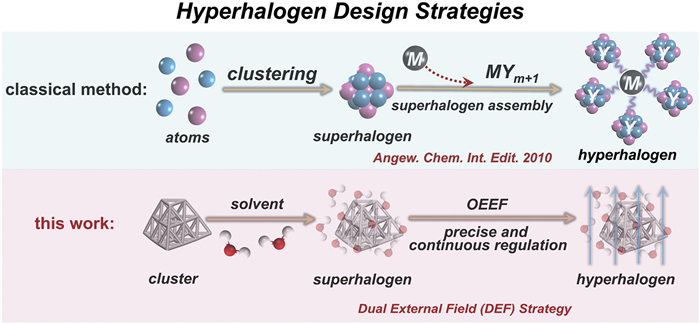
Figure 1.
Schematic description of the concept of the proposed DEF regulation strategy for constructing hyperhalogen. The classical formulation-based method is also included for comparison.
Beyond superhalogen assembly: Field-driven hyperhalogen design via dual-external-field cooperativity
Ao-Hua Wang , Jun Li , Shi-Hu Du , Jia Liu , Yao Zhang , Muhammad Bilal Ahmed Siddique , Jing Chen , Shi-Bo Cheng

Clusters are atomic, molecular, or ionic aggregates bound by physicochemical forces, occupying an intermediate scale between atoms/molucules and bulk matter with remarkable stability [1–7]. Recent advances in cluster science have enabled applications in nanomaterials, catalysis, sensors, and energy [8–13]. Certain clusters, termed "superatoms" [14–16], exhibit atomic-like electronic structures and chemical behaviors due to analogous electron shell configurations and spatial distributions. While the periodic table remains fundamental to understanding matter, superatoms suggest a paradigm shift from its traditional 2D framework to a 3D architecture, potentially revolutionising materials design. Superatom classifications rely on valence electron configurations and physicochemical properties, with superhalogens, which are theoretically proposed by Gutsev and Boldyrev (1981) and experimentally confirmed (Castleman et al., 2004) [14,17–19], being the most studied. Superhalogens not only contain highly stable cluster ions, but also exhibit higher vertical detachment energies (VDE) than halogen atoms (3.06–3.62 eV) [14,19]. Subsequent studies have expanded the superhalogen library, with diverse examples documented in numerous studies [20–32]. Specifically, superhalogens were originally defined as [MXk+1]¯ species, where M is a central atom in its maximum oxidation state (+k) coordinated by (k + 1) halogen ligands [19]. Subsequent theories have extended these concepts through electron counting rules (Jellium model [33], 18-electron/Wade-Mingos/Hückel rule [34–39]) to classify and design superhalogens. However, traditional design methods face two limitations: rigid electronic configurations requiring structural/electronic modifications, and practical constraints in experimental validation of conventional rules.
Another exciting development was the discovery of hyperhalogens, highly electronegative species pioneered by Kandalam, Ganteför, Jena, and colleagues [40]. These species were formed by replacing halogen atoms in conventional superhalogens with superhalogen ligands, resulting in electron-withdrawing capabilities exceeding those of their constituent superhalogens. Their general formula, MYm+1, where M is also a metal atom in its maximum valence state +m, and Y denotes a superhalogen ligand, provides a fundamental framework for rational design. While this classical strategy has enabled the design of novel hyperhalogens, its practical utility is limited by the inherent complexity of manipulating superhalogen assemblies, particularly in achieving precise cluster synthesis. Thus, developing a novel and simple strategy to construct hyperhalogens with greater compositional flexibility and to precisely regulate their electronic properties is both vital and valuable. To this end, recent advances in external-field regulation strategies (EFRS) may offer promising alternatives to conventional superatom design. Unlike traditional electron-counting approaches, EFRS modulate cluster behavior solely through environmental perturbations, preserving intrinsic properties such as composition and valence electron count, thereby establishing a non-invasive paradigm for superatom design. Previous results have demonstrated that a single external field can effectively enhance the electron attachment capability of metal clusters while maintaining their composition and structural stability. For instance, the OEEF has been proven to possess the capability to precisely and continuously regulate the electronic properties of gold clusters without significantly altering their geometrical configurations [13]. The solvent field can also boost anion stabilization without altering geometric/electronic configurations [20]. Consequently, these findings motivate a critical inquiry: Could synergistic application of multiple external fields enable novel hyperhalogen design, circumventing the limitations of traditional superhalogen assembly?
In this work, we propose a dual external field (DEF) synergistic strategy, which combines the solvent effect and an OEEF, to address this question. As illustrated in Fig. 1, the DEF approach involves two sequential steps: first, leveraging polar solvents to construct superhalogens, followed by applying the OEEF to facilitate hyperhalogen formation. To evaluate the feasibility of this strategy, we examine anionic Ag3, Ag5, and Ag19 clusters as model systems. Under DEF conditions, their VDE values exhibited significant enhancement. Furthermore, the universality of the DEF strategy was validated using an experimental synthesized Ag25(SH)18− cluster. Subsequent analysis of O2 activation on the truncated pyramidal Ag19− cluster revealed that the DEF induces pronounced electron polarization, creating distinct active sites.
We begin by modulating electronic properties of bare anionic clusters through systematic variation in solvent polarity and dielectric constants. We employ Ag3−, Ag5−, and Ag19− (model systems [41]) with distinct 1D, 2D, and 3D geometries (Fig. 2a) to probe dual regulatory effects of solvent fields and OEEF across dimensional regimes. Theoretical calculations reveal gas-phase VDE of Ag3− being about 2.39 eV, closely matching the experimentally measured value of 2.43 eV [42]. This demonstrates the reliability and accuracy of our theoretical level. Moreover, the theoretical VDE of Ag5− and Ag19− are about 2.23 and 2.94 eV, respectively. None of these VDE values reach the superhalogen threshold (3.06 eV), establishing a framework for exploring dual-field modulation in cluster anion behavior and superhalogen design. Here, Ag19−, a "magic number" cluster with closed electronic shells [41,42], was selected as the primary subject of investigation for its exceptional stability, which suppresses structural and electronic fluctuations. This stability enables controlled studies of external field effects while minimizing size-dependent structural artifacts. The 3D configuration of Ag19− provides intricate electronic and spatial features, allowing systematic interrogation of solvent fields and OEEF on electronic properties.
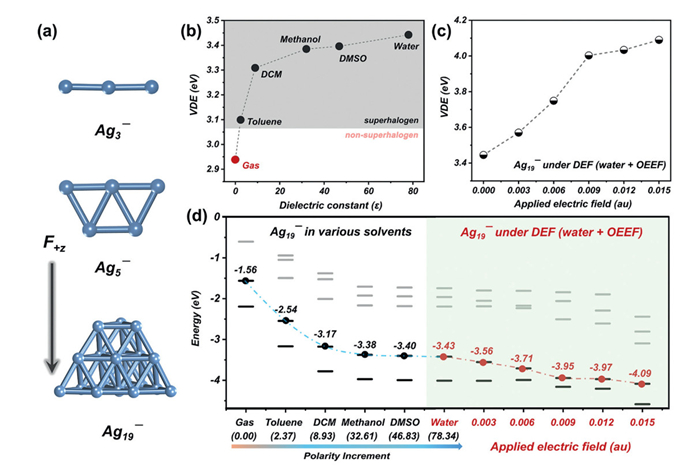
The modulating effects of solvent environments on the electronic properties of bare silver clusters were first investigated. Here, we use an implicit solvent model with the parameter settings given in the Supporting Information. A series of polar solvents, including toluene, dichloromethane (DCM), methanol, dimethylsulfoxide (DMSO), and water, with static dielectric constants (ɛ) ranging from 2.37 to 78.34 (Table S1 in Supporting information) were selected to systematically evaluate their effects on the VDE values of silver cluster anions. As illustrated in Fig. 2b, Figs. S3a and S4a (Supporting information), the calculated trends in VDE variations reveal a consistent solvent-induced enhancement. Notably, solvent environments markedly elevate VDEs across all clusters compared to their gas-phase counterparts. For instance, the gas-phase Ag19− exhibits a VDE of 2.94 eV. In toluene, this value increases to 3.10 eV, thereby classifying Ag19− as a superhalogen (Fig. 2b). This demonstrates that even weakly polar solvents can facilitate a transition in Ag19− from non-superhalogen to superhalogen behavior. In particular, when water is used as a solvent, the electron-binding capacity of Ag19− is significantly enhanced, reaching a VDE of 3.44 eV. Analogous solvent field modulation effects are observed in other model systems, i.e., Ag3− and Ag5−, demonstrating the generality of this phenomenon (Figs. S3 and S4 in Supporting information). These findings collectively establish the solvent field as a potent external field stimulus for activating superatomic characteristics in these cluster anions.
As part of our planned investigation, we next want to examine whether an additional external field (OEEF) can further elevate the VDE of these solvated cluster anions to hyperhalogen level. Detailed description about the OEEF calculations is summarized in the Supporting Information. Ag19− in water solvent, denoted as Ag19−@H2O, was selected as the model system. Given the intrinsic low symmetry of Ag19−, the field orientation was hypothesized to critically influence its VDE. Six OEEF directions (±x, ±y, and ±z) were systematically evaluated. Calculations revealed that ±x and ±y axes induce substantial structural deformations, resulting in non-convergent total energies. In contrast, the z-oriented OEEF significantly increased the VDE of Ag19−@H2O while preserving structural integrity, making it ideal for further study. Structural stability under z-axis OEEF (0.000–0.015 au) was confirmed by minimal Ag-Ag bond elongation (Fig. S1b and Table S2 in Supporting information). Concurrently, the energy gap between the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), termed the H-L gap, of Ag19−@H2O in aqueous solution (exceeding 1.00 eV, Fig. S2a in Supporting information) exceeded gas-phase value, indicating enhanced electronic stability. Analogous stability trends are also observed in Ag3− and Ag5− (Figs. S2b and c in Supporting information). Thus, these findings indicate that the OEEF not only preserves the geometrical stability of the studied silver clusters but also enhances their H-L gaps.
While the geometry of silver clusters remains largely unaffected by the application of an OEEF+z, their electronic properties undergo significant modification in aqueous environments. This is confirmed by the calculated VDE values of these silver anions under OEEF+z intensities ranging from 0.000 au to 0.015 au. As shown in Fig. 2c, the VDE of Ag19−@H2O increases markedly with OEEF application along +z, reaching a maximum enhancement at 0.015 au Specifically, the VDE rises from 3.44 eV in the absence of OEEF to 4.00 eV at an OEEF strength of 0.009 au, and further increases to 4.11 eV at 0.015 au. Notably, this value exceeds the VDEs of all halogen atoms in the periodic table, indicative of hyperhalogen behavior. Similar situations also exist in Ag3− and Ag5− (Figs. S3b and S4b). These findings highlight the role of the proposed DEF strategy, a synergistic combination of solvent effects and OEEF application, in tailoring cluster electronic properties to achieve hyperhalogen behavior. This approach represents a unconventional methodology in hyperhalogen design.
To elucidate the mechanisms contributing to the increase in the VDE of Ag19− in polar solvent and under an OEEF, we systematically analyzed the one-electron energy levels of Ag19− across varying solvent polarities and under OEEF in the most polar solvent, water. The HOMO energy of an anion is a key determinant of its outermost electron stability, with lower HOMO energies correlating to stronger electron-binding capacity. As shown in Fig. 2d, polar solvents induce a pronounced downward shift in the HOMO levels of Ag19−. Specifically, the HOMO level of Ag19− decreases from −1.56 eV in the gas phase to −2.54 eV in toluene, demonstrating the solvent field's significant role in modulating electron retention. This trend persists with increasing solvent polarity: in water, the HOMO level further declines to −3.43 eV. Application of an OEEF (up to 0.015 au) to Ag19−@H2O amplifies this effect, lowering the HOMO level to −4.09 eV. These results correlate strongly with the observed VDE enhancement across solvents and OEEF intensities. Similar behavior is observed for smaller clusters (Ag3− and Ag5−), where analogous HOMO energy trends emerge (Figs. S3c and S4c). Thus, the downward shift in the electronic spectrum is identified as the primary factor driving the transition from non-halogen to hyperhalogen behavior in the presence of dual external fields. Furthermore, it is worth noting that while the experimental implementation of the proposed OEEF methodology presents technical challenges, we outline several feasible experimental strategies. A primary approach involves preparing silver clusters between scanning tunneling microscopy (STM) tips or on the substrates, where the OEEF directionality can be optimized according to the spatial alignment of individual clusters. Notably, recent advancements in scanning tunneling microscopy break junction (STM-BJ) and mechanically controllable break junction (MCBJ) techniques [43–45] have demonstrated exceptional capability in achieving single-molecule-scale electrostatic catalysis under precisely controlled OEEFs. These methodologies offer promising avenues for directional manipulation of individual clusters or ligand-stabilized cluster frameworks via gold-cluster-gold junctions, thereby enabling controlled investigations of OEEF-induced cluster behavior through tunable field orientations. Alternatively, OEEF regulation may be achieved by immobilizing clusters on functionalized substrates followed by localized electric field application, providing complementary insights into field-dependent structural and electronic responses.
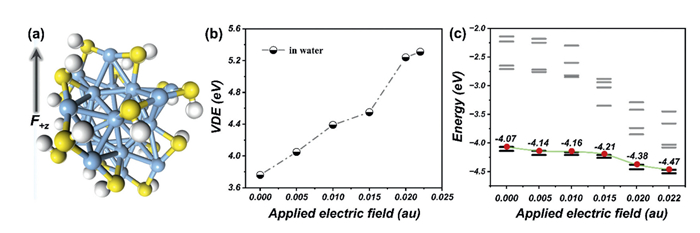
Additionally, the past two decades have witnessed significant advances in the chemical synthesis of atomically precise nanoclusters, with solvent environments playing a pivotal role in these processes. Solvent-mediated effects are critical for directing reaction pathways and stabilizing intermediates during synthesis. Moreover, naked nanoclusters exhibit inherent instability in solution due to their high surface reactivity and tendency to coalesce, necessitating the use of protective ligands in condensed-phase systems. While the aforementioned results demonstrate the regulation potential of the DEF strategy in simplified model clusters, extending this approach to practical ligand-protected systems could provide deeper and valuable mechanistic insights into DEF-assisted hyperhalogen design under realistic condensed-phase conditions. Thus, to validate the broader applicability of the DEF strategy, it is valuable to examine its performance not only in model systems but also in experimentally accessible nanoclusters, particularly those maintaining structural homology with Ag-based systems. Notably, the silver nanocluster Ag25(SH)18−, previously synthesized and characterized in experimental studies [46–48], serves as an ideal candidate. This cluster shares key features with the model Ag19− cluster, both are negatively charged and exhibit valence electron counts corresponding to the Jellium model's magic numbers (8e for Ag25(SH)18− and 20e for Ag19−). Its optimized anionic geometry is illustrated in Fig. 3a. The primary objective of this vestigation was to determine whether DEF strategy could enhance the VDE of a practical silver nanocluster to achieve hyperhalogen characteristics. Water was selected as the solvent due to its pronounced effect on elevating the VDE of naked silver cluster anions. The calculated VDE of Ag25(SH)18− in aqueous solution is approximately 3.76 eV (Fig. 3b), exhibiting superhalogen behavior. Notably, the VDE of Ag25(SH)18− increases monotonically with the OEEF strength. As shown in Fig. 3b, the VDE reaches 5.24 eV at an OEEF intensity of 0.022 au, marking its transition into a hyperhalogen state. Analysis of the HOMO levels under DEFs further reveals that these external fields can significantly stabilize the HOMO level of the nanocluster (Fig. 3c), thereby enhancing the anion's electron-binding capability and correlating directly with the observed VDE enhancement (Fig. 3b). These findings demonstrate that the DEF strategy can effectively regulate the electronic properties of practical ligand-protected nanoclusters to form hyperhalogen, extending its applicability beyond naked cluster anions. This highlights the feasibility and versatility of the DEF approach in tailoring nanocluster functionality.
Having established the efficacy of the DEF strategy in superatom design, we subsequently examined its synergistic interplay with induced polarization and electron transfer in the silver cluster. We first investigated charge redistribution in Ag19− under DEF conditions by partitioning it into three geometrically distinct regions (Fig. 4a). Natural bond orbital (NBO) [49–51] analysis revealed field strength-dependent charge transfer toward the electric field source, with the top layer (TL) accumulating 3.5 |e| excess charge at 0.015 au To highlight the significant charge transfer effect of the DEF strategy, comparative NBO charge analyses under isolated external fields, i.e., an OEEF in the gas phase (Fig. S5a in Supporting information) and the solvent field (Fig. S5b in Supporting information), were also performed, confirming DEF's synergistic enhancement of charge transfer efficiency. Additionally, given the critical role of electrostatic potential (ESP) distribution in cluster systems, we investigated its property for Ag19− under DEF conditions (Fig. S6b in Supporting information). To isolate the distinct contributions of DEF from those of a single OEEF, comparative ESP analyses were conducted under isolated OEEF conditions (gas phase, Fig. S6a in Supporting information). The results reveal pronounced electron density polarization under DEF: the field-proximal region exhibited a potential as low as −14.07 eV, whereas the distal region reached a positive potential of +7.22 eV (Fig. S6b). In contrast, gas-phase OEEF-induced polarization showed markedly reduced magnitude, with potentials of −3.96 eV (field-facing side) and −0.82 eV (opposite side) (Fig. S6a), indicating significantly weaker polarization. These findings substantiate that the DEF strategy enables superior ESP modulation in metal clusters compared to conventional single-field approaches. The enhanced modulation capacity stems from DEF's synergistic field interactions, which amplify charge redistribution beyond the limitations of isolated OEEF systems.
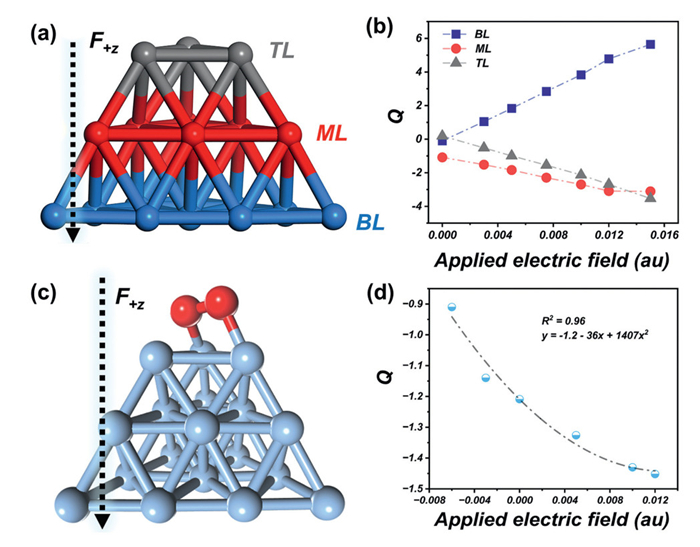
Subsequently, we investigated the impact of the DEF on the catalytic reactivity of Ag19−, given their pronounced influence on charge redistribution. Oxygen activation, a crucial process involving the conversion of O2 into reactive oxygen species, drives numerous biological and chemical transformations, with broad applications in organic synthesis, catalytic material development, and environmental remediation [52,53]. While advanced catalytic systems, including nanoparticles, nanoclusters, and single-atom catalysts, have been engineered to enhance oxygen activation, we focus here on DEF-regulated oxygen activation dynamics in Ag19−, motivated by the critical role of O–O bond activation. Structural optimization revealed the global minimum configuration of Ag19O2− (Fig. 4c). Under DEF conditions, we quantified two key parameters, which are O–O bond length, and adsorption energy (Ead) for the cluster, a thermodynamic stability metric. The Ead was calculated by using the following equation:
|
|
(1) |
The adsorption of O2 on Ag19− under DEF was systematically investigated with the F+ z field strength ranging from −0.006 au to 0.012 au. As summarized in Table S3 (Supporting information), the global minimum structure of Ag19O2− exhibits an O–O bond length of 1.49 Å and an adsorption energy of −0.42 eV. The F+ z field strength was analyzed in two regimes: Negative (−0.006 au to 0.000 au) and positive (0.000–0.012 au). In the positive regime, increasing F+ z strength induces progressive O–O bond elongation, reaching 1.53 Å at 0.012 au, a value approaching bond lengths typical of oxidized species, concurrent with enhanced O2 activation. Adsorption energy initially increases to −1.01 eV at 0.010 au before declining to −0.67 eV. Despite this non-monotonic behavior, adsorption stability remains superior to the zero-field configuration. Mechanistic analysis reveals DEF-driven charge polarization across the Ag19− cluster surface, where electrons redistribute from the bottom to the top region, facilitating electron transfer to O2 (Fig. 4b). Conversely, in the negative regime, O–O bond contraction, indicating strengthened bonding, and reduced electron transfer promote molecular O2 desorption. Notably, positive adsorption energies in this regime confirm thermodynamic instability. NBO analysis (Fig. 4d) quantifies DEF-modulated charge transfer: O2 gains 1.2–1.45 |e| in the positive F+ z range, while losing electrons (0.91 |e|) under negative fields. This demonstrates that positive F+ z fields drive electron accumulation at the cluster apex, populating the π* orbital of O2 to activate and elongate the O–O bond. Reverse field direction suppresses charge transfer, destabilizing adsorption. The charge distribution on O2 exhibits a quadratic dependence on F+ z field strength, as evidenced by the second-order polynomial fit (y = −1.2 − 36x + 1407x2). This equation highlights DEF's unique capacity for continuous electronic modulation, a critical advantage over conventional electron-accounting strategies. To assess universality, calculations in methanol solvent revealed analogous O–O bond length, adsorption energy, and charge-field correlations (Table S4 and Fig. S7 in Supporting information). This confirms the robustness of the DEF strategy across diverse solvent environments. Notably, field orientation modulates spatial charge redistribution patterns, potentially enabling stereoelectronic selectivity for targeted reaction pathways. Thus, this investigation explores a novel approach to creating highly reactive sites for O2 activation by leveraging the synergistic interaction between solvent field effects and OEEF. The Ag19− cluster analyzed in this work serves as a representative model system to demonstrate this synergistic mechanism. The underlying principle of electron density enhancement at active sites through this synergistic combination suggests broad applicability across diverse catalytic systems, independent of their specific chemical composition.
Additionally, to facilitate experimental validation of the DEF strategy proposed in this work, we calculated both photoelectron and UV–vis absorption spectra for the Agn− (n = 3, 5, 19) clusters (Figs. S8 and S9 in Supporting information). These spectral identifications may serve as valuable signatures for monitoring hyperhalogen formation in future experimental studies. For example, as shown in Fig. S8, the photoelectron spectra exhibit a systematic blue shift in peak positions with increasing OEEF intensity across all Ag clusters.
In summary, this study elucidates the synergistic modulation mechanism of solvent fields and OEEF on the electronic properties of silver cluster anions (Agn−) across varying dimensions using density-functional theory (DFT). By integrating solvent polarity and OEEF into a dual external field (DEF) strategy, we achieve precise control over Agn− electronic structures, surpassing conventional hyperhalogen design limitations, i.e. the superhalogen assembly. Notably, apart from the simplified Agn− systems, the DEF strategy demonstrates broader applicability in the experimentally synthesized Ag25 nanocluster to produce the hyperhalogen. The proposed DEF strategy enhances VDEs by driving charge redistribution through amplified intra-cluster electronic polarization, enabling targeted active site formation. Using 3D Ag19− as a model, we demonstrate DEF's critical role in O2 activation: solvent-OEEF synergy induces tunable O–O bond elongation (1.44–1.53 Å) and charge transfer (0.81–1.45 |e|), proportional to field strength. These effects dynamically regulate O2 activation efficiency, while preserving cluster geometry. Our findings establish a field-driven paradigm for hyperhalogen design that retains native cluster composition, offering a theoretical framework for engineering high-performance catalysts via active site control. We wish future research could extend the DEF strategy to diverse metal cluster systems, exploring its potential in design of functional nanomaterials.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Ao-Hua Wang: Writing – original draft, Investigation, Formal analysis. Jun Li: Validation, Formal analysis. Shi-Hu Du: Validation, Formal analysis. Jia Liu: Validation, Formal analysis. Yao Zhang: Validation, Formal analysis. Muhammad Bilal Ahmed Siddique: Validation, Formal analysis. Jing Chen: Validation, Formal analysis. Shi-Bo Cheng: Writing – review & editing, Supervision, Project administration, Funding acquisition, Conceptualization.
This study is based upon work supported by the National Natural Science Foundation of China (NSFC, Nos. 12474274, 92161101), the Innovation Project of Jinan Science and Technology Bureau (No. 2021GXRC032), the Natural Science Foundation of Shandong Province (No. ZR2024MA091). The scientific calculations in this paper have been done on the HPC cloud platform of Shandong University and the HPC platform for theoretical and computational chemistry of the School of Chemistry and Chemical Engineering of Shandong University.
Supplementary material associated with this article can be found, in the online version, at doi:
A.W. Castleman Jr., K.H. Bowen, J. Phys. Chem. 100 (1996) 12911–12944. doi: 10.1021/jp961030k
A.W. Castleman Jr., P. Jena, Proc. Natl. Acad. Sci. U. S. A. 103 (2006) 10554–10559. doi: 10.1073/pnas.0601780103
P. Jena, Q. Sun, Chem. Rev. 118 (2018) 5755–5870. doi: 10.1021/acs.chemrev.7b00524
G.D. Stein, Phys. Teach. 17 (1979) 503–512. doi: 10.1119/1.2340341
A.W. Castleman Jr., P. Jena, Proc. Natl. Acad. Sci. U. S. A. 103 (2006) 10552–10553. doi: 10.1073/pnas.0601783103
J. Li, X. Li, H.-J. Zhai, et al., Science (1979) 299 (2003) 864–867.
H. Fang, P. Jena, J. Phys. Chem. Lett. 7 (2016) 1596–1603. doi: 10.1021/acs.jpclett.6b00435
V. Bonačić-Koutecký, M. Perić, Ž. Sanader, J. Phys. Chem. Lett. 9 (2018) 2584–2589. doi: 10.1021/acs.jpclett.8b00819
P. Koirala, M. Willis, B. Kiran, et al., J. Phys. Chem. C 114 (2010) 16018–16024. doi: 10.1021/jp101807s
X.N. Li, L.X. Jiang, L.N. Wang, et al., J. Phys. Chem. Lett. 10 (2019) 7850–7855. doi: 10.1021/acs.jpclett.9b03056
Z.C. Wang, N.V. Tkachenko, L. Qiao, et al., Chem. Comm. 56 (2020) 6583–6586. doi: 10.1039/d0cc02525a
J. Zhao, N. Ma, T. Wang, et al., J. Mater. Chem. A 10 (2022) 21611–21621. doi: 10.1039/d2ta04855h
Y. Zhao, J. Wang, H.C. Huang, et al., J. Phys. Chem. Lett. 11 (2020) 1093–1099. doi: 10.1021/acs.jpclett.9b03794
D.E. Bergeron, A.W. Castleman Jr., T. Morisato, et al., Science (1979) 304 (2004) 84–87.
D.E. Bergeron, P.J. Roach, A.W. Castleman Jr., et al., Science (1979) 307 (2005) 231–235. doi: 10.1126/science.1105820
Z. Luo, A.W. Castleman Jr., Acc. Chem. Res. 47 (2014) 2931–2940. doi: 10.1021/ar5001583
S.N. Khanna, P. Jena, Phys. Rev. Lett. 69 (1992) 1664–1667. doi: 10.1103/PhysRevLett.69.1664
S.N. Khanna, P. Jena, Phys. Rev. B 51 (1995) 13705–13716. doi: 10.1103/PhysRevB.51.13705
G.L. Gutsev, A.I. Boldyrev, Chem. Phys. 56 (1981) 277–283. doi: 10.1016/0301-0104(81)80150-4
H. Wang, J. Li, J. Chen, et al., Chin. Chem. Lett. 34 (2023) 108222. doi: 10.1016/j.cclet.2023.108222
Y. Gao, S. Bulusu, X.C. Zeng, J. Am. Chem. Soc. 127 (2005) 15680–15681. doi: 10.1021/ja055407o
Q. Xue, M. Zhong, J. Zhou, et al., J. Mater. Chem. A 126 (2022) 3536–3542. doi: 10.1021/acs.jpca.2c02530
L.P. Ding, P. Shao, C. Lu, et al., Sci. Rep. 7 (2017) 45149. doi: 10.1038/srep45149
Y.J. Ko, H. Wang, K. Pradhan, et al., J. Chem. Phys. 135 (2011) 244312. doi: 10.1063/1.3671457
H.T. Nguyen, N.T. Cuong, N.T. Lan, et al., RSC Adv. 12 (2022) 13487–13499. doi: 10.1039/d1ra08527a
B. Yin, Q. Du, L. Geng, et al., J. Phys. Chem. Lett. 11 (2020) 5807–5814. doi: 10.1021/acs.jpclett.0c01643
B. Pathak, D. Samanta, R. Ahuja, et al., Chemphyschem. 12 (2011) 2423–2428. doi: 10.1002/cphc.201100320
I. Anusiewicz, S. Freza, P. Skurski, Inorg. Chem. 55 (2016) 10161–10169. doi: 10.1021/acs.inorgchem.6b01304
Q. Wang, Q. Sun, P. Jena, J. Chem. Phys. 131 (2009) 124301. doi: 10.1063/1.3236576
S. Smuczyńska, P. Skurski, Inorg. Chem. 48 (2009) 10231–10238. doi: 10.1021/ic901253r
F. Wudl, Acc. Chem. Res. 17 (1984) 227–232. doi: 10.1021/ar00102a005
C. Sikorska, P. Skurski, Inorg. Chem. 50 (2011) 6384–6391. doi: 10.1021/ic200945e
W.D. Knight, K. Clemenger, W.A. de Heer, et al., Phys. Rev. Lett. 52 (1984) 2141–2143. doi: 10.1103/PhysRevLett.52.2141
G.N. Lewis, J. Am. Chem. Soc. 38 (1916) 762–785. doi: 10.1021/ja02261a002
I. Langmuir, J. Am. Chem. Soc. 41 (1919) 868–934. doi: 10.1021/ja02227a002
I. Langmuir, Science (1979) 54 (1921) 59–67. doi: 10.1126/science.54.1386.59
K. Wade, J. Chem. Soc. D: Chem. Comm. (1971) 792–793.
D.M.P. Mingos, Nat. Phys. Sci. 236 (1972) 99–102. doi: 10.1038/physci236099a0
D.M.P. Mingos, Acc. Chem. Res. 17 (1984) 311–319. doi: 10.1021/ar00105a003
M. Willis, M. Götz, A.K. Kandalam, et al., Angew. Chem. Int. Ed. 49 (2010) 8966–8970. doi: 10.1002/anie.201002212
M.-S. Liao, J.D. Watts, M.-J. Huang, J. Phys. Chem. C 118 (2014) 21911–21927. doi: 10.1021/jp501701f
H. Handschuh, C.Y. Cha, P.S. Bechthold, et al., J. Chem. Phys. 102 (1995) 6406–6422. doi: 10.1063/1.469356
A.C. Aragonès, N.L. Haworth, N. Darwish, et al., Nature 531 (2016) 88–91. doi: 10.1038/nature16989
J. Bai, A. Daaoub, S. Sangtarash, et al., Nat. Mater. 18 (2019) 364–369. doi: 10.1038/s41563-018-0265-4
Z. Chen, L. Chen, J. Liu, et al., J. Phys. Chem. Lett. 10 (2019) 3453–3458. doi: 10.1021/acs.jpclett.9b00796
K.L.D.M. Weerawardene, C.M. Aikens, J. Phys. Chem. C 122 (2018) 2440–2447. doi: 10.1021/acs.jpcc.7b11706
C.P. Joshi, M.S. Bootharaju, M.J. Alhilaly, et al., J. Am. Chem. Soc. 137 (2015) 11578–11581. doi: 10.1021/jacs.5b07088
C.M. Aikens, J. Phys. Chem. C 112 (2008) 19797–19800. doi: 10.1021/jp8090914
A.E. Reed, R.B. Weinstock, F. Weinhold, J. Chem. Phys. 83 (1985) 735–746. doi: 10.1063/1.449486
A.E. Reed, F. Weinhold, J. Chem. Phys. 78 (1983) 4066–4073. doi: 10.1063/1.445134
J.P. Foster, F. Weinhold, J. Am. Chem. Soc. 102 (1980) 7211–7218. doi: 10.1021/ja00544a007
X. Xie, Y. Li, Z.-Q. Liu, et al., Nature 458 (2009) 746–749. doi: 10.1038/nature07877
M. Valden, X. Lai, D.W. Goodman, Science 281 (1998) 1647–1650. doi: 10.1126/science.281.5383.1647

Figure 1 Schematic description of the concept of the proposed DEF regulation strategy for constructing hyperhalogen. The classical formulation-based method is also included for comparison.

Figure 2 The (a) optimized global minima of Agn− (n = 3, 5, and 19), (b) theoretical VDE values of Ag19− in the gas phase and different solvents with a reference to the lower superhalogen threshold (3.06 eV for the iodine atom), (c) VDE values of Ag19− under varied OEEF strength, and (d) one-electron energy levels of Ag19− in various solvents and under different OEEF intensities (occupied MOs are indicated by solid black lines and unoccupied MOs are represented by gray lines).

Figure 3 The (a) optimized geometry of Ag25(SH)18−, (b) VDE values of Ag25(SH)18−@H2O under varied OEEF strength, and (c) one-electron energy levels of Ag25(SH)18−@H2O under different OEEF intensities.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:

 下载:
下载: