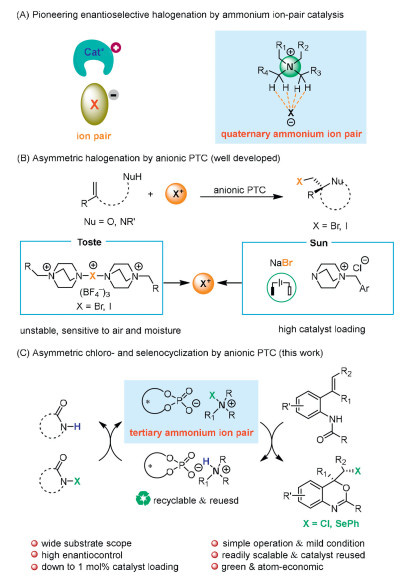
Scheme 1.
Asymmetric halogenation via anionic PTC strategies.
Asymmetric chloro– and selenocyclization of 2-alkenyl anilides enabled by tertiary ammonium salt catalysis
Xue Du , Ze-Hua Sun , Penglei Zhang , Li-Ping Xu , Xiaodong Xiong

Asymmetric halocyclization is an important class of transformation to construct valuable halogenated compounds which are widely present in natural products and biological molecules [1-5]. The halogen handles in these products were readily manipulated using well-developed methods to give useful and attractive building blocks [6-8]. Moreover, the insertion of chlorine into the particular site of bioactive molecule could provide favorable pharmacokinetic and pharmacological properties [9,10]. Since more than 200 drugs including chlorine were approved by FDA, the use of chlorine to the optimization of lead compounds has received great interest from pharmaceutical chemists [3]. Although great achievements have been made in the field of enantioselective chlorination with various organocatalytic strategies, low temperature and high catalyst loading are often required for excellent enantio– and diastereoselectivity [11-22].
Phase-transfer catalysis (PTC) is considered as one powerful and versatile methodology for organic synthesis because of the character of green chemistry and unique advantage over homogeneous alternatives [23]. The development of new ligands and catalyst variants for efficient PTC has received continuous attention from synthetic chemists [24,25]. Among the existing methods, quaternary ammonium salts with chiral cation as PTC catalysts are prevalent in chiral ion-pair catalysis. Based on this strategy, considerable effort has been devoted to develop several synthetic methods for asymmetric halogenation (Scheme 1A) [26-29]. However, this chiral cationic PTC system suffers from low enantioselectivity, harsh condition and limited reaction types. Recently, the asymmetric fluorination process represents a significant milestone in PTC by employing chiral phosphate anion to form an ion pair with the ammonium-based electrophilic fluorinating cation [30]. In this PTC system, nonpolar solvents and Selectfluor were required to exclude the undesired background reaction. Later, Toste et al. extended this strategy to other electrophiles for asymmetric transformation [31,32]. Unstable and insoluble bromo–, iodo analogues of Selectfluor were developed as electrophilic reagents for enantioselective halogenations under the chiral anion phase-transfer conditions (Scheme 1B) [33-40]. Very recently, Sun et al. demonstrated electricity-driven catalytic asymmetric bromocyclization with NaBr as bromine source with high catalyst loading by chiral phosphate anion PTC (Scheme 1B) [41]. As for the fledgling nature of this area, relatively large catalyst loading is required to achieve meaningful levels of enantioselectivity. Furthermore, efficient asymmetric conversions with chlorine and other electrophile have remained underdeveloped. The highly reactive nature of chloronium ions makes the development of enantioselective chlorination a formidable challenge by the strategy of anionic PTC [42-44].
Inspired by the previous work of highly ortho-chlorination of aniline and phenol, asymmetric bromination and iodination with secondary ammonium salt as catalyst [45-47], we rationalized that more bulky tertiary ammonium salt could exchange with the chlorine and selenylation source to generate stable ammonium ion-pair, employed as key stereodetermining intermediate for asymmetric induction [48]. This strategy would allow us to develop electrophilic halogen-initiated asymmetric cyclization to furnish chiral heterocycles. Herein, we are pleased to describe a mild and highly practical protocol for intramolecular asymmetric chlorocyclization of 2-alkenyl anilides via anionic PTC to provide expedient access to chiral chlorinated 4H-3,1-benzoxazine framework (Scheme 1C). However, in sharp contrast to the general success achieved for the bromocyclization, enantioselective chlorocyclization of 2-alkenyl anilides remains challenging [49]. A breakthrough was made by the Deng group, who developed asymmetric chlorocyclization utilizing cinchonidine-derived chiral esters as catalyst. However, high catalyst loading and harsh conditions were required, and substrate scope was restricted to 1,1-disubstituted alkenes in this protocol, which somehow limit its practical application [50]. The commonly motifs of 4H-3,1-benzoxazine are the key cores of bioactive natural products, agrochemicals, biological active pharmaceuticals, functional materials, chiral ligands and synthetic building blocks [51-54]. Hence, numerous endeavors have been made over years toward asymmetric synthesis of optical 4H-3,1-benzoxazine [55-58].
The chiral ammonium salts 1 are air and moisture-stable, nontoxic, easy to handle, which easily be prepared by the reaction between amines and chiral phosphoric acid (CPA) under mild conditions. At the initial stage of investigation, the ammonium salt 1 catalyzed chlorocyclization of aniline 2a was examined at room temperature using n-hexane and 1,3-dichloro-5,5-dimethylhydantoin (DCDMH) as the solvent and chlorinating reagent, respectively (Table 1). The reaction was sluggish in the absence of catalyst owing to the insolubility of DCDMH in nonpolar solvent (entry 1). The chlorination worked well using tertiary ammonium salt 1a as catalyst to provide desired product 3a in 96% yield and 95% ee (entry 2). Then different ammonium salts were tested for this chlorocyclization. A survey of different chiral counter anions revealed that H8-TRIP phosphate was the catalyst of choice. The more bulky ammonium salt 1d gave the best result (entries 2–5). However, poor enantioselectivity was obtained in the presence of secondary ammonium salt 1e (entry 6). Subsequent screening of more solvents failed to give better results (entries 7–9). Cinchonine-derived quaternary ammonium salt, CPA and its derivatives which previously used as typical organocatalysts for asymmetric halogenation were also evaluated for the chlorination of 2a [59-63]. It is interesting to realize that the catalytic power of tertiary ammonium salt 1 was much higher than that of some N-benzyl-cinchoninium bromide C1-C2, and quaternary ammonium salt C3 (entries 10–12). H8-TRIP, H8-TRIP-Na, co-catalyst mixture (1:1) of H8-TRIP with Lewis base LB1 and LB2 were examined in this reaction, poor conversions or enantioselectivities were observed (entries 13–16). Moderate reaction yield of 3a with good enantioselectivity was obtained when a catalyst blend (1:1) of H8-TRIP and LB3 was used (entry 17). The efficiency with tertiary ammonium phosphate 1d was found to be considerably higher than quaternary ammonium salt, phosphoric acid and its derivatives. These results highlighted the excellent catalytic performance and unique usefulness of the newly developed ammonium phosphate salt catalyst 1.
 DownLoad:
CSV
DownLoad:
CSV
 | ||||
| Entry | Catalyst | Solvent | Yield (%)b | ee (%)c |
| 1 | – | n-hexane | trace | – |
| 2 | 1a | n-hexane | 96 | 95 |
| 3 | 1b | n-hexane | 96 | 97 |
| 4 | 1c | n-hexane | 91 | 5 |
| 5 | 1d | n-hexane | 98 | 99 |
| 6 | 1e | n-hexane | 92 | 19 |
| 7 | 1d | CCl4 | 96 | 36 |
| 8 | 1d | Toluene | 95 | 32 |
| 9 | 1d | n-heptane | 96 | 98 |
| 10 | C1 | n-hexane | 34 | 28 |
| 11 | C2 | n-hexane | 25 | 21 |
| 12 | C3 | n-hexane | 34 | 79 |
| 13 | H8-TRIP | n-hexane | 4 | 28 |
| 14 | H8-TRIP-Na | n-hexane | 10 | 75 |
| 15 | H8-TRIP/LB1 | n-hexane | 68 | 6 |
| 16 | H8-TRIP/LB2 | n-hexane | 28 | 30 |
| 17 | H8-TRIP/LB3 | n-hexane | 55 | 86 |
| a Reactions were carried out with aniline 2a (0.05 mmol), catalyst (0.0025 mmol) and DCDMH (0.055 mmol) in the indicated solvent (2 mL) at 25 ℃ in the absence of light. b Isolated yield. c The ee values were determined by HPLC analysis on a chiral stationary phase. | ||||
With the optimized reaction conditions being established, we turned to evaluate the substrate scope of the tertiary ammonium salt catalyzed chlorocyclization, and the results are shown in Scheme 2. A wide range of o-vinylanilides were compatible in this reaction, giving the chlorocyclized products in high yields and excellent enantioselectivities. The compatibility of various substituents at the alkene moiety was then examined. The aliphatic substrates with cyclic and uncyclic substituents deliver the chlorinated products 3a-3c in excellent yields and enantioselectivity. Then the substrates with various substituents on the phenyl ring at the alkene moiety were explored. Both electron-donating groups and electron-withdrawing groups were tolerant in the reaction conditions, affording the corresponding cyclized compounds 3d-3i in up to 99% ee. It is noteworthy that the steric hindrance plays little influence on the chlorocyclization, excellent enantioselectivity was observed when the o-methyl group was substituted (3e). The 1-naphthyl-substituted substrate 2j gave rise to the chlorocyclized compound 3j in 93% yield and 99% ee, and the absolute configuration was confirmed by X-ray analysis. The substrates of different substituted aniline derivatives were further explored. Electron-donating groups such as methyl and phenyl and electron-withdrawing groups such as fluoride, chloride and bromide were well tolerated, providing the desired products 3k–3t in excellent results. The substrate 2u with other aromatic core was successfully evaluated for the chlorination to furnish 3u in excellent outcomes. Next, various amides were also explored for the scope compatibility of this chlorocyclization. The position and electron feature of the substituent to the aromatic ring had negligible impact on the enantioselectivity, the corresponding cyclized products 3v-3af were also obtained smoothly in up to 99% ee. Notably, the aliphatic amide can be applied in this reaction, providing the corresponding product 3ag in excellent yield and high enantioselectivity. Switching the benzene ring to other aromatic cores such as thiophene, pyridine and naphthalene gave the desired products 3ah-3aj in excellent yields and enantioselectivities. Furthermore, the internal and cyclic olefins were well amenable in the optimized condition, leading to the desired products 3ak-3am in high enantioselectivity and diastereoselectivity.
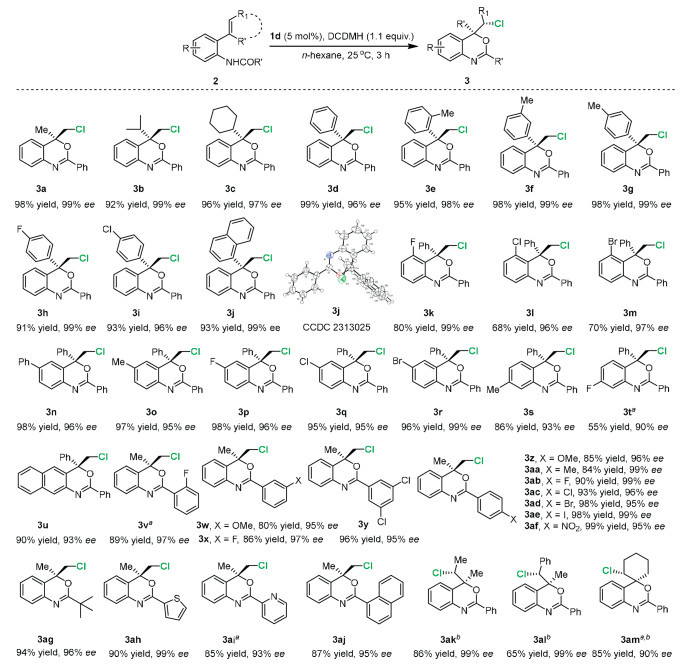
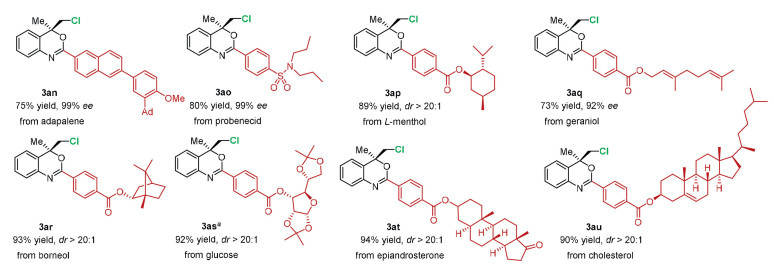
The late-stage functionalization of complex molecules illustrated the utility of this catalytic system (Scheme 3). The amides containing the motif of drug molecules derivatives successfully engage in this catalytic asymmetric chlorination to afford adapalene 3an and probenecid 3ao in generally good yields with excellent diastereomeric excess. Natural products derivatives containing menthol, borneol, geraniol, epiandrosterone and cholesterol worked well in the reactions, yielding the desired products 3ap-3au with excellent results. The modification of these important compounds, many of which contain sensitive alkene, ketal and carbonyl groups, suggest the good functional group tolerance of this method.
The recycling stability of this type of PTC catalyst was also investigated. This developed PTC protocol could be easily scaled up and the catalyst loading could be reduced to 1 mol% although a longer reaction time was applied. For instance, 4 mmol of aniline 2h was subjected to the reaction and 1.39 g (99% yield and 99% ee) of the desired chlorinated product 3h was obtained (Scheme 4A). After the completion of this chlorination, the tertiary ammonium salt catalyst 1d could be recovered by column chromatography on silica gel and the comparable results in terms of reaction conversion and enantioselectivity were obtained during the halogenation/catalyst recycling process for 3 times. These results indicating that the catalyst worked with negligible loss in both the activity and selectivity. Since the chlorine carrier hydantoin is insoluble in hexane, we attempted to isolate the product under column-free condition. Upon completion, the chlorocyclization was filtered and the insoluble hydantoin was washed with hexane to furnish the chlorinated compound 3h in 99% yield with purities of up to 99% (determined by 1H NMR) and 98% ee (Scheme 4B). The practical and environmental benign features of this catalytic system are attractive for the modern manufacture sectors.
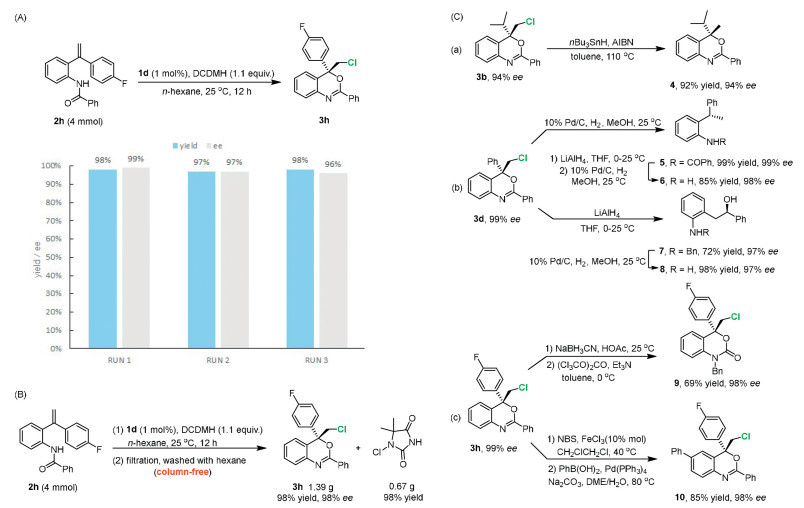
The corresponding chlorocyclized compounds are very useful building blocks for the synthesis of functional molecules (Scheme 4C). Dechlorination of 3b was achieved via nBu3SnH reduction, affording product 4 in high yield. The compound 3d underwent hydrogenation to give chiral compound 5 in 99% yield, followed by remove of the benzoyl group to provide diarylamine 6 in high enantioselectivity, which is useful in drug discovery and chiral catalyst synthesis [64,65]. Simple reduction of 3d by LiAlH4 readily furnished secondary alcohol 7 in 72% yield. Subsequent deprotection of the benzyl group of 7 with hydrogen to form chiral amino alcohol 8, which is used as an important intermediate for the synthesis of chiral ligand, base and benzoxazine nucleus [66]. Treatment of benzoxazine 3h with NaBH3CN in acetic acid yield amino alcohol intermediate, which cyclized with triphosgene in the base condition, providing 1,4-dihydro-2H-3,1-benzoxazine-2-one derivatives 9 in 69% yield without any loss of stereochemistry. 3h was subjected to the site-selective bromination and Suzuki coupling, affording the functionalized benzoxazine 10 in 85% yield and 98% ee.
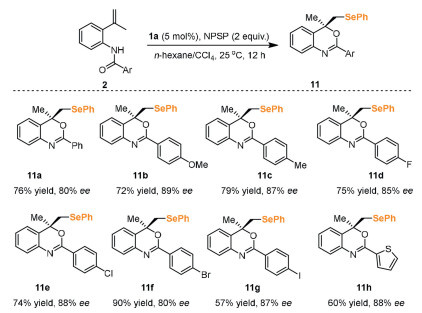
Further evaluation of chiral tertiary ammonium salt showed that catalyst 1a could catalyze the selenocyclization of o-vinyl anilides 2 with N-(phenylseleno)phthalimide (NPSP) as the electrophile. As shown in Scheme 5, the substrates 2 with diverse functionalities were proved compatible under this catalytic protocol and could react smoothly with the selenium reagent NPSP to furnish the corresponding selenylated compounds 11 in moderate yields and good enantioselectivities.
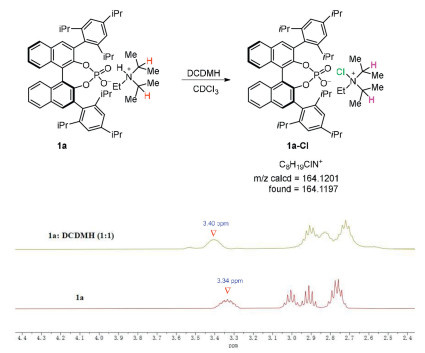
To get a better understanding of the catalytic system, we attempted to probe the active species (Scheme 6). Upon mixing 1a and DCDMH in CDCl3, a new proton signal at 3.40 ppm appeared which might be due to species 1a-Cl, further evidenced using ESI high-resolution mass spectrometry. This active species 1a-Cl was vital for the asymmetric induction which the phosphate anion interacts with tertiary ammonium cation tightly via electrostatic and multiple electropositive hydrogens interact with the phosphate oxygens.
To gain insights in the reaction mechanism and the origins of enantioselectivity, we have performed density functional theory studies. The ammonium salt 1d, aniline 2a, and DCDMH were used as the catalyst, substrate, and chlorinating reagent, respectively, in our calculations. As shown in Fig. 1, the complexation of 1d and DCDMH would lead to IN1 and IN2, in which the chlorine has bound to nitrogen in the latter. This step is slightly endergonic by 4.1 kcal/mol and there are multiple C-H…O hydrogen bonding interactions between the phosphate anion and the ammonium cation. Following that, the binding of substrate 2a would form isomeric species IN3 and IN3’, which are exergonic by 1.4 and 5.5 kcal/mol, respectively. Starting from IN3, the asymmetric chlorocyclization occurs via transition state TS4 to generate IN5. This process has an activation free energy barrier of 15.0 kcal/mol and is exergonic by 31.7 kcal/mol (with respect to IN3’). On the other hand, from IN3’, the minor pathway takes place through TS4’ to provide IN5’. This pathway has an activation free energy barrier of 16.1 kcal/mol and is exergonic by 34.9 kcal/mol. By comparison, the pathway leading to the major enantiomer (via TS4) is 1.1 kcal/mol more favorable than that leading to the minor enantiomer (via TS4’), which is consistent with experiment (i.e., 99% ee). By detailed analyses on the selectivity-determining transition state, we found that there was favorable hydrogen bonding interaction between phosphate oxygen and the ammonium C-H bond (O…H = 2.24 Å) in TS4. In addition, there were much pronounced steric repulsions between phosphate anion and the aniline substrate part in TS4’ (i.e., the shortest H…H distances are 2.23 and 2.26 Å). Therefore, the hydrogen bonding interaction and the steric effect account for the observed high enantioselectivity.
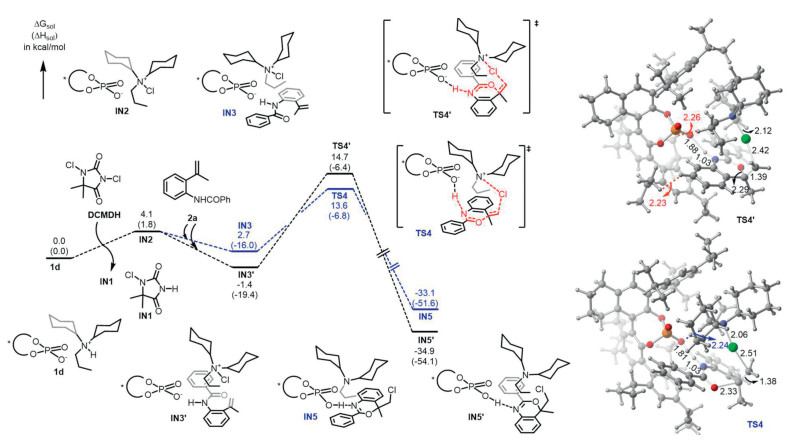
In our previous ion pair catalysis work [47], the strong bidentate noncovalent interaction of the hydrogen bonding (N-H…O) and ion pair interaction between phosphate anion and secondary ammonium cation was responsible for the asymmetric induction of bromination and iodation with high directionality, and the hydrogen bonding of C-H…O (2.66 Å) was not observed. However, as shown in TS4, the phosphate anion interacts with tertiary ammonium cation tightly via strong attractive force between ammonium cation and phosphate ion, and multiple electropositive hydrogens interact with the phosphate oxygens (C-H…O = 2.24, 2.51, 2.54 Å) in this work. Consequently, the resulting highly directional noncovalent interaction would result in effective asymmetric induction for the chlorocyclization.
In conclusion, we have developed the chiral anion PTC for asymmetric chlorination and selenylation of o-vinylanilides in high enantioselectivities under mild reaction conditions. This catalytic protocol is easily scale up and catalyst loading could be further reduced to 1 mol%. The robust tertiary ammonium salt could be easily recovered and reused for 3 times without notable loss of catalytic performance. The features of excellent reactivity, selectivity, stability and recyclability, and column-free purification of this chiral anion PTC system are significant for practical application in organic synthesis. In addition, computational mechanistic studies were used to elucidate the origin of the stereoselectivity. Further investigation on the application of this PTC system to other asymmetric reactions is underway.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Xue Du: Methodology, Investigation, Formal analysis. Ze-Hua Sun: Software, Formal analysis. Penglei Zhang: Investigation, Formal analysis. Li-Ping Xu: Writing – review & editing, Software, Formal analysis. Xiaodong Xiong: Writing – review & editing, Writing – original draft, Supervision, Resources, Funding acquisition, Formal analysis, Conceptualization.
This work was supported by the Nation Natural Science Foundation of China (No. 22061026), Jiangxi Provincial Natural Science Foundation (Nos. 20202BAB203004, 20212ACB213007), the start-up Fund of Nanchang University, the Youth Innovation Team Program in Colleges and Universities of Shandong Province (No. 2022KJ228), and the Qilu Young Scholar of Shandong University for financial support.
Supplementary material associated with this article can be found, in the online version, at doi:
K.C. Nicolaou, Angew. Chem. Int. Ed. 48 (2009) 660–719. doi: 10.1002/anie.200801695
S. Bras̈e, A. Encinas, J. Keck, Chem. Rev. 109 (2009) 3903–3990. doi: 10.1021/cr050001f
B.R. Smith, C. Eastman, J.T. Njardarson, J. Med. Chem. 57 (2014) 9764–9773. doi: 10.1021/jm501105n
J. Yan, Z. Zhou, W. Xie, et al., Org. Chem. Front. 9 (2022) 499–516. doi: 10.1039/d1qo01395e
S. Liu, B. -Q. Zhang, J. Deng, et al., Adv. Synth. Catal. 364 (2022) 3974–4005. doi: 10.1002/adsc.202200611
T. Akiyama, K. Mori, Chem. Rev. 115 (2015) 9277–9306. doi: 10.1021/acs.chemrev.5b00041
J. Bariwal, E.V.D. Eycken, Chem. Soc. Rev. 42 (2013) 9283–9303. doi: 10.1039/c3cs60228a
R. Martin, S.L. Buchwald, Acc. Chem. Res. 41 (2008) 1461–1473. doi: 10.1021/ar800036s
H. Sun, C.E. Keefer, D.O. Scott, Drug Metab. Lett. 5 (2011) 232–242. doi: 10.2174/187231211798472575
Y. Wang, C. Bi, P.S. Baran, et al., Nat. Chem. 16 (2024) 1539–1545. doi: 10.1038/s41557-024-01539-4
H. Wack, A.E. Taggi, T. Lectka, et al., J. Am. Chem. Soc. 123 (2001) 1531–1532. doi: 10.1021/ja005791j
M. Marigo, S. Bachmann, K.A. Jørgensen, et al., Angew. Chem. Int. Ed. 43 (2004) 5507–5510. doi: 10.1002/anie.200460462
M.P. Brochu, S.P. Brown, D.W.C. MacMillan, J. Am. Chem. Soc. 126 (2004) 4108–4109. doi: 10.1021/ja049562z
D.C. Whitehead, R. Yousefi, A. Jaganathan, B. Borhan, J. Am. Chem. Soc. 132 (2010) 3298–3300. doi: 10.1021/ja100502f
W. Zheng, Z. Zhang, M.J. Kaplan, J.C. Antilla, J. Am. Chem. Soc. 133 (2011) 3339–3341. doi: 10.1021/ja109824x
Q. Yin, S.G. Wang, S.L. You, et al., Chem. Sci. 6 (2015) 4179–4183. doi: 10.1039/C5SC00494B
W. -J. Chung, C.D. Vanderwal, Angew. Chem. Int. Ed. 55 (2016) 4396–4434. doi: 10.1002/anie.201506388
K. Shibatomi, K. Kitahara, S. Iwasa, et al., Nat. Commun. 8 (2017) 15600–15606. doi: 10.1038/ncomms15600
S. Ponath, M. Menger, M. Christmann, et al., Angew. Chem. Int. Ed. 57 (2018) 11683–11687. doi: 10.1002/anie.201806261
G.F. Yang, Y. Yuan, Z. Tang, et al., J. Am. Chem. Soc. 145 (2023) 5439–5446. doi: 10.1021/jacs.2c13758
G. Hutchinson, C. Alamillo-Ferrer, J. Burés, J. Am. Chem. Soc. 143 (2021) 6805–6809. doi: 10.1021/jacs.1c02997
Z. Li, B. Wang, J. Sun, et al., J. Am. Chem. Soc. 146 (2024) 2779–2788. doi: 10.1021/jacs.3c12826
T. Hashimoto, K. Maruoka, Chem. Rev. 107 (2007) 5656–5682. doi: 10.1021/cr068368n
R.J. Phipps, G.L. Hamilton, F.D. Toste, Nat. Chem. 4 (2012) 603–614. doi: 10.1038/nchem.1405
G. Pupo, V. Gouverneur, J. Am. Chem. Soc. 144 (2022) 5200–5213. doi: 10.1021/jacs.2c00190
M. Wang, L.X. Gao, S.B. Zhang, et al., J. Org. Chem. 69 (2004) 2874–2876. doi: 10.1021/jo035719e
X. Zeng, C. Miao, W. Sun, et al., Chem. Commun. 49 (2013) 2418–2420. doi: 10.1039/c2cc38436a
M. Majdecki, P. Grodek, J. Jurczak, J. Org. Chem. 86 (2021) 995–1001. doi: 10.1021/acs.joc.0c02486
P. Zebrowski, I. Eder, M. Waser, et al., ACS Org. Inorg. Au 2 (2022) 34–43. doi: 10.1021/acsorginorgau.1c00025
V. Rauniyar, A.D. Lackner, G.L. Hamilton, F.D. Toste, Science 334 (2011) 1681–1684. doi: 10.1126/science.1213918
Y.M. Wang, J. Wu, F.D. Toste, et al., J. Am. Chem. Soc. 134 (2012) 12928–12931. doi: 10.1021/ja305795x
E. Miller, S. Kim, F.D. Toste, et al., J. Am. Chem. Soc. 142 (2020) 8946–8952. doi: 10.1021/jacs.0c02331
W. Xie, G. Jiang, D. Ma, et al., Angew. Chem. Int. Ed. 52 (2013) 12924–12927. doi: 10.1002/anie.201306774
F. Romanov-Michailidis, L. Guénée, A. Alexakis, Org. Lett. 15 (2013) 5890–5893. doi: 10.1021/ol402981z
H. Liu, G. Jiang, W. Xie, et al., Org. Lett. 16 (2014) 1908–1911. doi: 10.1021/ol5004109
Z. Xia, J. Hu, W. Xie, et al., Org. Lett. 18 (2016) 80–83. doi: 10.1021/acs.orglett.5b03303
K. Yoshida, K. Okada, H. Ueda, H. Tokuyama, Angew. Chem. Int. Ed. 59 (2020) 23089–23093. doi: 10.1002/anie.202010759
H. Wang, H. Zhong, X. Jiang, et al., Adv. Synth. Catal. 362 (2020) 5328–5362.
H.J. Long, Y.L. Li, J. Deng, et al., Org. Lett. 21 (2021) 8153–8157. doi: 10.1021/acs.orglett.1c02817
J. Lusi-Barrera, S. Rodriguez, J.L. Vicario, et al., Chem. Eur. J. 28 (2022) e202202267. doi: 10.1002/chem.202202267
X. Tan, Q. Wang, J. Sun, Nat. Commun. 14 (2023) 357–365.
G.A. Olah, J.M. Bollinger, Y.K. Mo, J.M. Brinich, J. Am. Chem. Soc. 94 (1972) 1164–1168. doi: 10.1021/ja00759a022
G. Olah, P.W. Westerman, E.G. Melby, Y.K. Mo, J. Am. Chem. Soc. 96 (1974) 3565–3573. doi: 10.1021/ja00818a033
B.K. Ohta, R.E. Hough, J.W. Schubert, Org. Lett. 9 (2007) 2317–2320. doi: 10.1021/ol070673n
X. Xiong, Y.Y. Yeung, Angew. Chem. Int. Ed. 55 (2016) 16101–16105. doi: 10.1002/anie.201607388
X. Xiong, Y.Y. Yeung, ACS Catal. 8 (2018) 4033–4043. doi: 10.1021/acscatal.8b00327
Z. Chen, Z. Wu, X. Xiong, et al., ACS Catal. 14 (2024) 14387–14398. doi: 10.1021/acscatal.4c04829
D. Qian, J. Sun, Chem. Eur. J. 25 (2019) 3740–3751. doi: 10.1002/chem.201803752
X. Chen, X. Li, W.M. He, et al., Org. Chem. Front. 11 (2024) 5553–5557. doi: 10.1039/d4qo01068j
Q. Xie, H.J. Long, J. Deng, et al., J. Org. Chem. 85 (2020) 1882–1893. doi: 10.1021/acs.joc.9b02395
S.J. Hays, B.W. Caprathe, J.C. Jaen, et al., J. Med. Chem. 41 (1998) 1060–1067. doi: 10.1021/jm970394d
P. Zhang, E.A. Terefenko, Z. Zhang, et al., J. Med. Chem. 45 (2002) 4379–4382. doi: 10.1021/jm025555e
P. Zhang, E.A. Terefenko, J. Yardley, et al., Bioorg. Med. Chem. Lett. 12 (2002) 787–790. doi: 10.1016/S0960-894X(02)00025-2
D.S. Zinad, A. Mahal, M.R.F. Pratama, et al., Chem. Biol. Drug Des. 95 (2020) 16–47. doi: 10.1111/cbdd.13633
D. Zhao, M. Fañanás-Mastral, B.L. Feringa, et al., Chem. Sci. 5 (2014) 4216–4220. doi: 10.1039/C4SC01940G
S. Rajkumar, M. Tang, X. Yang, Angew. Chem. Int. Ed. 59 (2020) 2333–2337. doi: 10.1002/anie.201913896
R.F. Cao, L. Yu, Z.M. Chen, et al., Org. Lett. 24 (2022) 4093–4098. doi: 10.1021/acs.orglett.2c01731
C. Li, X. Xiang, X.Q. Dong, et al., Org. Lett. 25 (2023) 1172–1177. doi: 10.1021/acs.orglett.3c00148
U. Hennecke, C, H. Müller, R. Fröhlich, Org. Lett. 13 (2011) 860–863. doi: 10.1021/ol1028805
S.E. Denmart, M.T. Burk, Org. Lett. 14 (2012) 256–259. doi: 10.1021/ol203033k
G.X. Li, Q.Q. Fu, Z. Tang, et al., Tetrahedron: Asymmetry 23 (2012) 245–251. doi: 10.1016/j.tetasy.2012.02.016
Z. Shen, X. Pan, X. Xie, et al., Chem. Sci. 6 (2015) 6986–6990. doi: 10.1039/C5SC02485D
T. Zheng, R. Chen, Y.Y. Yeung, et al., Chem. 9 (2023) 1255–1269. doi: 10.1016/j.chempr.2023.01.016
W.J. Moree, B.F. Li, S. Malany, et al., J. Med. Chem. 52 (2009) 5307–5310. doi: 10.1021/jm900933k
A. Albright, D. Edding, R.E. Gawley, et al., J. Org. Chem. 76 (2011) 7341–7351. doi: 10.1021/jo2012434
A. Sollsdié-Cavallo, P. Lupattelli, N.D. Blasio, et al., J. Org. Chem. 71 (2006) 9891–9894. doi: 10.1021/jo0617969

Scheme 2 Substrate scope of the catalytic chlorocyclization of anilines. Reactions were carried out with substrate 2 (0.1 mmol), catalyst 1d (5 mol%) and DCDMH (0.11 mmol) in n-hexane (4 mL) for 3 h in the absence of light at 25 ℃. The yields were isolated yields. a Catalyst 1a was used. bdr > 20:1.

Scheme 3 The functionalization of complex molecules derived from pharmaceuticals and natural products. Reactions were carried out with substrate 2 (0.1 mmol), catalyst 1d (5 mol%) and DCDMH (0.11 mmol) in n-hexane (4 mL) for 3 h in the absence of light at 25 ℃. The yields were isolated yields. a The reaction was conducted with catalyst 1b at 0 ℃.

Scheme 4 Scale-up the chlorocyclization and late-stage modification. (A) Reaction scale-up and catalyst recycling. (B) Column-free asymmetric chlorocyclization. (C) Synthetic application.

Scheme 5 Asymmetric selenocyclization of aniline. Reactions were carried out with substrate 2 (0.1 mmol), catalyst 1a (5 mol%), and NPSP (0.2 mmol) in n-hexane/CCl4 (4 mL, 2:1, v/v) for 12 h in the absence of light at 25 ℃. The yields were isolated yields.

Figure 1 The calculated reaction free energy profile and the selectivity-determining transition states (bond distances are in Å).
Table 1. Optimization of the reaction conditions.a
| ||||
| Entry | Catalyst | Solvent | Yield (%)b | ee (%)c |
| 1 | – | n-hexane | trace | – |
| 2 | 1a | n-hexane | 96 | 95 |
| 3 | 1b | n-hexane | 96 | 97 |
| 4 | 1c | n-hexane | 91 | 5 |
| 5 | 1d | n-hexane | 98 | 99 |
| 6 | 1e | n-hexane | 92 | 19 |
| 7 | 1d | CCl4 | 96 | 36 |
| 8 | 1d | Toluene | 95 | 32 |
| 9 | 1d | n-heptane | 96 | 98 |
| 10 | C1 | n-hexane | 34 | 28 |
| 11 | C2 | n-hexane | 25 | 21 |
| 12 | C3 | n-hexane | 34 | 79 |
| 13 | H8-TRIP | n-hexane | 4 | 28 |
| 14 | H8-TRIP-Na | n-hexane | 10 | 75 |
| 15 | H8-TRIP/LB1 | n-hexane | 68 | 6 |
| 16 | H8-TRIP/LB2 | n-hexane | 28 | 30 |
| 17 | H8-TRIP/LB3 | n-hexane | 55 | 86 |
| a Reactions were carried out with aniline 2a (0.05 mmol), catalyst (0.0025 mmol) and DCDMH (0.055 mmol) in the indicated solvent (2 mL) at 25 ℃ in the absence of light. b Isolated yield. c The ee values were determined by HPLC analysis on a chiral stationary phase. | ||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们


 下载:
下载: