. S11A (Supporting information), ML162 alone was not sufficient to effectively suppress tumor growth. However, when administered in combination with C7, there was a significant inhibition of tumor growth, and no significant changes in the body weight of the mice were noted during the administration period. The tumor growth inhibition ratio (TGI) was calculated based on tumor weight. The C7 (10 mg/kg) + ML162 (1 mg/kg) group performed a TGI of 89.9%; the C7 (5 mg/kg) + ML162 (1 mg/kg) group performed a TGI of 70.8% and the iFSP1 (10 mg/kg) + ML162 (1 mg/kg) group performed a TGI of 65.1%. An essential observation lay in the prominent synergism revealed by this combination, affirming the presence of the synthetic antitumor effects between FSP1 degradation and GPX4 inhibition in vivo.
Additionally, hearts, kidneys, livers, lungs, spleens, and tumors were collected for further pathology and biochemical criterion analysis. Hematoxylin and eosin (H & E) staining of the organs suggested that there was no significant morphological change between the vehicle group and drug-treated groups (Fig. S11B in Supporting information), indicating a praisable biosecurity of C7. Furthermore, the median lethal dose (LD50) of C7 was measured and found to be 201.4 mg/kg (Table S1 in Supporting information). Moreover, compared with the vehicle group, the FSP1 level was obviously decreased in tumor issues after the treatment of C7 (Fig. S11C in Supporting information) by immunohistochemistry (IHC) analysis of tumors. These results proved that C7 was a safe and efficacious degradation agent of FSP1.
Given the excellent biological efficacy of C7, we further investigated its pharmacokinetic (PK) properties in rats (Table S2 in Supporting information). The data indicated that C7 exhibits a favorable PK profile, with a half-life (t1/2) of 2.55 h following a single intravenous administration. However, the PK properties of C7 were found to be lower with intraperitoneal injection compared to intravenous administration, resulting in a bioavailability of only 15.7%. Although the bioavailability of C7 via intraperitoneal injection was low, its significant antitumor effect in vivo highlighted the potential value of the FSP1 degrader in hepatoma therapeutics.
Summarily, based on the importance of FSP1 in the ferroptosis defense mechanism, we hypothesized that rapid degradation of FSP1 would have advantages over simply inhibiting its activity. Taking advantage of the PROTAC platform, we designed the VHL-recruiting and CRBN-recruiting degraders for the FSP1 using iFSP1 analogue that targets the quinone-binding pocket of FSP1. Among these, C7, as the first FSP1 PROTAC degrader, displayed the most efficient FSP1 degradation potency and was picked for biological activity studies in HepG2 cells. Mechanistically, C7 played a pivotal role in the recruitment of CRBN to the proximity of FSP1, thereby facilitating the formation of a FSP1-C7-CRBN ternary complex. This complex subsequently led to the down-regulation of FSP1 levels via the Ub-proteasome pathway. Furthermore, as the first FSP1 PROTAC degrader, C7 could significantly enhance the sensitivity of HepG2 cells to ML162 (GPX4 inhibitor). The combination of C7 and ML162 significantly upregulated the ferroptosis signaling pathway, resulting in the failure of the ferroptosis defense mechanism and the accumulation of LPO. Moreover, the combination of C7 and ML162 significantly inhibited tumor growth without obvious toxicity in the HepG2 xenograft model. All the results indicated that C7 can be used as a chemical probe for FSP1-related disease exploration, and that PROTAC degraders of FSP1 function as potent sensitizers of GPX4 inhibitors to induce ferroptosis, thus representing a promising strategy for hepatoma treatment.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Jiangmin Zhu: Writing – review & editing, Writing – original draft, Project administration, Formal analysis, Data curation, Conceptualization. Qimei Tan: Writing – review & editing, Project administration, Data curation. Shiying Fan: Writing – review & editing, Data curation. Yalin Li: Validation, Data curation. Ling Zhu: Software, Formal analysis. Lihong Hong: Formal analysis, Data curation. Yuxia Wang: Data curation. Chuzhen Zhang: Software, Data curation. Chen Chen: Supervision, Formal analysis. Lingyi Kong: Supervision, Resources. Jianguang Luo: Writing – review & editing, Supervision, Resources.
This research was financially supported by the 111 Project from Ministry of Education of China and the State Administration of Foreign Export Affairs of China (No. B18056), and the "Double First-Class" University Project (No. CPU2022QZ28).
Supplementary material associated with this article can be found, in the online version, at doi:
G. Lei, L. Zhuang B. Gan, Nat. Rev. Cancer 22 (2022) 381–396. doi: 10.1038/s41568-022-00459-0
L. Zhao, X. Zhou, F. Xie, et al., Cancer Commun. 42 (2022) 88–116. doi: 10.1002/cac2.12250
T. Nakamura, M. Conrad, Nat. Chem. Biol. 26 (2024) 1407–1419. doi: 10.1038/s41556-024-01425-8
J. Huang, X. Ma, Z. Liao, et al., Chin. J. Nat. Med. 21 (2023) 775–788.
Q. Zhou, Y. Meng, D. Li, et al., Signal Transduct. Target Ther. 9 (2024) 55.
B.R. Stockwell, Cell 185 (2022) 2401–2421. doi: 10.1016/j.cell.2022.06.003
W. Li, L. Liang, S. Liu, et al., Trends Mol. Med. 29 (2023) 753–764. doi: 10.1016/j.molmed.2023.05.013
Y. Ye, A. Chen, L. Li, et al., Kidney Int. 102 (2022) 1259–1275. doi: 10.1016/j.kint.2022.07.034
H. Liu, F. Forouhar, T. Seibt, et al., Nat. Chem. Biol. 18 (2021) 91–100.
J.K. Eaton, L. Furst, R.A. Ruberto, et al., Nat. Chem. Biol. 16 (2020) 497–506. doi: 10.1038/s41589-020-0501-5
E. Karaj, S.H. Sindi, N. Kuganesan, et al., J. Med. Chem. 65 (2022) 11788–11817. doi: 10.1021/acs.jmedchem.2c00909
K. Bersuker, J.M. Hendricks, Z. Li, et al., Nature 575 (2019) 688–692. doi: 10.1038/s41586-019-1705-2
S. Doll, F.P. Freitas, R. Shah, et al., Nature 575 (2019) 693–698. doi: 10.1038/s41586-019-1707-0
M. Yang, M.G. Tsui, J.K.W. Tsang, et al., Cell Death Dis. 13 (2022) 468. doi: 10.1038/s41419-022-04924-4
T. Nakamura, E. Mishima, N. Yamada, et al., Nat. Struct. Mol. Biol. 30 (2023) 1806–1815. doi: 10.1038/s41594-023-01136-y
D. Liang, Y. Feng, F. Zandkarimi, et al., Cell 186 (2023) 2748–2764. doi: 10.1016/j.cell.2023.05.003
J.M. Hendricks, C.E. Doubravsky, E. Wehri, et al., Cell Chem. Biol. 30 (2023) 1090–1103. doi: 10.1016/j.chembiol.2023.04.007
T. Nakamura, C. Hipp, A. Santos Dias Mourão, et al., Nature 619 (2023) 371–377. doi: 10.1038/s41586-023-06255-6
H. Qiu, S. Huang, Y. Liu, et al., Acta Pharm. Sin. B 14 (2024) 2581–2597. doi: 10.1016/j.apsb.2024.03.015
A.C. Lai, C.M. Crews, Nat. Rev. Drug. Disc. 16 (2016) 101–114.
A. Kaneshige, L. Bai, M. Wang, et al., Nat. Chem. Biol. 19 (2023) 703–711. doi: 10.1038/s41589-022-01248-4
Q. Gong, J. Song, Y. Song, et al., Chin. Chem. Lett. 36 (2025) 110456. doi: 10.1016/j.cclet.2024.110456
D. Chirnomas, K.R. Hornberger, C.M. Crews, Nat. Rev. Clin. Oncol. 20 (2023) 265–278. doi: 10.1038/s41571-023-00736-3
S. Khan, X. Zhang, D. Lv, et al., Nat. Med. 25 (2019) 1938–1947. doi: 10.1038/s41591-019-0668-z
Y. Yang, H. Xie, X. Yu, et al., Chin. Chem. Lett. 35 (2024) 109570. doi: 10.1016/j.cclet.2024.109570
Y. Liu, Y. Liu, K. Yang, et al., Chin. Chem. Lett. 35 (2024) 109264. doi: 10.1016/j.cclet.2023.109264
W.S. Yang, R. SriRamaratnam, M.E. Welsch, et al., Cell 156 (2014) 317–331. doi: 10.1016/j.cell.2013.12.010
B. Hassannia, P. Vandenabeele, T. Vanden Berghe, Cancer Cell 35 (2019) 830–849.

Scheme 1 The synthesis of the target compounds. Reagents and conditions: (ⅰ) K2CO3, DMF, 90 ℃, 12 h; (ⅱ) sodium ascorbate, CuSO4, DMF/H2O, r.t., 24 h.

Figure 1 Compound C7 induced FSP1 degradation via the Ub-proteasome system. (A) Western blot (WB) analysis of FSP1 level after treatment with indicated concentrations of C7 for 24 h in HepG2 cells. (B) WB analysis of FSP1 level after treatment with C7 (1 µmol/L) at indicated time points in HepG2 cells. (C) WB analysis of FSP1 level after treatment with indicated compounds in HepG2 cells for 24 h; C7 (1 µmol/L) and MG132 (10 µmol/L). (D) HepG2 cells were treated with indicated concentrations of C7 for 24 h. The total proteins were immunoprecipitated with anti-FSP1 and immunoblotted with anti-Ub antibodies. Analysis results represented mean ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 vs. control group. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; IP, immunoprecipitation; IB, immunoblotting.

Figure 2 Compound C7 sensitized HepG2 cells to ML162. (A) The percentage of cell viability after treatment with indicated compounds for 24 h in HepG2. (B) The percentage of cell viability after treatment with C7 (1 µmol/L) and ML162 (100 nmol/L) at indicated time points in HepG2 cells. (C) CLSM imaging of intracellular LPO level after treatment with indicated compounds for 24 h in HepG2 cells. Scale bar: 100 µm. (D) The intracellular MDA level and (E) 4-HNE level after treatment with indicated compounds for 24 h in HepG2 cells. Analysis results represented mean ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 vs. control group.

Figure 3 In vivo antitumor activity of C7/ML162 in the mouse HepG2 xenograft models. Change of (A) tumor volume, (B) tumor weight and (C) body weight was exhibited after treated with vehicle or compounds. Analysis results represented mean ± SD (n = 6). **P < 0.01, ***P < 0.001 vs. control group.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
 下载:
下载: