CAS Center for Excellence in Nanoscience, CAS Key Laboratory for Biomedical Effects of Nanomaterials and Nanosafety, National Center for Nanoscience and Technology (NCNST), Beijing 100190, China
b.
Sino-Danish College, Sino-Danish Center for Education and Research, University of Chinese Academy of Sciences, Beijing 100049, China
c.
Center of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
d.
University of Chinese Academy of Sciences, Beijing 100049, China
* Corresponding author. E-mail address: wanghao@nanoctr.cn (H. Wang). 1 These authors contributed equally to this work.
Received Date:
07 December 2024 Accepted Date:
24 April 2025 Revised Date:
23 April 2025 Available Online:
15 May 2026
Abstract:
Target therapy represents a paradigm shift to a precise and personalized approach. Unlike the great success of antibody-drug conjugate (ADC) in clinical practice, peptide-drug conjugate (PDC) with good tissue penetration and drug loading capacity exhibits poor stability, quick blood clearance and cellular internalization that limit their translation. In this study, a feasible approach for constructing an in vivo self-assembling peptide-drug conjugate (sPDC) was proposed by rationally designing the combination of tumor-specific targeting peptide module, responsive self-assembling peptide module, and therapeutic drug. Two optimized sPDCs (sPDC1 and sPDC2) capable of specifically targeting human epidermal growth factor receptor 2 (HER2) on the surface of tumors were reported. sPDCs could selectively target HER2-positive tumors and effectively kill HER2 overexpressing tumor cells. In addition, weak but significant efficacy of sPDCs was also observed in HER2-negative tumors, which was likely by-stander effect due to the release of monomethyl auristatin E (MMAE) in the tumor microenvironment. Finally, in HER2-positive xenograft mouse models, sPDC1 showed superior therapeutic efficacy over the clinical HER2-targeted therapeutic agents trastuzumab and lapatinib, and roughly equivalent therapeutic efficacy compared with RC48 even in large tumor-bearing mouse models. Therefore, sPDC1 was promising to serve as a lead compound for further clinical development for oncology therapy.
Human epidermal growth factor receptor 2 (HER2), also known as ErbB2, c-erbB2 or HER2/neu, is a member of the epidermal growth factor receptor (EGFR) family of receptor tyrosine kinases, which includes HER1 (or EGFR), HER2, HER3, and HER4. HER2 is unique within this family as it lacks a natural ligand and instead initiates signaling cascades through homodimerization or heterodimerization with other EGFR family members. This process activates a range of signal transduction pathways essential for tumor cell processes such as growth, apoptosis, adhesion, migration, and differentiation [1]. HER2 is an ideal target for targeted cancer therapy due to its prevalent overexpression on the surface of cells such as breast and gastric cancers [2].
Antibody-drug conjugates (ADCs) represent a groundbreaking advancement in oncology, combining the precise targeting capability of monoclonal antibodies with the potent cytotoxic effects of small molecule drugs. Although ADCs have been successfully used in clinical settings, many challenges remain. Key challenges include limited tumor penetration due to the large size of conventional antibodies, significant systemic toxicities arising from premature payload release, and heterogeneous expression of target antigens within tumors. Complex manufacturing processes and stability issues further add to the challenges [3–5].
To overcome the ADCs' limitations, a novel approach is to replace the traditional monoclonal antibodies (mAbs) structure by functional peptides with small molecular weight, leading to the development of peptide-drug conjugates (PDCs). PDCs consist of three main components: The cell-targeting peptide, the linker, and the cytotoxic payload [6,7]. Despite conceptual similarities to ADCs, PDCs differ significantly in structure and nature [8]. The smaller size of PDCs may overcome the poor tumor penetration often observed with the larger ADCs, which enhances the ability to target and kill solid tumors more effectively [9]. Peptides in PDCs have fewer epitopes and are less likely to be recognized by the immune system as foreign entities, reducing the risk of immune responses compared to ADCs [10]. Furthermore, PDCs are metabolized by the kidneys, whereas ADCs are primarily metabolized by the liver. Hepatotoxicity is a concern with ADCs due to their non-specific uptake by the liver and reticuloendothelial system. In contrast, the rapid renal clearance of PDCs minimizes the exposure of non-target tissues to the cytotoxic payload, thereby reducing systemic toxicity [11]. PDCs also offer greater versatility in drug conjugation, enabling the attachment of various cytotoxic agents and improving the overall therapeutic index. These benefits make PDCs a kind of promising alternative to ADCs, though challenges such as poor stability, low oral bioavailability and short circulation half-life still need to be addressed [12].
Self-assembling peptide drug delivery technology has also demonstrated significant advantages in tumor-targeted therapy [13–16]. By designing specific biological stimuli in the tumor microenvironment (e.g., enzyme activities, pH changes, and reduction reactions) as well as exogenous stimuli (e.g., light, magnetic fields, and ultrasound), in situ self-assembly allows the formation of nanostructures in vivo. These nanostructures, including nanoparticles, nanofibers, and etc., ensure precise and controlled release of the drug through intelligent response properties, which not only improves drug enrichment and retention time at the tumor site, but also penetrates deeply into the tumor tissue and enhances therapeutic efficacy while minimizing systemic toxicity. Our research group has been working on the development of in vivo self-assembly peptide strategies, which has been demonstrated by many studies to be a promising approach for various diagnostic and therapeutic applications [17–22].
In this study, we employed a rational modular design to develop two in vivo self-assembly PDCs (sPDCs) targeting HER2. sPDCs consist of three modules: (ⅰ) a targeting module that ensures the precise recognition and binding of HER2 on the surface of tumor cells; (ⅱ) a self-assembling module that allows sPDCs upon binding to cellular receptors to promote the in situ formation of nanoaggregates, thereby enhancing the accumulation and retention of sPDCs at the tumor site [21,23]; and classic (ⅲ) linker and drug module, comprising a cathepsin B-sensitive linker and the chemotherapeutic agent monomethyl auristatin E (MMAE).
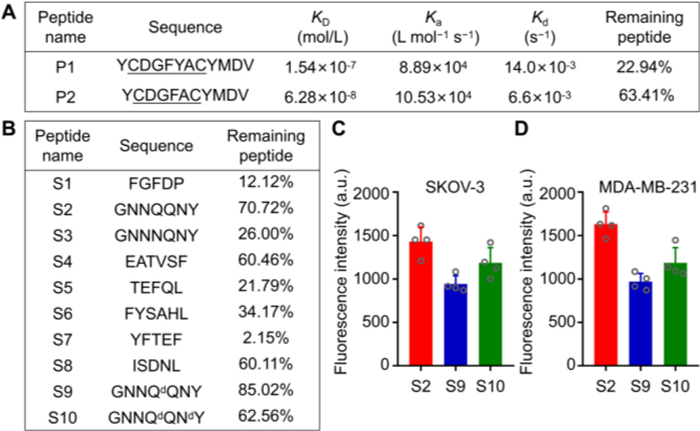
Firstly, the targeting peptides YCDGFYACYMDV (P1) and YCDGFYACYMDV (P2) capable of specifically binding to HER2 were synthesized and characterized (Figs. S1 and S2 in Supporting information) [20,24,25]. The binding affinity of the peptides to HER2 protein was assessed using surface plasmon resonance (SPR) and the results showed that P2 exhibited a higher affinity for HER2 protein (1.54 × 10−7 mol/L vs. 6.28 × 10−8 mol/L). The in vitro stability of P1 and P2 was evaluated in rat plasma at 37 ℃, and the relative amount of remaining peptides was estimated after 30 min using liquid chromatography-mass spectrometry (LC-MS). P2 showed better in vitro plasma stability than P1 (Fig. 1A). Therefore, P2 was chosen as targeting sequence for the sPDCs.
Figure 1
Figure 1.
Preparation and evaluation of targeting and self-assembling peptides. (A) The design of the targeting peptides: The binding affinity analysis to HER2 protein and in vitro stability in rat plasma for 30 min at 37 ℃. (B) The design of the self-assembling peptides: the in vitro stability in rat plasma for 30 min at 37 ℃. (C, D) The amount of cellular uptake of self-assembling peptides in HER2-positive human ovarian cancer cell lines (SKOV-3, C) and in HER2-negative breast cancer lines (MDA-MB-231, D) after 2 h at 37 ℃. Data were presented as the mean ± standard deviation (SD) (n = 4).
Secondly, eight representative peptides (S1–S8) with self-assembling capabilities were selected as the assembly modules of sPDCs (Figs. S3–S10 in Supporting information) [26–28]. The stability characteristics of self-assembling peptides were evaluated by incubating them in rat plasma at 37 ℃ for 30 min and measurements of remaining peptides. The results revealed that S2 (GNNQQNY) exhibited better in vitro plasma stability with 70.72% remaining (Fig. 1B). To further increase the stability of peptides in the blood circulation, the amino acids in the susceptible sites of S2 were replaced by d-amino acids to form GNNQdQNY (S9) and GNNQdQNdY (S10) (Figs. S11 and S12 in Supporting information) [29], and the effect of d-amino acid substitution on stability and cellular uptake as well were subsequently examined. It was found that the introduction of d-amino acid substitutions enhanced the S9's stability in rat plasma. However, d-amino acid substitution led to a decrease in cellular uptake (Figs. 1C and D, Figs. S13 and S14 in Supporting information). After comprehensively evaluating the effect of d-amino acid substitution on in vitro stability and cell entry efficiency, S2 (GNNQQNY) was ultimately chosen as the self-assembling motif for our following sPDCs construction.
Subsequently, MMAE was chosen as the payload for sPDCs due to its potent cytotoxic properties, which effectively inhibit cell division by disrupting microtubule dynamics, leading to robust apoptosis induction in cancer cells. Its excellent stability enables conjugation with peptides, ensuring targeted delivery and controlled release within the tumor microenvironment. Additionally, MMAE is hydrophobic and membrane-permeable, once released it can diffuse into neighboring tumor cells with low or heterogeneous expression of antigens to extend the killing range [30,31]. Finally, each sPDC contains a valine-citrulline dipeptide motif that ensures great stability in plasma and as well as efficient recognition and cleavage by cathepsin B, a protease found in abundance in lysosomes [32]. This targeted release mechanism is designed to release the active pharmaceutical ingredient after it has been internalized by cancer cells, maximizing therapeutic efficacy while minimizing potential off-target effects. Further considering the limitations of sPDC such as poor stability and rapid elimination, an oligoethylene glycol chain with four repeating units is introduced to sPDC to enhance its pharmacokinetic properties [33,34]. Additionally, given the crucial role of histidine in lysosomal escape by leveraging the proton sponge effect, three histidine residues (H3) were incorporated into sPDC to improve lysosomal escape [35,36]. Finally, two sPDCs (sPDC1 and sPDC2) were synthesized (Figs. S15 and S16 in Supporting information).
The efficacy of sPDC is significantly influenced by the systemic stability of the drug-peptide linkage before internalization [37]. High plasma stability is essential for maintaining therapeutic efficacy and safety, as it ensures that the drug remains intact and active in the bloodstream, reaching the target site at effective concentrations. Therefore, prior to initiating an in vivo evaluation of therapeutic effects, the stability of two sPDCs was initially assessed in mouse plasma in vitro. The results showed that sPDC1 was much more stable in mouse plasma than sPDC2 (Fig. S17 in Supporting information). The expected half-life of sPDC1 and sPDC2 in mouse plasma is 6.8 and 2.5 h, respectively.
We then initiated the tumor cell killing studies of sPDCs in HER2-positive breast cancer cell lines (HCC-1954), human ovarian cancer cell lines (SKOV-3), and HER2-negative breast cancer lines (MDA-MB-231) and non-small cell lung cancer cell lines (NCI H838) (Figs. S18A–D in Supporting information). sPDC1 and sPDC2 demonstrated significantly high cytotoxicity on HER2-positive cell lines in a dose-dependent manner, with inhibitory concentration at 50% (IC50) values of 216.0 and 256.0 nmol/L in HCC-1954 cell lines, and 240.8 and 440.0 nmol/L in SKOV-3 cell lines. In addition, sPDC1 and sPDC2 also exhibited some cytotoxic effects on HER2-negative cell lines. When sPDCs concentrations was < 250.0 nmol/L, sPDC1 and sPDC2 showed almost no cytotoxicity on MDA-MB-231 and NCI-H838 cell lines. Their cytotoxic effects became evident when the concentrations were > 250.0 nmol/L, with IC50 values of 1391.0 and 1386.0 nmol/L in MDA-MB-231 cell lines, and 2789.0 and 2153.0 nmol/L in NCI-H838 cell lines (Fig. S18E in Supporting information).
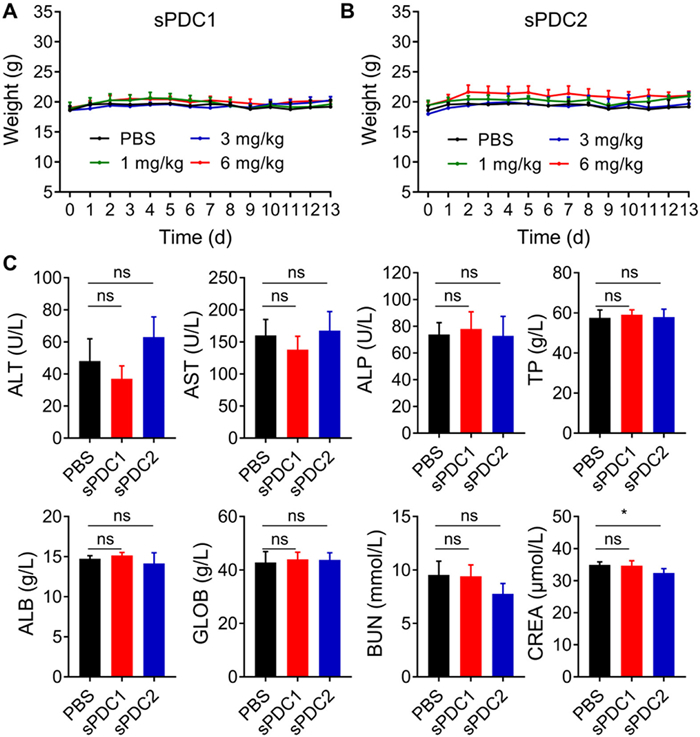
Next, toxicity assay was performed in healthy BALB/c nude mice after a single intravenous (i.v.) injection of different doses (low, medium and high doses were 1, 3 and 6 mg/kg, respectively) of sPDC1 or sPDC2 on day 0. All the animal experimental procedures conducted were approved by the Animal Research Committee at the National Center for Nanoscience and Technology, China (permit No. NCNST 21–2108–0603). The results demonstrated that sPDCs treatment did not cause weight loss in mice (Figs. 2A and B). Hematological parameters of mice from the highest dose (6 mg/kg) groups were evaluated after 2 weeks of sPDC1 or sPDC2 treatment. There were no significant changes in the blood parameters compared to the phosphate buffered saline (PBS) treated control group (Fig. 2C). Hematoxylin and eosin (H&E) staining of major organs (heart, liver, spleen, lung and kidney) also showed that there was no apparent toxicity in both sPDCs-treated experimental groups (Fig. S19 in Supporting information).
Figure 2
Figure 2.
Systemic single dose toxicity study of sPDC1 and sPDC2. Body weight analysis of healthy mice after i.v. administration of low (1 mg/kg), medium (3 mg/kg) and high (6 mg/kg) concentrations of sPDC1 (A) and sPDC2 (B) on day 0. (C) Haematological analysis of healthy mice two weeks after i.v. administration of sPDC1 (6 mg/kg) and sPDC2 (6 mg/kg). Healthy mice treated with PBS were used as the control group. Statistics significance was determined by one-way ANOVA. Data are shown as mean ± SD (n = 4). *P < 0.05. ns, not significant; ALT, alanine aminotransferase; AST, aspartate aminotransferase; ALP, alkaline phosphatase; TP, total protein; ALB, albumin; GLOB, globulin; BUN, blood urea nitrogen; CREA, creatinine.
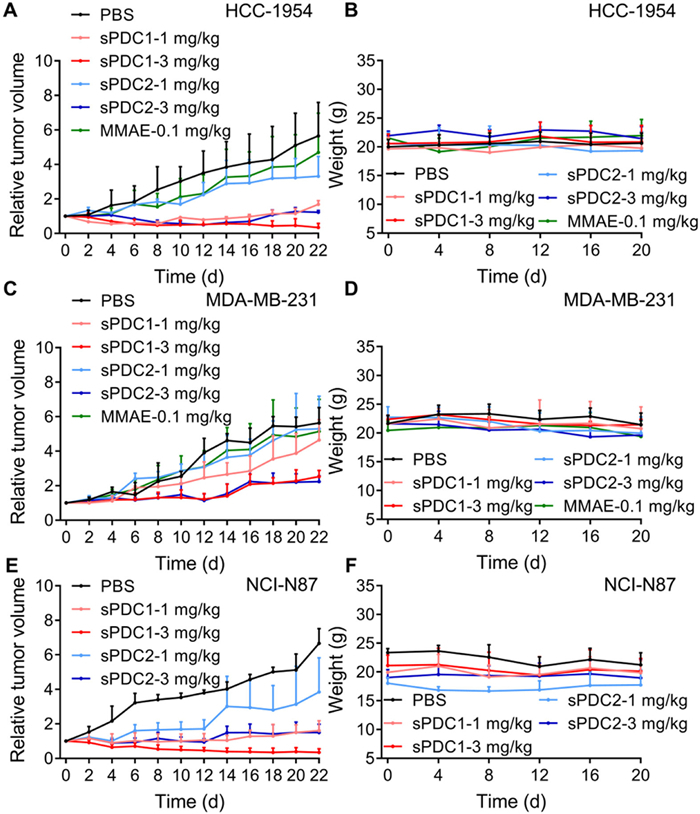
The anticancer efficacy of sPDC1 and sPDC2 were assessed in vivo using a variety of cell line-derived xenograft (CDX) tumor models. First, the HCC-1954 (HER2-positive breast cancer) and MDA-MB-231 (HER2-negative breast cancer) xenografts in BALB/c nude mice were established, and then these tumor-bearing mice were i.v. administrated with 1 or 3 mg/kg of either sPDC1 or sPDC2 every other day for a total of three treatments. Treatment with 0.1 mg/kg MMAE and PBS were used as positive and negative control groups, respectively. As depicted in Figs. 3A and B, the i.v. administration of sPDC1 at doses of 3 mg/kg resulted in significant and sustained regression of tumors in mice bearing HCC-1954 xenografts, with a tumor growth inhibition (TGI) rate of 116.5%. Although mice received only three i.v. injections of sPDC1, tumor regression was sustained over an extended observation period. In the group of 1 mg/kg sPDC1 and 3 mg/kg sPDC2, there was also observed suppression of the ongoing tumor volume growth, with the 82.2% and 95.0% TGI rate, respectively. Mice treated with 1 mg/kg sPDC2 or 0.1 mg/kg MMAE exhibited very limited tumor inhibition. The major organs (heart, liver, spleen, lung and kidney) of the mice in each group were collected for H&E staining three weeks after the first injection. Upon histological examination, no significant alterations or pathological lesions were found, indicating that sPDCs did not induce significant systemic toxicity (Fig. S20 in Supporting information).
Figure 3
Figure 3.In vivo therapeutic efficacy of sPDCs in different xenograft mouse models. Relative tumor volume (A) and weight (B) in HCC-1954 xenograft mouse models after treatment. Mice were i.v. administrated with 0.1 mg/kg MMAE, 1 or 3 mg/kg sPDC1, 1 or 3 mg/kg sPDC2 every other day for 3 times. Relative tumor volume (C) and weight (D) in MDA-MB-231 xenograft mouse models after treatment. Mice were i.v. administrated with 0.1 mg/kg MMAE, 1 or 3 mg/kg sPDC1, 1 or 3 mg/kg sPDC2 every other day for 3 times. Relative tumor volume (E) and weight (F) in NCI-N87 xenograft mouse models after treatment. Mice were i.v. administrated with 1 or 3 mg/kg sPDC1, 1 or 3 mg/kg sPDC2 every other day for 3 times. Mice treated with PBS were used as the control group. All treatments were initiated on day 0. Data are shown as mean ± SD (n = 3).
In the MDA-MB-231 xenografts, i.v. administration of 1 mg/kg sPDC1 or sPDC2 resulted in a very limited inhibition of tumor volume in mice. Treatment with 3 mg/kg of sPDC1 or sPDC2 were still effectively inhibited tumor growth compared to the PBS control group. The TGI rate was 72.3% and 71.1%, respectively (Figs. 3C and D). The inhibitory effect of sPDCs on HER2-negative tumors may be attributed to its cleavable linker, which, under tumor-relevant pH conditions, is efficiently cleaved by cathepsin B secreted by tumor cells into the extracellular environment, thereby releasing the active MMAE toxin [38]. H&E staining of major organs revealed that the mice treated with 3 mg/kg sPDC1, 1 mg/kg sPDC2, or 3 mg/kg sPDC2 exhibited splenic atrophy characterized by the degeneration of the white pulp three weeks later, but no related pathological changes were observed in the heart, liver, lung or kidney (Fig. S21 in Supporting information). The lack of specific targets in the HER2-negative tumor models may alter the distribution and metabolism of sPDCs, leading to nonspecific effects on the spleen. In contrast, the sPDCs exhibited good safety in HER2-positive tumor mice model, probably due to the high selectivity and affinity to HER2, which reduces nonspecific binding and toxicity to normal tissues.
To test whether the sPDCs are effective in inhibiting different types of tumors, in next therapeutic efficacy study of sPDCs, mice bearing NCI-N87 xenografts (HER2-positive gastric cancer) were i.v. administrated with sPDC1 or sPDC2 at doses of 1 or 3 mg/kg, respectively, every other day for a total of three times. All experimental groups demonstrated a significant capacity to delay or inhibit the growth of tumor. Notably, the 3 mg/kg dose of sPDC1 exhibited a sustained and pronounced inhibition of tumor growth with a 110.1% inhibition rate (Figs. 3E and F). The above in vivo studies revealed that sPDC1 showed higher efficacy than that of sPDC2, probably due to the high plasma stability, which was utilized for further in vivo evaluation.
Given the effective tumor suppression observed with 3 mg/kg of sPDC1 in HER2-positive cancer both HCC-1954 and NCI-N87 xenografts, we tried to optimize the dosing frequency by study the antitumor efficacy of various dosing frequencies at this dose. Mice receiving 3 mg/kg of sPDC1 via i.v. injection either every other day or every 3 days for 3 times demonstrated comparable tumor suppression efficacy, both of which were superior to that of mice receiving injections every 7 days for 3 times (Fig. S22A in Supporting information). Moreover, more multiple i.v. administrations either every other day for 12 times or every 3 days for 8 times to the mice led to a near-total eradication of the tumors, but were accompanied by a 15.0% and 12.2% reduction in body weight, respectively (Fig. S22B in Supporting information). No significant weight loss was observed in the other experimental groups. Based on the above experimental results, the i.v. administration of 3 mg/kg for once every 3 days and a total of 3 times exhibited the balance of efficacy and toxicity.
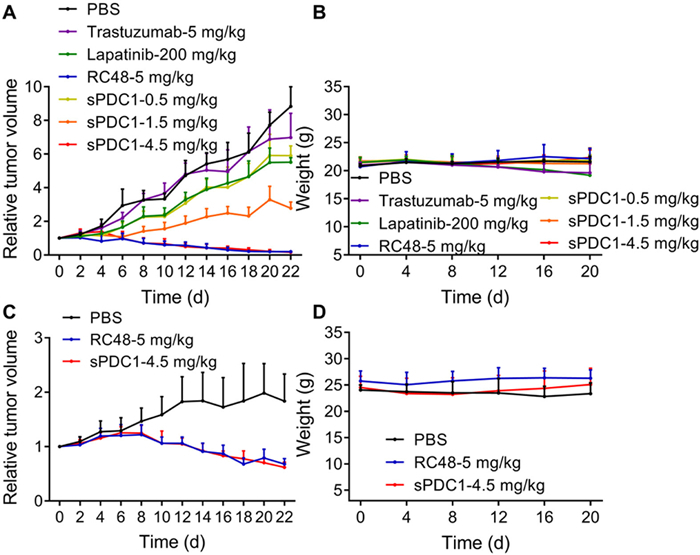
To further validate the preclinical therapeutic efficacy of sPDC1, a comparative analysis was conducted between multiple doses (0.5, 1.5 and 4.5 mg/kg) of sPDC1 and the clinical HER2-targeting agents trastuzumab (5 mg/kg), lapatinib (200 mg/kg) and RC48 (5 mg/kg) in NCI-N87 xenograft mice (Fig. 4A). It was found that the administration of 1.5 mg/kg sPDC1 effectively suppressed tumor growth, outperforming the inhibitory effects of trastuzumab (5 mg/kg), and lapatinib (200 mg/kg) on the tumors. Furthermore, the treatment of 4.5 mg/kg of sPDC1 demonstrated significantly superior efficacy compared to trastuzumab (5 mg/kg) and lapatinib (200 mg/kg), which was equivalent to that of the marketed ADC, RC48. The potential toxicity was assessed by analyzing variations in the body weight of mice (Fig. 4B), and the results exhibited that the weight loss in trastuzumab and lapatinib groups, while no weight loss was found in sPDC1 groups. Moreover, the inhibitory effect of sPDC1 was validated on large-volume tumors (tumor volume at the start of treatment is approximately 700–800 mm3) in NCI-N87 xenograft mice. The potency of sPDC1, administered at a dose of 4.5 mg/kg every 3 days for 3 times, was found to be comparable to that of the commercially available drug, RC48 in inhibiting tumor growth (Figs. 4C and D).
Figure 4
Figure 4.In vivo efficacy comparison of sPDC1 with clinical HER2 targeted agents in NCI-N87 xenograft mouse models. Relative tumor volume (A) and weight (B) in NCI-N87 xenograft mouse models after treatment (tumor volume at the start of treatment is approximately 100 mm3). Mice were i.v. administrated with 5 mg/kg trastuzumab every 7 days for 3 times, 5 mg/kg RC48 every 7 days for 3 times, 0.5 mg/kg sPDC1 every 3 days for 3 times, 1.5 mg/kg sPDC1 every 3 days for 3 times, 4.5 mg/kg sPDC1 every 3 days for 3 times, and 200 mg/kg lapatinib was given by gavage daily for 22 days. Relative tumor volume (C) and weight (D) in NCI-N87 xenograft mouse models after treatment (tumor volume at the start of treatment is approximately 700–800 mm3). Mice were i.v. administrated with 5 mg/kg RC48 every 7 days for 3 times, 4.5 mg/kg sPDC1 every 3 days for 3 times. Mice treated with PBS were used as the control group. All treatments were initiated on day 0. Data are shown as mean ± SD (n = 6).
In summary, we designed two HER2-targeting sPDCs, which selectively targeted HER2-positive tumors and exhibited significant therapeutic efficacy in both in vitro and in vivo experiments. In addition, sPDCs also exhibited weak but significant efficacy in HER2-negative tumors, which was probably due to the extracellular release of MMAE caused by the cleavage of the linker by cathepsin B secreted by tumor cells. Furthermore, in HER2-positive xenograft tumor models, sPDC1 showed superior therapeutic efficacy compared with the clinical HER2-targeted therapeutic agents trastuzumab and lapatinib, and roughly equivalent therapeutic efficacy compared with RC48. Finally, the sPDC1 maintained tumor suppression effects similar to RC48 even in large tumors. These findings exhibited the potential of sPDC1 as a lead compound for further clinical development, providing a promising new option for HER2-targeted therapy.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDC0290200), National Key R&D Program of China (No. 2022YFA1205700) and Noncommunicable Chronic Diseases-National Science and Technology Major Project (No. 2023ZD0508000).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.111257.
M.M.C. Van Der Lee, P.G. Groothuis, R. Ubink, et al., Mol. Cancer Ther. 14 (2015) 692–703.
Figure 1
Preparation and evaluation of targeting and self-assembling peptides. (A) The design of the targeting peptides: The binding affinity analysis to HER2 protein and in vitro stability in rat plasma for 30 min at 37 ℃. (B) The design of the self-assembling peptides: the in vitro stability in rat plasma for 30 min at 37 ℃. (C, D) The amount of cellular uptake of self-assembling peptides in HER2-positive human ovarian cancer cell lines (SKOV-3, C) and in HER2-negative breast cancer lines (MDA-MB-231, D) after 2 h at 37 ℃. Data were presented as the mean ± standard deviation (SD) (n = 4).
Figure 2
Systemic single dose toxicity study of sPDC1 and sPDC2. Body weight analysis of healthy mice after i.v. administration of low (1 mg/kg), medium (3 mg/kg) and high (6 mg/kg) concentrations of sPDC1 (A) and sPDC2 (B) on day 0. (C) Haematological analysis of healthy mice two weeks after i.v. administration of sPDC1 (6 mg/kg) and sPDC2 (6 mg/kg). Healthy mice treated with PBS were used as the control group. Statistics significance was determined by one-way ANOVA. Data are shown as mean ± SD (n = 4). *P < 0.05. ns, not significant; ALT, alanine aminotransferase; AST, aspartate aminotransferase; ALP, alkaline phosphatase; TP, total protein; ALB, albumin; GLOB, globulin; BUN, blood urea nitrogen; CREA, creatinine.
Figure 3In vivo therapeutic efficacy of sPDCs in different xenograft mouse models. Relative tumor volume (A) and weight (B) in HCC-1954 xenograft mouse models after treatment. Mice were i.v. administrated with 0.1 mg/kg MMAE, 1 or 3 mg/kg sPDC1, 1 or 3 mg/kg sPDC2 every other day for 3 times. Relative tumor volume (C) and weight (D) in MDA-MB-231 xenograft mouse models after treatment. Mice were i.v. administrated with 0.1 mg/kg MMAE, 1 or 3 mg/kg sPDC1, 1 or 3 mg/kg sPDC2 every other day for 3 times. Relative tumor volume (E) and weight (F) in NCI-N87 xenograft mouse models after treatment. Mice were i.v. administrated with 1 or 3 mg/kg sPDC1, 1 or 3 mg/kg sPDC2 every other day for 3 times. Mice treated with PBS were used as the control group. All treatments were initiated on day 0. Data are shown as mean ± SD (n = 3).
Figure 4In vivo efficacy comparison of sPDC1 with clinical HER2 targeted agents in NCI-N87 xenograft mouse models. Relative tumor volume (A) and weight (B) in NCI-N87 xenograft mouse models after treatment (tumor volume at the start of treatment is approximately 100 mm3). Mice were i.v. administrated with 5 mg/kg trastuzumab every 7 days for 3 times, 5 mg/kg RC48 every 7 days for 3 times, 0.5 mg/kg sPDC1 every 3 days for 3 times, 1.5 mg/kg sPDC1 every 3 days for 3 times, 4.5 mg/kg sPDC1 every 3 days for 3 times, and 200 mg/kg lapatinib was given by gavage daily for 22 days. Relative tumor volume (C) and weight (D) in NCI-N87 xenograft mouse models after treatment (tumor volume at the start of treatment is approximately 700–800 mm3). Mice were i.v. administrated with 5 mg/kg RC48 every 7 days for 3 times, 4.5 mg/kg sPDC1 every 3 days for 3 times. Mice treated with PBS were used as the control group. All treatments were initiated on day 0. Data are shown as mean ± SD (n = 6).

 DownLoad:
DownLoad:

 下载:
下载:





 下载:
下载:
