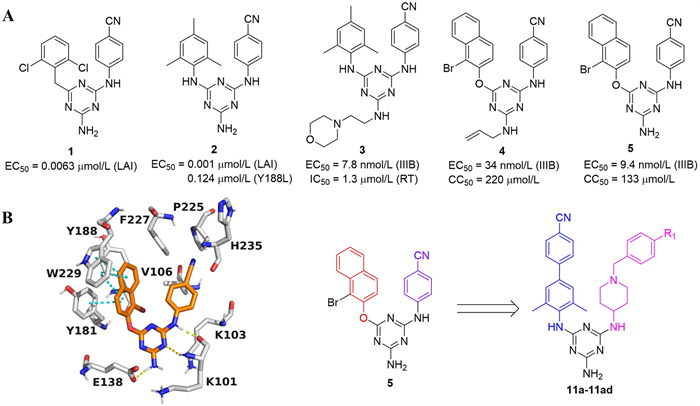
Figure 1.
(A) The chemical structures of triazines 1–5. (B) The docking analysis of 5 (PDB code: 2zd1) and design of target compounds via rational structure-based drug design strategy.
Rational structure-based design and optimization of next-generation biphenyl-piperidine-triazine derivatives as potent non-nucleoside reverse transcriptase inhibitors
Kun Zhang , Li-Min Zhao , Tianhao Xing , Yueyue Bu , Qingyun Wang , Christophe Pannecouque , Erik De Clercq , Angela Corona , Laura Dettori , Enzo Tramontano , Shuai Wang , Fen-Er Chen

As one of the major global epidemic diseases, acquired immunodeficiency syndrome (AIDS) caused by human immunodeficiency virus (HIV) infection is a persistent threat to human health [1,2]. Reverse transcriptase (RT) is a key enzyme in HIV life cycle, which is responsible for the reverse transcription of single-stranded RNA into double-stranded DNA [3–5]. Non-nucleoside reverse transcriptase inhibitors (NNRTIs) are an important component of active antiviral therapy (ART) and widely applied in the treatment of AIDS [6–9]. Although ART has achieved remarkable success in suppressing HIV to undetectable levels, the virus cannot be eradicated and patients require lifelong ART administration [2,10,11]. Moreover, it has been reported that some NNRTIs have side effects and poor oral pharmacokinetic parameters [12–15]. Worst of all, the continuous emergence of resistant strains further limits the clinical effectiveness of these drugs [16–18]. Therefore, the discovery of novel NNRTIs that feature low toxicity, high efficiency, and anti-resistance activity is still urgent.
In 2001, Janssen and co-workers reported the discovery of a triazine-containing NNRTI, namely compound 1. This compound exhibited potent inhibitory activity against HIV-1 (50% effective concentration (EC50) = 0.0063 µmol/L, Fig. 1A) [19]. Over the subsequent decades, several series of optimizations and structural modifications based on compound 1 were carried out for a continuous and in-depth exploration [20–26]. Compound 2 was acquired through the modification of the linker and the aromatic ring at the C-4 position of triazine. It demonstrated potent inhibitory activity against HIV-1 (EC50 = 0.001 µmol/L). Nevertheless, its activity against the Y188L mutant strain declined sharply (EC50 = 0.124 µmol/L). Subsequently, by targeting the entrance channel of the non-nucleoside reverse transcriptase inhibitor binding pocket (NNIBP) and modifying the C-6 position of triazine, compound 3 was discovered (EC50 = 0.0078 µmol/L) [27]. To enhance the π-π stacking interactions between the ligand and the highly conserved hydrophobic region (W229), the phenyl group was substituted with a naphthyl moiety, affording compound 4. This compound exhibited moderate anti-HIV-1 activity (EC50 = 34 nmol/L) along with low cytotoxicity (50% cytotoxic concentration (CC50) = 220 µmol/L) [28]. Removal of the allyl group yielded compound 5, which had enhanced antiviral activity (EC50 = 9.4 nmol/L) against the wild-type (WT) HIV-1 [29]. Nonetheless, this analog showed unsatisfactory activity against mutant strains [30–32]. Thus, the development of novel triazine-based derivatives with increased antiviral activity against both WT and mutant strains continues to be a critical priority.
Analysis of the molecular modeling investigation indicated that compound 5 adopted a “horseshoe-shaped” conformation within the binding pocket (Fig. 1B). The β-naphthyl fragment occupied the hydrophobic area and formed multiple π-H interactions with W229 and a π-π interaction with Y181. The triazine core and the NH at the C-2 position formed two key hydrogen bonds with E138 and K101 respectively, and the NH2 group formed an extra hydrogen bond with E138. The p-cyanophenyl group in the solvent-protein region did not interact with surrounding amino acids. To enhance the anti-resistance efficacy, a series of novel biphenyl-piperidine-triazine derivatives were designed and evaluated for their anti-HIV-1 activity. First, the β-naphthyl fragment was replaced with the biphenyl fragment to enhance hydrophobic interactions and π-π stacking length with surrounding residues. Then, a hydrophilic piperidyl moiety was introduced into the right wing to replace the p-cyanophenyl group, aiming to strengthen the interaction of the ligand with the solvent-protein region.
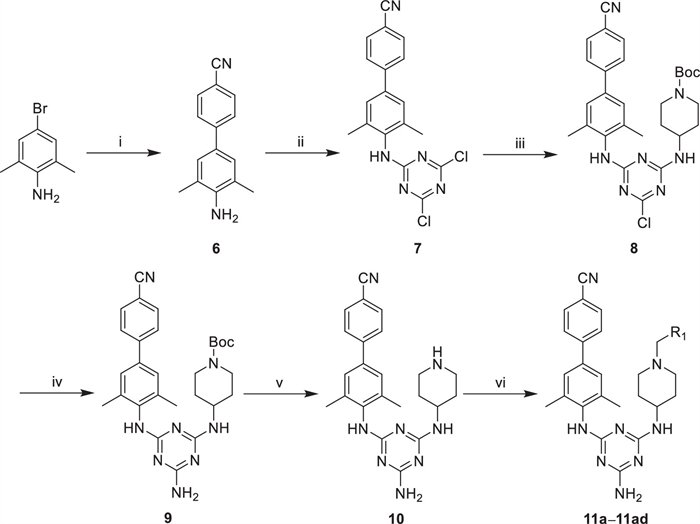
The synthetic routes of 11a–11ad are illustrated in Scheme 1. Specially, 4-bromo-2,6-dimethylaniline was chosen as starting material, and biphenyl fragment (6) was synthesized via Suzuki reaction [33,34]. Subsequently, 6 reacted with cyanuric chloride to generate 7. This intermediate was then treated with 4-amino-1-Boc-piperidine to yield 8. Then, an excess of NH3·H2O was employed to replace Cl atom in a sealed tube at 100 ℃, and intermediate 9 was gained after 12 h. Following this, 10 was synthesized by deprotecting the Boc group [35,36]. Finally, 11a–11ad were obtained via the nucleophilic substitution reaction.
The antiviral activity and cytotoxicity of the triazine derivatives 11a–11ad were assessed in MT-4 cell cultures infected with the WT HIV-1 IIIB strain, utilizing the testing method we have previously described [37]. The detailed data provided in Table 1. All these compounds exhibited notable antiviral activity, with EC50 values ranging from 0.002 µmol/L to 1.026 µmol/L. What is more, approximately half of the compounds demonstrated single-digit nanomolar activity against HIV-1 WT, especially compounds 11a, 11e, 11j, 11p–11w, and 11z–11ac, which had greater potency than lead compound 5 (EC50 = 0.002–0.008 vs. 0.009 µmol/L). Among them, compound 11s emerged as the most outstanding one, boasting an EC50 value of 0.002 µmol/L, which surpassed not only the lead compound 5 but also the benchmark antiretroviral drugs NVP, EFV, and ETR (EC50 = 0.007, 0.209, 0.004, and 0.003 µmol/L, respectively). Moreover, 11s (CC50 = 19.69 µmol/L) also showcased lower cytotoxicity than these drugs (NVP, CC50 > 15.02 µmol/L; EFV, CC50 > 6.38 µmol/L; ETR, CC50 > 4.59 µmol/L).
 DownLoad:
CSV
DownLoad:
CSV
| Compd. | R1 | EC50 (µmol/L)a | CC50 (µmol/L)b | SIc | Compd. | R1 | EC50 (µmol/L) | CC50 (µmol/L) | SI |
| 11a | 4-Phenyl-CN | 0.008 ± 0.008 | 11.12 ± 1.64 | 1436 | 11r | 4-Phenyl-SO2CH3 | 0.005 ± 0.001 | 4.98 ± 2.52 | 1027 |
| 11b | 3-Phenyl-CN | 0.080 ± 0.063 | 8.90 ± 1.20 | 111 | 11s | 4-Phenyl-CONH2 | 0.002 ± 0.001 | 19.69 ± 1.50 | 7438 |
| 11c | 2-Phenyl-CN | 0.040 ± 0.034 | 116.5 ± 6.07 | 2891 | 11t | 3-Phenyl-CONH2 | 0.006 ± 0.001 | 16.77 ± 2.52 | 2762 |
| 11d | 4-Phenyl-F | 0.009 ± 0.004 | 20.58 ± 9.57 | 2281 | 11u | 4-Phenyl-CONHCH3 | 0.008 ± 0.003 | 20.76 ± 3.63 | 2367 |
| 11e | 4-Phenyl-Cl | 0.008 ± 0.003 | 34.55 ± 11.24 | 4527 | 11v |  |
0.004 ± 0.001 | 14.73 ± 2.28 | 3399 |
| 11f | 4-Phenyl-CF3 | 0.086 ± 0.072 | 107.9 ± 44.61 | 1255 | 11w |  |
0.008 ± 0.003 | 17.99 ± 4.61 | 2291 |
| 11g | 4-Phenyl-OCF3 | 0.033 ± 0.013 | >211.6 | >6392 | 11x |  |
0.018 ± 0.014 | 18.41 ± 1.53 | 998 |
| 11h | 4-Phenyl-CH3 | 0.300 ± 0.274 | 23.91 ± 6.18 | 80 | 11y |  |
0.015 ± 0.011 | 16.18 ± 3.03 | 1107 |
| 11i | 2,4-Phenyl-2-F | 0.097 ± 0.086 | 8.86 ± 0.25 | 92 | 11z |  |
0.008 ± 0.003 | 9.79 ± 10.53 | 1464 |
| 11j | 3,4-Phenyl-2-F | 0.008 ± 0.003 | 71.31 ± 8.42 | 8877 | 11aa | 2-Pyridyl | 0.006 ± 0.002 | 20.87 ± 3.04 | 3769 |
| 11k | 4-Phenyl-COOH | 0.223 ± 0.033 | >227.8 | >1021 | 11ab | 3-Pyridyl | 0.004 ± 0.002 | 18.83 ± 2.72 | 4759 |
| 11l | 4-Phenyl-CH2COOH | 1.026 ± 0.226 | 209.7 ± 9.26 | 204 | 11ac | 4-Pyridyl | 0.003 ± 0.001 | 8.650 ± 2.870 | 3302 |
| 11m | 4-Phenyl-COOCH3 | 0.028 ± 0.012 | 52.23 ± 9.31 | 1872 | 11ad | 2-Naphthyl | 0.096 ± 0.139 | 83.83 ± 69.58 | 878 |
| 11n | 3-Phenyl-COOH | 1.014 ± 0.268 | >227.8 | >225 | NVP | - | 0.209 ± 0.091 | >15.02 | >72 |
| 11o | 4-Phenyl-B(OH)2 | 0.009 ± 0.004 | 14.51 ± 1.95 | 1569 | EFV | - | 0.004 ± 0.002 | >6.34 | >1425 |
| 11p | 3-Phenyl-B(OH)2 | 0.006 ± 0.002 | 13.36 ± 2.59 | 2166 | ETR | - | 0.003 ± 0.001 | >4.59 | >1340 |
| 11q | 4-Phenyl-SO2NH2 | 0.008 ± 0.004 | 15.20 ± 2.33 | 1674 | |||||
| a EC50: concentration of compound required to achieve 50% protection of MT-4 cell cultures against HIV-1-induced cytopathicity, as determined using the MTT method. b CC50: concentration required to reduce the viability of mock-infected cell cultures by 50%, as determined using the MTT method. c SI: the ratio of CC50/EC50. |
|||||||||
Preliminary investigations into the structure-activity relationships (SARs) have illuminated that the characteristics of substituents and their positions on the benzene ring play a pivotal role in determining antiviral activity. Generally, para-substituents are more favorable for enhancing antiviral activity compared to ortho- and meta-substituents (11a vs. 11b–11c, 11k vs. 11n, 11s vs. 11t). In terms of electronic effects, electron-donating substituents, like the methyl group in compound 11h (EC50 = 0.300 µmol/L), caused a marked reduction in anti-HIV-1 activity. In contrast, electron-withdrawing substituents in compounds 11a–11g conferred relatively high anti-HIV-1 activity (EC50 = 0.008–0.086 µmol/L). Regarding multiple substitutions, double-substitution patterns, particularly at the 2,4-positions, did not lead to significant improvement in activity. For example, compound 11d (EC50 = 0.009 µmol/L) showed higher activity than 11i (EC50 = 0.097 µmol/L). The influence of the COOH group on antiviral activity was quite interesting. The presence of a COOH group alone led to a notable decrease in activity (11k, EC50 = 0.223 µmol/L). However, the effect could be modulated by the introduction of a methyl group. Inserting a methyl group between the COOH and the phenyl ring further diminished the activity (11l, EC50 = 1.026 µmol/L), while adding a CH3 group at the end of the COOH group resulted in approximately a ten-fold increase in activity (11m, EC50 = 0.028 µmol/L).
Notably, the incorporation of other hydrophilic motifs significantly enhanced the antiviral activity (11o–11t, EC50 = 0.002–0.009 µmol/L). Among them, the order of their contribution to activity was CONH2 > SO2CH3 > SO2NH2 > B(OH)2. Subsequently, we modified the CONH2 group with an array of diverse amino fragments. Although these derivatives were effective (11u–11y, EC50 = 0.004–0.018 µmol/L), none of them outperformed compound 11s. Moreover, heterocyclic rings, specifically pyridyl substituents (11aa–11ac, EC50 = 0.003–0.006 µmol/L), were more advantageous than the 2-naphthyl group (11ad, EC50 = 0.096 µmol/L). In particular, 11ac demonstrated potent antiviral activity (EC50 = 0.003 µmol/L), surpassing 5 and NVP, and being comparable to EFV and ETR.
Representative compounds 11p, 11r–11t, 11v, and 11aa–11ac were selected to evaluate their inhibitory potential against five mutant strains (L100I, K103N, Y181C, Y188L, and E138K). The corresponding results are presented in Table 2. Favorably, these chosen compounds exhibited a spectrum of inhibitory effects against the clinical mutants. Of particular note was their activity against the K103N mutant, which is the most commonly transmitted drug-resistant strain in clinical settings [2]. Among the tested compounds, compound 11s, which had previously demonstrated the most potent activity against the WT strain, also displayed commendable anti-HIV-1 activity against the mutant strains. It achieved EC50 values of 0.021 µmol/L for the L100I mutant, 0.003 µmol/L for the K103N mutant, 0.013 µmol/L for the Y181C mutant, 0.073 µmol/L for the Y188L mutant, and 0.004 µmol/L for the E138K mutant. Significantly, 11s exhibited greater potency against the Y181C and E138K mutations compared to the reference NVP and ETR. Furthermore, compounds 11r, 11s, 11v, and 11ac demonstrated single-digit nanomolar antiviral activity against the K103N mutation (EC50 = 0.003–0.005 µmol/L). In the case of the L100I and Y188L mutants, although 11s and 11v were slightly less potent than ETR, they also significantly exceeded NVP and EFV. Overall, these results offer promising prospects for the development of next-generation antiretroviral therapies with enhanced efficacy against both WT and mutant strains of HIV-1.
DownLoad:
CSV
| Compd. | EC50 (µmol/L)a | ||||
| L100I | K103N | Y181C | Y188L | E138K | |
| 11p | 0.206 ± 0.145 | 0.020 ± 0.004 | 0.027 ± 0.007 | 0.304 ± 0.137 | 0.029 ± 0.015 |
| 11r | 0.029 ± 0.012 | 0.005 ± 0.001 | 0.011 ± 0.002 | 0.157 ± 0.079 | 0.008 ± 0.002 |
| 11s | 0.021 ± 0.090 | 0.003 ± 0.000 | 0.013 ± 0.003 | 0.073 ± 0.002 | 0.004 ± 0.001 |
| 11t | 0.135 ± 0.060 | 0.024 ± 0.014 | 0.109 ± 0.080 | 0.513 ± 0.185 | 0.024 ± 0.019 |
| 11v | 0.012 ± 0.004 | 0.005 ± 0.002 | 0.015 ± 0.005 | 0.068 ± 0.018 | 0.023 ± 0.018 |
| 11aa | 0.174 ± 0.105 | 0.010 ± 0.003 | 0.036 ± 0.004 | 0.307 ± 0.080 | 0.011 ± 0.001 |
| 11ab | 0.211 ± 0.076 | 0.012 ± 0.007 | 0.038 ± 0.017 | 0.545 ± 0.342 | 0.01 ± 0.003 |
| 11ac | 0.046 ± 0.032 | 0.005 ± 0.003 | 0.011 ± 0.001 | 0.076 ± 0.006 | 0.011 ± 0.004 |
| NVP | 2.050 ± 1.896 | 5.197 ± 2.693 | 5.768 ± 2.976 | > 15.02 | 0.157 ± 0.058 |
| EFV | 0.030 ± 0.015 | 0.084 ± 0.033 | 0.008 ± 0.003 | 12.67 ± 0.127 | 0.007 ± 0.003 |
| ETR | 0.007 ± 0.005 | 0.003 ± 0.001 | 0.016 ± 0.005 | 0.020 ± 0.008 | 0.009 ± 0.003 |
| a EC50: concentration of compound required to achieve 50% protection of MT-4 cell cultures against HIV-1-induced cytopathicity, as determined using the MTT method. | |||||
The inhibitory activity against WT HIV-1 RT enzymes of compounds 11a–11ad was assessed and the data presented in Table S1 (Supporting information). Compounds 11a–11ad displayed moderate RT inhibitory activity (IC50 = 0.20–1.70 µmol/L). Gratifyingly, approximately half of these compounds demonstrated superior inhibitory activity compared with NVP (IC50 = 0.75 µmol/L). Of particular significance, compounds 11s, 11ab, and 11ac also emerged as the most potent inhibitors of RT. Notably, the RT inhibitory activity of 11ab was 3.7-fold higher than that of NVP. These results suggest that compounds 11a–11ad effectively target the RT enzyme.
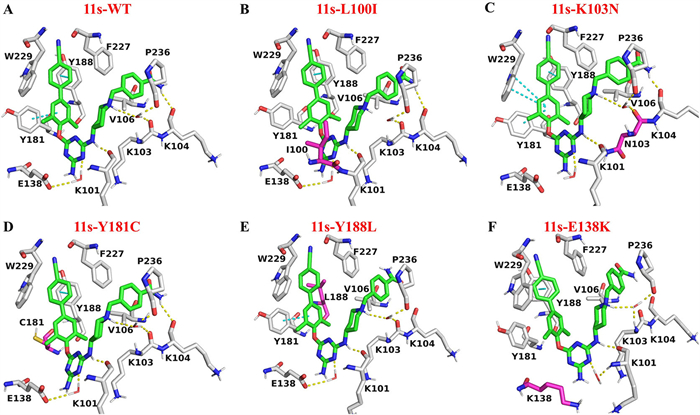
To gain a deeper understanding of the binding mechanisms of compound 11s with HIV-1 RT, molecular docking studies were carried out using Schrödinger Maestro 12.8. As depicted in Fig. 2A, compound 11s was docked into the NNIBP, assuming a “horseshoe-shaped” conformation similar to that of the lead compound 5 and ETR. The biphenyl moiety of 11s effectively inserted into the aromatic hydrophobic region and formed two π-π interactions with the Y181 and Y188 residues. These interactions not only enhanced the hydrophobic effect but also extended the length of the π-π stacking, thus validating the initial hypothesis regarding the design of the compound. At the core of the molecule, the N-1 position of the triazine ring participated in a water-mediated hydrogen bond with the E138 residue. Meanwhile, the adjacent NH group donated a hydrogen atom to form a crucial hydrogen bond with the backbone of the K101 residue. On the right wing, the piperidine linker acted as a bridge, positioning the terminal aryl motif within the groove surrounded by K104, V106, F227, and P236. Moreover, the protonated nitrogen atom of the piperidine formed a water-mediated bridge with the backbones of K103 and P236. Additionally, the terminal CONH₂ substituent extended towards the solvent-exposed surface of the RT protein and formed an additional hydrogen bond with the backbone of K104. The binding model of 11s with the mutant RT proteins was found to be highly similar to that with the WT RT. They all adopted the characteristic “horseshoe-shaped” conformations and maintained the crucial hydrogen bond interaction with the backbone of K101. In the cases of the L100I, Y181C, and Y188L mutants, the mutated residues led to the reduction or loss of one or more hydrogen bonds or π-π interactions with compound 11s, which, in turn, resulted in a decrease in their antiviral efficacy. Notably, for the K103N mutant, although the water-mediated hydrogen bond between the N-1 position of the triazine ring and E138 was weakened, the dimethyl-phenyl moiety formed two additional π-π interactions with W229. These new interactions effectively compensated for the loss of the previous hydrogen bond. Intriguingly, the E138K mutation caused the loss of a water-bridge between 11s and K138. However, a new water-mediated hydrogen bond was formed between 11s and K101, offsetting the negative impact of the mutation.
The potent inhibition of cytochrome P450 (CYP) enzymes, particularly CYP2C9 and CYP2C19, by RPV and ETR has been well-documented, which leads to severe drug-drug interactions [7,38,39]. In light of these concerns, the inhibitory potential of compound 11s against six major CYP isoforms was systematically evaluated. As presented in Table S2 (Supporting information), compound 11s demonstrated negligible inhibitory activity against CYP1A2, CYP2D6, CYP3A4-M, and CYP3A4-T isoforms (IC50 > 50 µmol/L). Moreover, 11s exerted only mild inhibitory effects on CYP2 sub-types. The IC50 values were determined to be 12.6 µmol/L for CYP2C9 and 14.7 µmol/L for CYP2C19. These values are significantly lower than those associated with ETR and RPV, suggesting that 11s has a reduced propensity to interfere with the normal function of these CYP enzymes. Collectively, these findings indicate that compound 11s presents a diminished risk of drug-drug interactions compared to RPV and ETR.
A significant number of drugs have been withdrawn from the market because of their high human ether-a-go-go-related gene (hERG) inhibitory activity [40]. RPV, for instance, has been linked to cardiotoxicity owing to its hERG-related side effects (IC50 = 0.50 µmol/L) [18]. Therefore, the hERG inhibitory activity of 11s was evaluated and cisapride was used as a reference compound [41]. Encouragingly, compound 11s exhibited a notably lower inhibitory effect on the hERG channel (IC50 = 15.979 µmol/L), which is substantially higher than those of cisapride and RPV (Fig. S1 in Supporting information). This outcome strongly suggests that compound 11s poses a low risk of inducing cardiotoxic side effects.
Next, we evaluated metabolic stability of 11s across five species. Table S3 (Supporting information) presents the metabolic stability data, which reveal a pronounced variation among the species (t1/2 = 19.4–87.2 min). Notably, 11s displayed the most extended t1/2 (87.2 min) in mouse liver microsomes, accompanied by an exceptionally low intrinsic clearance rate (24 mL min−1 gprot−1) and a commendable metabolic fraction (MF = 64.9%). Even though the metabolic stability of 11s in human and rat showed a slight decrease, the half-life and metabolic fractions remained more favorable compared to RPV (t1/2 = 41.4 min, MF = 27.9% (human); t1/2 = 19.4 min, MF = 29.1% (rat) vs. t1/2 = 16.03 min, MF = 13.02% (human); t1/2 = 7.29 min, MF = 13.36% (rat)) [42]. Collectively, these findings suggest that 11s exhibits markedly enhanced metabolic stability, particularly when juxtaposed with the commercially available drug RPV.
Compound 11s was proceeded to conduct pharmacokinetic (PK) studies in Sprague-Dawley rats using both single-dose intravenous (i.v.) and oral (p.o.) administrations (Table S4 in Supporting information). However, the half-life and oral bioavailability of compound 11s remained poor (t1/2 = 1.64 h, F = 1.52%).
A single-dose acute toxicity study was carried out to comprehensively evaluate the in vivo safety profile of compound 11s. Animal care and use protocols were approved by the Fudan University Committee on Animal Care (No. 2020CHEM-JS-009). As shown in Figs. S2A and B (Supporting information), there was neither mortality nor any significant alterations in body weight within the experimental group. This lack of adverse effects on survival and weight suggests that, at this dosage, 11s does not induce immediate and obvious systemic toxicity. To further assess the potential impact of 11s on vital organs, a histopathological examination was performed. The organs under scrutiny included the heart, liver, spleen, lungs, kidneys, and brain. Fig. S2C (Supporting information) presents the results of hematoxylin-eosin (HE) staining, a standard technique for visualizing tissue morphology. The staining results clearly demonstrated the absence of significant lesions or organ damage in the treated group. This indicates that 11s does not cause detectable structural harm to these crucial organs at the tested dosage. Collectively, these findings provide strong evidence that compound 11s is non-toxic at a dosage of 2000 mg/kg in mice.
In the ceaseless battle against the ever-evolving threat of HIV, the pursuit of more effective and safer antiretroviral agents remains a paramount objective. To this end, structure-based drug design strategy was rationally implemented, with the aim of augmenting the anti-resistance efficacy of the previously identified naphthyl-triazine compound 5. This strategic initiative led to the rational design of a novel series of biphenyl-piperidine-triazine-containing NNRTIs. Several of these newly designed compounds demonstrated an extraordinary single-digit nanomolar antiviral potency against both the WT HIV-1 and five clinically relevant mutant strains. Among these compounds, 11s emerged as a true champion. It exhibited unparalleled efficacy against WT HIV-1, with an EC50 value of 2 nmol/L, and also showed remarkable activity against the five mutant strains, with EC50 values ranging from 0.003 µmol/L to 0.073 µmol/L. These results are significantly superior to those of the lead compound 5. When compared with the existing drugs ETR and RPV, compound 11s achieved a 4-fold reduction in cytotoxicity. The metabolic profile of 11s is equally impressive. It demonstrated exceptional stability, with an elimination half-life (t1/2 = 41.4 min) more than double that of RPV (t1/2 = 16.03 min). This extended half-life implies less frequent dosing, which can significantly improve patient compliance and the overall effectiveness of treatment. The comprehensive safety evaluation of 11s further solidifies its potential as an antiretroviral agent. It showed minimal interference with CYP enzymes, low cardiac ion channel activity and no observable acute toxicity, suggesting a much lower risk profile for therapeutic applications. In conclusion, these promising characteristics of the biphenyl-piperidine-triazine derivatives, particularly compound 11s, significantly elevate their development potential as next-generation NNRTIs. This not only provides new hope for HIV patients but also represents a major step forward in the global fight against this devastating disease.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Kun Zhang: Writing – original draft, Formal analysis, Data curation. Li-Min Zhao: Writing – original draft, Formal analysis, Data curation. Tianhao Xing: Writing – original draft, Data curation. Yueyue Bu: Writing – original draft, Data curation. Qingyun Wang: Writing – original draft, Data curation. Christophe Pannecouque: Writing – review & editing, Resources, Formal analysis. Erik De Clercq: Writing – review & editing, Project administration. Angela Corona: Writing – review & editing, Resources. Laura Dettori: Writing – original draft, Data curation. Enzo Tramontano: Writing – review & editing, Supervision. Shuai Wang: Writing – review & editing, Supervision, Project administration, Funding acquisition, Conceptualization. Fen-Er Chen: Writing – review & editing, Supervision, Conceptualization.
This work was financially supported by National Natural Science Foundation of China (No. 82304297) and the State Key Laboratory of Natural and Biomimetic Drugs (No. K202408).
Supplementary material associated with this article can be found, in the online version, at doi:
World Health Organization, HIV and AIDS (2024)
S. Gao, L. Song, Y. Cheng, et al., Acta. Pharm. Sin. B 13 (2023) 2747–2764. doi: 10.1016/j.apsb.2023.01.003
K. Zhang, Y.J. Zhang, M. Li, et al., Med. Res. Rev. 45 (2024) 426–483. doi: 10.1016/j.isci.2025.114409
L. Zhao, C. Pannecouque, E. Clercq, et al., Acta. Pharm. Sin. B 13 (2023) 4906–4917. doi: 10.1016/j.apsb.2023.07.008
D. Feng, H. Lin, L. Jiang, et al., Eur. J. Med. Chem. 246 (2023) 114957. doi: 10.1016/j.ejmech.2022.114957
X. Jin, S. Wang, L. Zhao, et al., Acta. Pharm. Sin. B 13 (2023) 1192–1203. doi: 10.1016/j.apsb.2022.08.017
Y. Sang, C. Pannecouque, E. De Clercq, et al., Acta. Pharm. Sin. B13 (2023) 3054–3066. doi: 10.1016/j.apsb.2023.03.022
Q. Hao, X. Ling, C. Pannecouque, et al., Chin. Chem. Lett. 34 (2023) 107663. doi: 10.1016/j.cclet.2022.07.006
L. Zhao, C. Pannecouque, E. De Clercq, et al., Chin. Chem. Lett. 34 (2023) 108261. doi: 10.1016/j.cclet.2023.108261
J. Ananworanich, B. Hirschel, Expert. Rev. Anti. Infect. Ther. 3 (2014) 51–60.
T. Sibiya, T. Ghazi, A. Chuturgoon, Nutrients 14 (2022) 3076. doi: 10.3390/nu14153076
R. Schrijvers, Expert. Opin. Pharmacother. 14 (2013) 1087–1096. doi: 10.1517/14656566.2013.787411
A. Stockdale, S. Khoo, Curr. Opin. HIV. AIDS 17 (2022) 4–14. doi: 10.1097/coh.0000000000000709
T. Lane, V. Makarov, J.A.E. Nelson, et al., J. Med. Chem. 66 (2023) 6193–6217. doi: 10.1021/acs.jmedchem.2c02055
L. Ding, C. Zhuang, F. Chen, Med. Res. Rev. 41 (2021) 1255–1290. doi: 10.1002/med.21760
G. Li, Y. Wang, E. De Clercq, Acta. Pharm. Sin. B 12 (2022) 1567–1590. doi: 10.1016/j.apsb.2021.11.009
Y. Sun, Z. Zhou, D. Feng, et al., J. Med. Chem. 65 (2022) 15608–15626. doi: 10.1021/acs.jmedchem.2c00576
W. Huang, C. Pannecouque, E. De Clercq, et al., J. Med. Chem. 67 (2024) 19889–19904. doi: 10.1021/acs.jmedchem.4c02413
D. Ludovici, R. Kavash, M. Kukla, et al., Bioorg. Med. Chem. Lett. 11 (2001) 2229–2234. doi: 10.1016/S0960-894X(01)00411-5
B. Liu, Y. Lee, J. Zou, et al., Bioorg. Med. Chem. Lett. 20 (2010) 6592–6596. doi: 10.1016/j.bmcl.2010.09.034
W. Lee, K. Frey, R. Gallardo-Macias, et al., Bioorg. Med. Chem. Lett. 25 (2015) 4824–4827. doi: 10.1016/j.bmcl.2015.06.074
X. Chen, P. Zhan, X. Liu, et al., Bioorg. Med. Chem. 20 (2012) 3856–3864. doi: 10.1007/s10773-012-1271-y
W. Jorgensen, M. Bollini, V. Thakur, et al., J. Am. Chem. Soc. 133 (2011) 15686–15696. doi: 10.1021/ja2058583
K. Jin, M. Liu, C. Zhuang, et al. Acta. Pharm. Sin. B 10 (2020) 344–357. doi: 10.1016/j.apsb.2019.09.007
B. Viira, A. Selyutina, A.T. Garcia-Sosa, et al., Bioorg. Med. Chem. 24 (2016) 2519–2529. doi: 10.1016/j.bmc.2016.04.018
B. Huang, Z. Zhou, D. Kang, et al., Expert. Opin. Ther. Pat. 27 (2017) 9–15. doi: 10.1080/13543776.2017.1262349
X. Chen, Q. Meng, L. Qiu, et al., Chem. Biol. Drug. Des. 86 (2014) 122–128.
Y. Xiong, H. Hu, F. Chen, et al., Acta. Pharm. Sin. 44 (2009) 145–149.
Y. Xiong, F. Chen, J. Balzarini, E. et al. Eur. J. Med. Chem. 43 (2008) 1230–1236. doi: 10.1016/j.ejmech.2007.08.001
B. Meng, Z. Zhuo, H. Yu, et al., Chin. Chem. Lett. 35 (2024) 108827. doi: 10.1016/j.cclet.2023.108827
D. Feng, F. Wei, Y. Sun, et al., Chin. Chem. Lett. 32 (2021) 4053–4057. doi: 10.1016/j.cclet.2021.02.033
S. Han, Y. Lei, C. Pannecouque, et al., Chin. Chem. Lett. 31 (2020) 764–768. doi: 10.1016/j.cclet.2019.11.020
Q. Deng, Q. Zheng, B. Zuo, et al. Green Synth. Catal. 1 (2020) 75–78.
B. Gong, H. Zhu, Y. Liu, et al. Green Synth. Catal. 3 (2022) 110–115. doi: 10.1016/j.gresc.2023.04.003
J. Wang, K. Zhao, K. Zhang, et al., Bioorg. Chem. 147 (2024) 107340. doi: 10.1016/j.bioorg.2024.107340
K. Zhang, K. Hu, Q. Li, et al., J. Med. Chem. 66 (2023) 7221–7242. doi: 10.1021/acs.jmedchem.2c01675
Y. Sang, C. Pannecouque, E. Clercq, et al., Bioorg. Chem. 111 (2021) 104905. doi: 10.1016/j.bioorg.2021.104905
Y. Sang, C. Pannecouque, E. Clercq, et al. Acta. Pharm. Sin. B 13 (2023) 3054–3066. doi: 10.1016/j.apsb.2023.03.022
D. Kang, J. Yang, L. Kong, et al., Viruses 14 (2022) 2390. doi: 10.3390/v14112390
S. Kalyaanamoorthy, K.H. Barakat, Med. Res. Rev. 38 (2017) 525–555. doi: 10.1002/med.21445
S. Mohammad, Z. Zhou, Q. Gong, C.T. January, Am. J. Physiol. 273 (1997) H2534–H2538. doi: 10.1152/ajpheart.1997.273.5.h2534
W. Huang, C. Pannecouque, E. Clercq, et al., J. Med. Chem. 67 (2024) 17568–17584. doi: 10.1021/acs.jmedchem.4c01571

Figure 1 (A) The chemical structures of triazines 1–5. (B) The docking analysis of 5 (PDB code: 2zd1) and design of target compounds via rational structure-based drug design strategy.

Scheme 1 Reagents and conditions: (ⅰ) 4-cyanophenylboronic acid, Pd(dppf)Cl2, Cs2CO3, 1,4-dioxane:H2O = 2:1, N2, reflux, 12 h; (ⅱ) cyanuric chloride, K2CO3, 1,4-dioxane, r.t., 2 h; (ⅲ) 4-amino-1-Boc-piperidine, K2CO3, 1,4-dioxane, r.t., 3 h; (ⅳ) NH3·H2O, tetrahydrofuran (THF), 100 ℃, 12 h; (ⅴ) CF3COOH (TFA), dichloromethane (DCM), r.t., 0.5 h; (ⅵ) corresponding benzyl bromide or chloride, K2CO3, N,N-dimethylformamide (DMF), r.t., 2–6 h.

Figure 2 The docking results of 11s with HIV-1 RT. (A) 11s with WT, (B) 11s with L100I, (C) 11s with K103N, (D) 11s with Y181C, (E) 11s with Y188L, and (F) 11s with E138K. The protein carbon atoms were depicted in gray, mutant residues were depicted in purple and 11s was presented in green. Dashed yellow lines represent the hydrogen-bonding interactions. Dashed pink lines represent the π-π interaction.
Table 1. The anti-HIV-1 activity of 11a–11ad against WT HIV-1 in MT-4 cells.
| Compd. | R1 | EC50 (µmol/L)a | CC50 (µmol/L)b | SIc | Compd. | R1 | EC50 (µmol/L) | CC50 (µmol/L) | SI |
| 11a | 4-Phenyl-CN | 0.008 ± 0.008 | 11.12 ± 1.64 | 1436 | 11r | 4-Phenyl-SO2CH3 | 0.005 ± 0.001 | 4.98 ± 2.52 | 1027 |
| 11b | 3-Phenyl-CN | 0.080 ± 0.063 | 8.90 ± 1.20 | 111 | 11s | 4-Phenyl-CONH2 | 0.002 ± 0.001 | 19.69 ± 1.50 | 7438 |
| 11c | 2-Phenyl-CN | 0.040 ± 0.034 | 116.5 ± 6.07 | 2891 | 11t | 3-Phenyl-CONH2 | 0.006 ± 0.001 | 16.77 ± 2.52 | 2762 |
| 11d | 4-Phenyl-F | 0.009 ± 0.004 | 20.58 ± 9.57 | 2281 | 11u | 4-Phenyl-CONHCH3 | 0.008 ± 0.003 | 20.76 ± 3.63 | 2367 |
| 11e | 4-Phenyl-Cl | 0.008 ± 0.003 | 34.55 ± 11.24 | 4527 | 11v | |
0.004 ± 0.001 | 14.73 ± 2.28 | 3399 |
| 11f | 4-Phenyl-CF3 | 0.086 ± 0.072 | 107.9 ± 44.61 | 1255 | 11w | |
0.008 ± 0.003 | 17.99 ± 4.61 | 2291 |
| 11g | 4-Phenyl-OCF3 | 0.033 ± 0.013 | >211.6 | >6392 | 11x | |
0.018 ± 0.014 | 18.41 ± 1.53 | 998 |
| 11h | 4-Phenyl-CH3 | 0.300 ± 0.274 | 23.91 ± 6.18 | 80 | 11y | |
0.015 ± 0.011 | 16.18 ± 3.03 | 1107 |
| 11i | 2,4-Phenyl-2-F | 0.097 ± 0.086 | 8.86 ± 0.25 | 92 | 11z | |
0.008 ± 0.003 | 9.79 ± 10.53 | 1464 |
| 11j | 3,4-Phenyl-2-F | 0.008 ± 0.003 | 71.31 ± 8.42 | 8877 | 11aa | 2-Pyridyl | 0.006 ± 0.002 | 20.87 ± 3.04 | 3769 |
| 11k | 4-Phenyl-COOH | 0.223 ± 0.033 | >227.8 | >1021 | 11ab | 3-Pyridyl | 0.004 ± 0.002 | 18.83 ± 2.72 | 4759 |
| 11l | 4-Phenyl-CH2COOH | 1.026 ± 0.226 | 209.7 ± 9.26 | 204 | 11ac | 4-Pyridyl | 0.003 ± 0.001 | 8.650 ± 2.870 | 3302 |
| 11m | 4-Phenyl-COOCH3 | 0.028 ± 0.012 | 52.23 ± 9.31 | 1872 | 11ad | 2-Naphthyl | 0.096 ± 0.139 | 83.83 ± 69.58 | 878 |
| 11n | 3-Phenyl-COOH | 1.014 ± 0.268 | >227.8 | >225 | NVP | - | 0.209 ± 0.091 | >15.02 | >72 |
| 11o | 4-Phenyl-B(OH)2 | 0.009 ± 0.004 | 14.51 ± 1.95 | 1569 | EFV | - | 0.004 ± 0.002 | >6.34 | >1425 |
| 11p | 3-Phenyl-B(OH)2 | 0.006 ± 0.002 | 13.36 ± 2.59 | 2166 | ETR | - | 0.003 ± 0.001 | >4.59 | >1340 |
| 11q | 4-Phenyl-SO2NH2 | 0.008 ± 0.004 | 15.20 ± 2.33 | 1674 | |||||
| a EC50: concentration of compound required to achieve 50% protection of MT-4 cell cultures against HIV-1-induced cytopathicity, as determined using the MTT method. b CC50: concentration required to reduce the viability of mock-infected cell cultures by 50%, as determined using the MTT method. c SI: the ratio of CC50/EC50. |
|||||||||
 下载: 导出CSV
下载: 导出CSV
Table 2. The antiviral activity of selected compounds toward HIV-1 mutants.
| Compd. | EC50 (µmol/L)a | ||||
| L100I | K103N | Y181C | Y188L | E138K | |
| 11p | 0.206 ± 0.145 | 0.020 ± 0.004 | 0.027 ± 0.007 | 0.304 ± 0.137 | 0.029 ± 0.015 |
| 11r | 0.029 ± 0.012 | 0.005 ± 0.001 | 0.011 ± 0.002 | 0.157 ± 0.079 | 0.008 ± 0.002 |
| 11s | 0.021 ± 0.090 | 0.003 ± 0.000 | 0.013 ± 0.003 | 0.073 ± 0.002 | 0.004 ± 0.001 |
| 11t | 0.135 ± 0.060 | 0.024 ± 0.014 | 0.109 ± 0.080 | 0.513 ± 0.185 | 0.024 ± 0.019 |
| 11v | 0.012 ± 0.004 | 0.005 ± 0.002 | 0.015 ± 0.005 | 0.068 ± 0.018 | 0.023 ± 0.018 |
| 11aa | 0.174 ± 0.105 | 0.010 ± 0.003 | 0.036 ± 0.004 | 0.307 ± 0.080 | 0.011 ± 0.001 |
| 11ab | 0.211 ± 0.076 | 0.012 ± 0.007 | 0.038 ± 0.017 | 0.545 ± 0.342 | 0.01 ± 0.003 |
| 11ac | 0.046 ± 0.032 | 0.005 ± 0.003 | 0.011 ± 0.001 | 0.076 ± 0.006 | 0.011 ± 0.004 |
| NVP | 2.050 ± 1.896 | 5.197 ± 2.693 | 5.768 ± 2.976 | > 15.02 | 0.157 ± 0.058 |
| EFV | 0.030 ± 0.015 | 0.084 ± 0.033 | 0.008 ± 0.003 | 12.67 ± 0.127 | 0.007 ± 0.003 |
| ETR | 0.007 ± 0.005 | 0.003 ± 0.001 | 0.016 ± 0.005 | 0.020 ± 0.008 | 0.009 ± 0.003 |
| a EC50: concentration of compound required to achieve 50% protection of MT-4 cell cultures against HIV-1-induced cytopathicity, as determined using the MTT method. | |||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载: