zmsu@nenu.edu.cn (Z. Su). 1 These authors contributed equally to this work.
Received Date:
05 December 2024 Accepted Date:
22 April 2025 Revised Date:
26 February 2025 Available Online:
15 January 2026
Abstract:
Integration of single-atom catalysts (SACs) onto metal-organic frameworks (MOFs) with porous channels has garnered significant interest in the field of CO2 reduction. However, MOFs are usually bulky can impede the diffusion of intermediates with substrates and maximizing catalytic site utilization remains a challenge. In this study, we utilized firstly the post-synthetic single-atom chelation sites on zirconium-based metal-organic cages (Zr-MOCs) to anchor cobalt (Co) atom to synthesize single-dispersible ZrT-1-NH2-IS-Co molecular cages for CO2 photoreduction. Experimental results demonstrate that ZrT-1-NH2-IS-Co exhibits impressive catalytic performance, achieving syngas yields of up to 30.9 mmol g-1 h-1, ranking among the highest values of reported crystalline porous catalysts. Mechanistic insights reveal the newly introduced metal serving as the catalytic site and *COOH acts as a crucial intermediate in the CO2 reduction process. Furthermore, the successful synthesis of ZrT-1-NH2-IS-Ni and ZrT-1-NH2-IS-Mn show the universality of the modification strategies, with their CO2 catalytic activity surpassing that of ZrT-1-NH2.
Photocatalytic conversion of carbon dioxide (CO2) into valuable chemicals and fuels is widely recognized as a sustainable approach to achieving carbon neutrality [1–6]. However, the inherent thermodynamic and kinetic stability of CO2 molecules poses a significant obstacle to their efficient reduction [7–9]. In response to this challenge, numerous research groups have dedicated efforts to designing effective catalysts for CO2 reduction. While many efficient photocatalysts have been successfully developed, the majority are still constrained by key factors such as limited catalytic unit activity, low utilization of catalytic sites, and poorly defined catalytic centers [10–13]. Therefore, there is a pressing need to develop catalysts that exhibit high activity, optimal utilization of catalytic sites, and well-defined catalytic centers.
Single-atom catalysts (SACs) are highly regarded among catalysts for their unique features, such as fully exposing active sites, efficiently utilizing metal atoms, and facilitating light-induced electron transport [14–20]. The integration of SACs into metal-organic frameworks (MOFs) has garnered significant interest in the realm of CO2 reduction. This interest stems from their porous channels can provide shelter for CO2 molecules, as well as the well-defined structures allow a better study of structure-activity relationships [21,22]. However, they are usually bulky can impede the diffusion of intermediates with substrates, potentially impacting catalytic site utilization [23–27]. Exploring the incorporation of SACs into low-dimensional porous metal-organic materials shows promise. Metal-organic cages (MOCs) are particularly appealing due to their structural resemblance to MOFs and the presence of capping groups that prevent extensive network formation, resulting in low-dimensional metal-organic materials [28,29]. Thanks to their excellent dispersibility in solvents, MOCs have emerged as a prominent player in catalysis in recent years [30,31]. This unique property allows them to combine the benefits of homogeneous materials (e.g., easy access to active sites) with those of heterogeneous materials (e.g., recyclability). Consequently, the exploration of SACs based on metal-organic cages is significant and valuable for CO2 reduction. However, the related research is still in its infancy.
Herein, we successfully synthesize SAC (ZrT-1-NH2-IS-Co) by utilizing the post-synthetic single-atom chelation sites on Zr-MOCs to anchor Co, and explored their performance in CO2 reduction. Photoelectrochemical analyses reveal that the introduction of single-metal enhances electron transport and charge separation. Catalytic experiments show the incorporation of single-atom resulted in a significant improvement in the photoreduction of CO2 by Zr-MOCs. Notably, ZrT-1-NH2-IS-Co exhibits the highest yield of 30.9 mmol g-1 h-1 for syngas production. The mechanistic studies indicate that the newly introduced metal served as catalytic sites, with *COOH identified as a key intermediate in the CO2 reduction process. Furthermore, the successful introduction of Ni and Mn metals into Zr-MOCs demonstrated the universality of the synthesis strategy. This work offers valuable insights for the design of SACs on MOC catalysts aimed at efficient CO2 conversion.
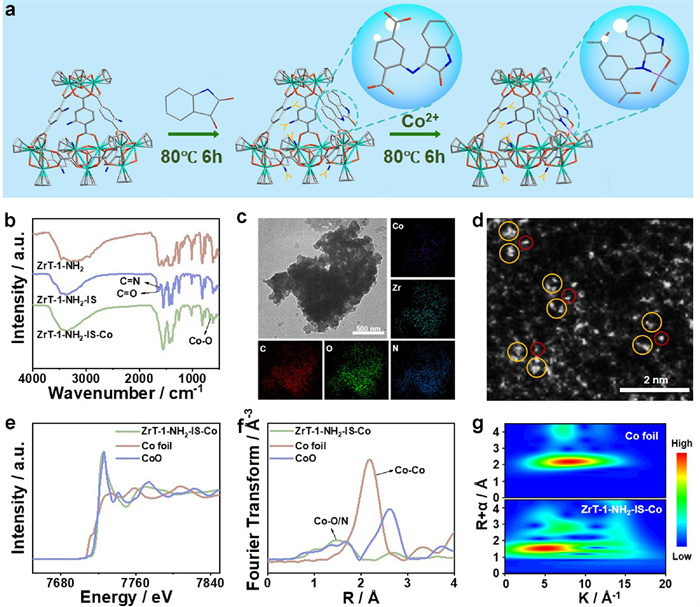
ZrT-1-NH2-IS-Co was synthesized via a stepwise approach, and the detailed synthesis method is shown in the experimental section. First, a zirconium-oxygen-cluster nanocage (ZrT-1-NH2) containing an amino group was synthesized by a simple solvothermal method from zirconocene dichloride and 2-aminoterephthalic acid (Fig. S1 in Supporting information) [32]. Then, ZrT-1-NH2 and isatin were synthesized as zirconium-oxygen-cluster nanocages with heterometallic coordination sites (ZrT-1-NH2-IS) via Schiff base reaction. Finally, a novel zirconium-oxygen-cluster nanocage, named ZrT-1-NH2-IS-Co, was synthesized by introducing metal cobalt (Co) (Fig. 1a).
Figure 1
Figure 1.
(a) Schematic of the synthesis of ZrT-1-NH2-IS-Co. (b) FT-IR spectra of ZrT-1-NH2, ZrT-1-NH2-IS and ZrT-1-NH2-IS-Co. (c) TEM and mapping images of ZrT-1-NH2-IS-Co. (d) HAADF-STEM image of ZrT-1-NH2-IS-Co. (e) Normalized Co K-edge XANES spectrum of ZrT-1-NH2-IS-Co. (f) k3-weighted FT-EXAFS spectrum of ZrT-1-NH2-IS-Co. (g) The wavelet transform of ZrT-1-NH2-IS-Co.
The crystal structure of ZrT-1-NH2 was examined using single-crystal X-ray diffraction (SC-XRD), and the result indicates the successful synthesis of zirconium-oxygen-cluster nanocages. The powder X-ray diffraction (PXRD) pattern demonstrates that the peaks of ZrT-1-NH2 match the simulated peaks extremely well, suggesting the high crystallinity and purity of the sample (Fig. S2 in Supporting information). Successful preparation of ZrT-1-NH2-IS can be confirmed by Fourier-transform infrared (FT-IR) spectroscopy. Compared with ZrT-1-NH2, the new peaks at 1660 cm-1 and 1626 cm-1 are attributed to C=O and C=N bonds in ZrT-1-NH2-IS, confirming the satisfactory synthesis of ZrT-1-NH2-IS (Fig. 1b). The successful synthesis of ZrT-1-NH2-IS was further validated through proton nuclear magnetic resonance (1H NMR) and mass spectrometry (MS) (Figs. S3-S6 in Supporting information) [32]. Mass spectrometry suggested the potential introduction of five isatin molecules into ZrT-1-NH2. When Co is introduced, both C=O and C=N bonds almost disappear and the new peak belonging to a characteristic peak of Co-O bonding at 669 cm-1 appears, indicating the successful anchor of the second metal (Fig. 1b) [33–35]. Moreover, the uniform distribution of Co and Zr elements on transmission electron microscopy (TEM) mapping images further demonstrates the incorporation of Co into Zr-MOCs (Fig. 1c). X-ray photoelectron spectroscopy (XPS) spectra were employed to assess the coordination environment of the elements. The XPS spectra of C 1s, O 1s, and Zr 3d in ZrT-1-NH2-IS-Co exhibit negligible changes compared to ZrT-1-NH2, indicating that the post-synthesis modifications in ZrT-1-NH2 did not alter the structure of the original Zr-MOC (Figs. S7 and S8 in Supporting information). The XPS spectrum of Co 2p, which displays peaks at 781.7 and 797.7 eV, is attributed to CoⅡ peaks, suggesting that cobalt exists in a single-atom state (Fig. S8c) [36,37]. In addition, high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image observes the presence of single atoms (Fig. 1d). X-ray absorption fine structure spectral (XAFS) was employed to fully characterize the electronic state and coordination environment of Co within the cage. The X-ray absorption near-edge spectroscopy (XANES) analysis of ZrT-1-NH2-IS-Co shows that Co is mainly present as CoⅡ, which is assigned to the closer proximity of the XANES spectrum to CoO (Fig. 1e). Furthermore, the absence of the Co-Co bond peak (2.18 Å) in the extended X-ray absorption fine structure spectrum (EXAFS) and the characteristic peak (1.51 Å) is located near the Co-O (1.68 Å), which is attributed to the Co-O/N bond. These indicate that Co exists in a single-atom state (Fig. 1f). The wavelet transform (WT) diagram of ZrT-1-NH2-IS-Co demonstrates a WT intensity maximum of about 5.0 Å-1 (Co-O/N), further proving the existence of Co single atom (Fig. 1g) [38].
Thermogravimetric analysis (TGA) of ZrT-1-NH2, ZrT-1-NH2-IS and ZrT-1-NH2-IS-Co reveals the first weight loss at 170 ℃, which can be attributed to the loss of solvent molecules in the sample pores, and remains good from 170 ℃ to 380 ℃, beyond which there is a significant weight loss and the skeleton of the sample begins to collapse. The results imply that ZrT-1-NH2-IS and ZrT-1-NH2-IS-Co retained the favorable thermal stability of ZrT-1-NH2 (Fig. S9 in Supporting information).
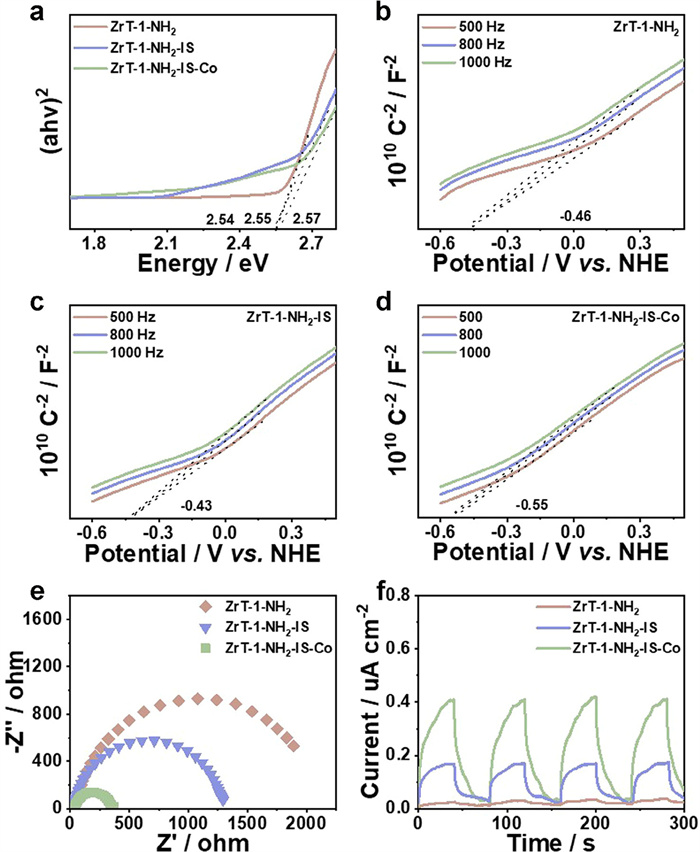
As shown in Fig. S10 (Supporting information), the ultraviolet-visible (UV–vis) diffuse reflectance spectra of ZrT-1-NH2, ZrT-1-NH2-IS and ZrT-1-NH2-IS-Co absorbed in both the UV and visible regions. Furthermore, the broad absorption band of ZrT-1-NH2-IS-Co in the range of 500–750 nm may originate from the 3d-4d electron transitions of Co [4]. The band gaps of materials were investigated by UV–vis diffuse reflectance spectroscopy (DRS) based on the Tauc plot equation. The band gap energies (Eg) of ZrT-1-NH2, ZrT-1-NH2-IS and ZrT-1-NH2-IS-Co were assessed to be 2.55, 2.54 and 2.57 eV, respectively, demonstrating typical semiconductor properties (Fig. 2a) [39]. The Mott-Schottky (MS) plots were used to speculate on the energy level structure of MOCs. The flat-band (FB) potentials of ZrT-1-NH2, ZrT-1-NH2-IS and ZrT-1-NH2-IS-Co are −0.46, −0.43 and −0.55 V vs. normal hydrogen electrode (NHE) (Figs. 2b-d). At the same time, the materials are all n-type semiconductors and are confirmed due to the positive slope of the MS curve. For n-type semiconductors, the conduction band (CB) is 0.1 V lower relative to FB, therefore the conduction band (CB) of ZrT-1-NH2, ZrT-1-NH2-IS and ZrT-1-NH2-IS-Co were −0.56, −0.53 and −0.65 V vs. NHE [40]. According to the formula ECB = EVB - Eg, the valence band (VB) potentials of ZrT-1-NH2, ZrT-1-NH2-IS and ZrT-1-NH2-IS-Co were obtained as 1.99, 2.01 and 1.92 V vs. NHE. The energy level structures of the materials suggest that ZrT-1-NH2-IS-Co can reduce CO2 to prepare syngas which is consistent with the catalytic results (CO2-CO = −0.53 V vs. NHE, H+-H2 = −0.42 V vs. NHE) (Fig. S11 in Supporting information) [41].
Figure 2
Figure 2.
(a) Tauc plots of ZrT-1-NH2, ZrT-1-NH2-IS and ZrT-1-NH2-IS-Co. Mott-Schottky plots of ZrT-1-NH2 (b), ZrT-1-NH2-IS (c) and ZrT-1-NH2-IS-Co (d). (e) EIS Nyquist plots of ZrT-1-NH2, ZrT-1-NH2-IS and ZrT-1-NH2-IS-Co. (f) Transient photocurrent response of ZrT-1-NH2, ZrT-1-NH2-IS and ZrT-1-NH2-IS-Co.
It is well known that the photogenerated charge transfer and separation efficiencies of the samples have a crucial impact on the photocatalytic efficiency, therefore electrochemical impedance spectroscopy (EIS) and photocurrent experiments were performed. Compared with ZrT-1-NH2 and ZrT-1-NH2-IS, ZrT-1-NH2-IS-Co demonstrates the smallest charge transport resistance, indicating the highest charge separation efficiency (Fig. 2e) [42]. Photocurrent experiments reveal that ZrT-1-NH2-IS-Co has higher current density, which implies enhanced spatial separation of photogenerated charge carriers within the material (Fig. 2f). These findings suggest that Co introduction facilitates charge separation and photoelectron transport.
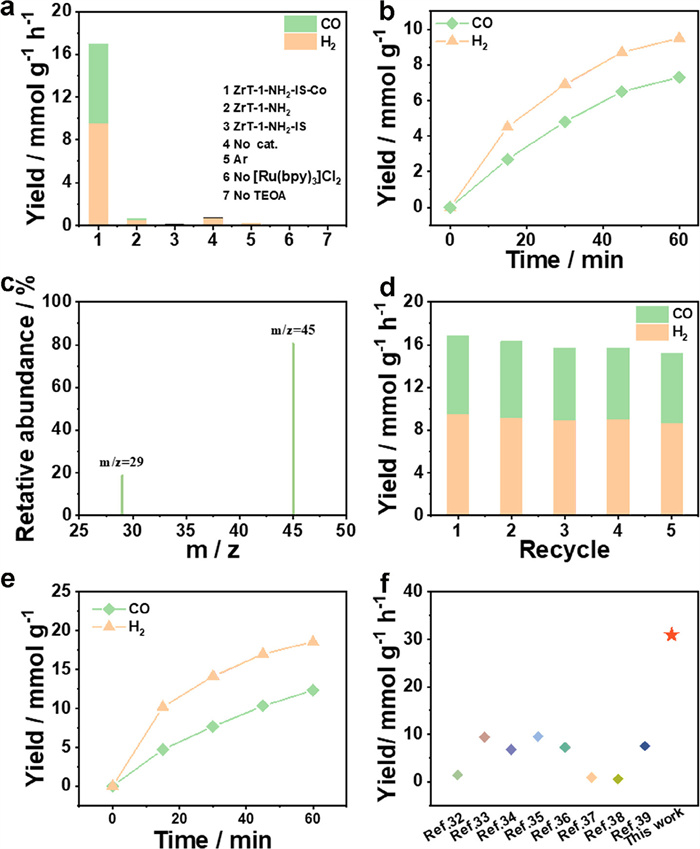
To investigate the photocatalytic performance of ZrT-1-NH2-IS-Co, photocatalytic CO2 reduction experiments were executed in a 10 mL reactor (CO2-saturated). Sample, Ru(bpy)3Cl2 as a photosensitizer, and TEA/TEOA as a sacrificial reagent was dispersed in a reactor with MeCN, and then irradiated by applying a 300 W xenon lamp at 420 nm for 1 h. To determine the optimum catalytic system, the type and quantity of sacrificial reagents and the ratio of dispersant to sacrificial reagents were optimized (Figs. S12 and S13 in Supporting information). Finally, 1 mg catalyst, 2 mL TEOA, 7 mg Ru(bpy)3Cl2 and 4 mL MeCN were used as the optimal catalytic system for further study. Gaseous products are detected by GC, and liquid products are detected by IC. As a control, the photocatalytic properties of ZrT-1-NH2 and ZrT-1-NH2-IS were investigated under the same reaction conditions. In Fig. 3a, compared to ZrT-1-NH2 and ZrT-1-NH2-IS, which have almost no CO2 reduction activity, ZrT-1-NH2-IS-Co exhibits excellent catalytic activity for the preparation of syngas (7.3 mmol g-1 h-1 CO yield and 9.56 mmol g-1 h-1 H2 yield), suggesting that the newly introduced Co serves as catalytic sites for the photocatalytic reduction of CO2 to syngas.
Figure 3
Figure 3.
(a) Photocatalytic performances under different reaction conditions. (b) Trend of CO2 reduction of ZrT-1-NH2-IS-Co over time. (c) The result of the isotopic experiment. (d) Reproducibility experiment of ZrT-1-NH2-IS-Co. (e) Trend in CO2 reduction of monodisperse ZrT-1-NH2-IS-Co over time. (f) The syngas yields compared with other reported literature.
In the absence of photosensitizers, light and catalysts, the carbon products are almost negligible, proving that the catalytic process is photosensitizing and the catalytic site is on the catalyst (Fig. 3a). As illustrated in Fig. 3b, the CO concentration exhibited a time-dependent trend during the first 60 min of the reaction. However, the CO concentration slowly increased above 45 min, and the combined catalysts maintained effective catalytic activity even after several cycling experiments, which may be due to the deactivation of the photosensitizer.
Comparative experiment under argon gas (Ar) atmosphere and isotope labeling experiment were performed in order to evidence that CO originates from the photocatalytic reduction of CO2. The reduction products were analyzed by gas chromatography-mass spectrometry (GC−MS). There is negligible CO production in the Ar control experiment, suggesting that CO originates from CO2 reduction (Fig. 3a). At the same time, the 13CO signal (m/z = 29) was observed in the isotope experiment, further confirming that the carbon origin of CO (Fig. 3c).
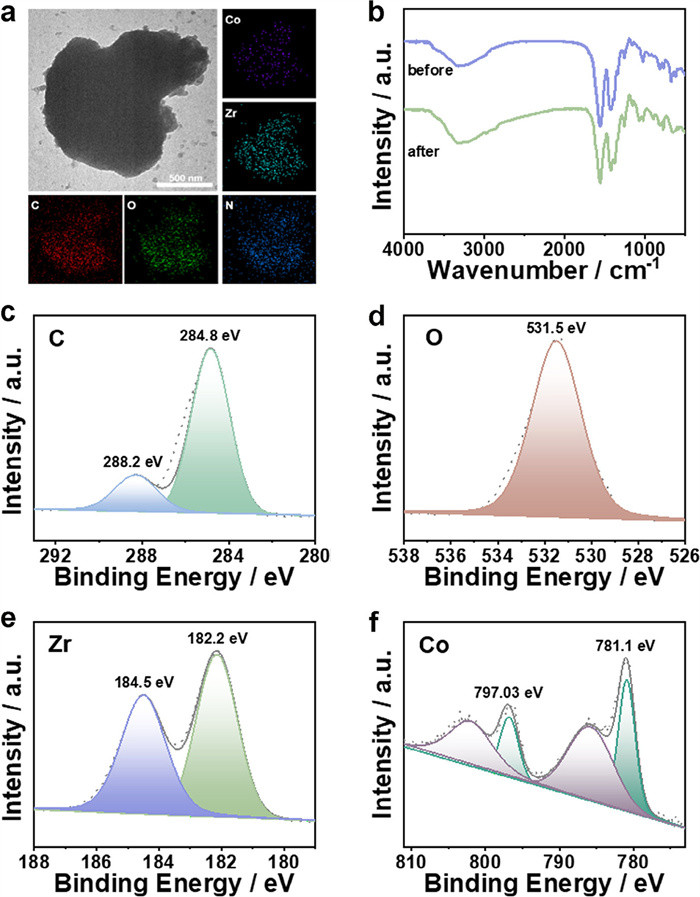
The recoverability and stability of catalysts are important parameters considered for industrial applications, so cycling experiments were executed to examine the stability of ZrT-1-NH2-IS-Co. After five consecutive cycling experiments, ZrT-1-NH2-IS-Co remained appealing catalytic activity (Fig. 3d). In addition, TEM, FT-IR and XPS tests were conducted to characterize the post-catalytic sample. The TEM and mapping plots indicate that the Co and Zr distribution in the material remained uniform, and the morphology of the reacted ZrT-1-NH2-IS-Co samples remained unchanged, demonstrating the high stability of ZrT-1-NH2-IS-Co (Fig. 4a). The post-catalytic FT-IR spectrum and XPS spectra had negligible changes, further confirming the durability of the catalysts (Figs. 4b-f).
Figure 4
Figure 4.
(a) Post-catalytic TEM and mapping images of ZrT-1-NH2-IS-Co. (b) Pre-catalytic and post-catalytic FT-IR spectra of ZrT-1-NH2-IS-Co. Post-catalytic XPS of ZrT-1-NH2-IS-Co (c) C 1s, (d) O 1s, (e) Zr 3d, (f) Co 2p.
The outstanding dispersion of ZrT-1-NH2-IS-Co in MeOH offers an attractive platform to study the effect of single ZrT-1-NH2-IS-Co cage on the activity of photocatalytic CO2 reduction [23]. Experiments were performed in a 6 mL system (2 mL TEOA, 2 mL MeCN and 2 mL MeOH). The ultraviolet-visible diffuse reflectance spectra exhibited almost identical strong of ZrT-1-NH2-IS-Co in MeCN and MeCN/MeOH, indicating that the catalyst is stable in MeOH solution. The catalytic results reveal more efficient catalytic activity with a syngas yield of 30.9 mmol g-1 h-1, which is nearly twice the yield of the directly dispersed catalyst. The ZrT-1-NH2-IS-Co has been significantly greater than that of most crystalline porous photocatalysts, which can be assigned to the introduction of single-atom sites in the molecular cage and the monodispersion of the catalyst in the solvent exposing more catalytically active sites (Figs. 3e and f and Figs. S14 and S15 in Supporting information) [43–50].
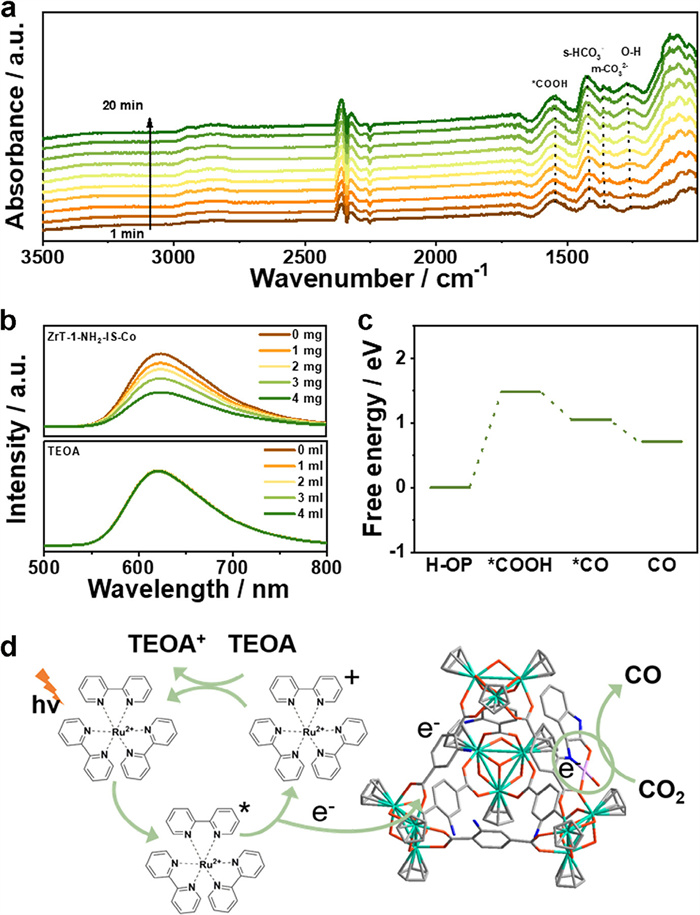
To better reveal the key intermediates in the photocatalytic CO2 reduction process, the in-situ Fourier-transform infrared (FT-IR) test was executed. The catalytic system was in a CO2-saturated atmosphere, and when light was introduced, obvious new absorption peaks were generated, which enhanced with time. The peak at 1545 cm-1 is attributed to *COOH, implying that *COOH is a key intermediate in the conversion of CO2 to CO (Fig. 5a) [33].
Figure 5
Figure 5.
(a) In situ infrared spectrum of CO2 reduction on ZrT-1-NH2-IS-Co. (b) Emission spectra of [Ru(bpy)3]2+ in the varying amounts of ZrT-1-NH2-IS-Co and TEOA. (c) Gibbs free energy diagram for CO2 reduction of ZrT-1-NH2-IS-Co. (d) Possible mechanisms of ZrT-1-NH2-IS-Co for photocatalytic CO2 reduction.
To deeply explore the photocatalytic mechanism, electron transfer was first evaluated during CO2RR. Various quantities of ZrT-1-NH2-IS-Co and TEOA were added to the acetonitrile solution with Ru(bpy)3Cl2. The fluorescence intensity gradually becomes weaker with the addition of different masses of ZrT-1-NH2-IS-Co. Differently, as the amount of TEOA increased, the fluorescence intensity remained essentially unchanged (Fig. 5b). These findings verify that the excitation electrons from Ru(bpy)3Cl2 are transferred straight to ZrT-1-NH2-IS-Co rather than to TEOA, which suggests the existence of an oxidative burst mechanism between ZrT-1-NH2-IS-Co and Ru(bpy)3Cl2 [51]. Based on the above studies, the lowest unoccupied molecular orbital (LUMO) energy levels and the highest occupied molecular orbital (HOMO) of Ru(bpy)3Cl2 are −1.25 V and 1.24 V, respectively. It is noteworthy that the conduction-band (CB) potential of the ZrT-1-NH2-IS-Co is −0.65 V, which is lower than the LUMO energy level of Ru(bpy)3Cl2, suggesting that the excitation electrons from Ru(bpy)3Cl2 can be efficiently transferred to the catalyst to trigger the CO2 and H+ reduction (Fig. S16 in Supporting information).
Density functional theory (DFT) was further employed for the evaluation of catalytic sites. Previous work by Qi et al. has demonstrated that Zr in Zr-MOC does not serve as a catalytic center for CO2 reduction, thus, we focused on the evaluation of the anchored Co site (Fig. S17 in Supporting information) [23]. The Gibbs free energies of the CO2 reduction intermediates are shown in Fig. 5c. The formation of CO2-*COOH is a rate-limiting step in the catalytic reaction, whereas *COOH-*CO and the subsequent desorption of *CO to produce CO are exothermic processes [37]. DFT calculation suggests that the Co site anchored within Zr-MOC can function as an effective catalytic site for CO2 reduction. Moreover, when combined with data from catalytic experiments, it is evident that Co on the Zr-MOC cage acts as the catalytic center. Therefore, a plausible catalytic mechanism is proposed in which Ru(bpy)3Cl2 is excited by light irradiation to produce Ru(bpy)32+. In the process, electrons are transferred from Ru(bpy)3Cl2 to Co, and CO2 and H+ are reduced to CO and H2. Finally, Ru(bpy)33+ is reduced to Ru(bpy)32+ by TEOA, completing the whole cycle (Fig. 5d).
To illuminate the universality of the post-synthetic strategy in single-atom anchor on zirconium-MOC, Ni and Mn were tried. The FT-IR spectrum demonstrates the successful synthesis of the materials, designated as ZrT-1-NH2-IS-Ni and ZrT-1-NH2-IS-Mn (Fig. S18 in Supporting information). The TGA of ZrT-1-NH2-IS-Ni and ZrT-1-NH2-IS-Mn indicates satisfactory stability of their structures (Fig. S19 in Supporting information). Application of photoelectrochemical characterization to evaluate the photovoltaic properties of materials and explore their potential in the preparation of syngas by photocatalytic CO2RR. The results display that both ZrT-1-NH2-IS-Ni and ZrT-1-NH2-IS-Mn possess effective charge separation efficiencies and electron transfer performance (Figs. S20-S22 in Supporting information). The photocatalytic properties of ZrT-1-NH2-IS-Ni and ZrT-1-NH2-IS-Mn were investigated under the optimal catalytic conditions (2 mL TEOA, 2 mL MeCN and 2 mL MeOH), and the results illustrate that both ZrT-1-NH2-IS-Ni and ZrT-1-NH2-IS-Mn have appealing catalytic activity for the photocatalytic CO2RR for the preparation of syngas with yields of 21.8 and 18.6 mmol g-1 h-1, respectively (Figs. S23-S25 in Supporting information).
In conclusion, by utilizing the step-wise post-synthetic strategy, single-atom sites of Co were anchored on Zr-MOCs (ZrT-1-NH2-IS-Co) for the first. Photoelectrochemical analysis revealed that ZrT-1-NH2-IS-Co displayed enhanced electron transport properties and charge separation efficiency. The experimental findings demonstrate that ZrT-1-NH2-IS-Co exhibits remarkable catalytic activity with a high yield of up to 30.9 mmol g-1 h-1 in the reduction of CO2 to produce syngas. This achievement ranks among the highest in crystalline porous materials. Meanwhile, Co was confirmed to be the catalytic site for the preparation of syngas. Additionally, we successfully synthesized ZrT-1-NH2-IS-Ni and ZrT-1-NH2-IS-Mn using a similar modification strategy, both of which displayed excellent catalytic properties for CO2 conversion. This research offers valuable insights into the development of SACs on metal-organic cage catalysts for efficient CO2 conversion.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was financially supported by the NSFC of China (Nos. 22175033, 22371033, 22371032), Jilin Provincial Department of Science and Technology (No. 20230508108RC), the Fundamental Research Funds for the Central Universities Excellent Youth Team Program (No. 2412023YQ001), and the Postdoctoral Fellowship Program of CPSF (No. GZC20230939).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.111253
.
[1]
E.V. Kondratenko, G. Mul, J. Baltrusaitis, G.O. Larrazabal, J. Perez-Ramirez, Energy Environ. Sci. 6 (2013) 3112–3135. doi: 10.1039/c3ee41272e
R. Paul, R. Das, N. Das, et al., Angew. Chem. Int. Ed. 62 (2023) e202311304.
[50]
P.M.M. Stanley, A.Y.Y. Su, V. Ramm, et al., Adv. Mater. 35 (2023) 2207380.
[51]
M. Dong, Q. Pan, F. Meng, et al., J. Colloid Interface Sci. 662 (2024) 807–813.
Figure 1
(a) Schematic of the synthesis of ZrT-1-NH2-IS-Co. (b) FT-IR spectra of ZrT-1-NH2, ZrT-1-NH2-IS and ZrT-1-NH2-IS-Co. (c) TEM and mapping images of ZrT-1-NH2-IS-Co. (d) HAADF-STEM image of ZrT-1-NH2-IS-Co. (e) Normalized Co K-edge XANES spectrum of ZrT-1-NH2-IS-Co. (f) k3-weighted FT-EXAFS spectrum of ZrT-1-NH2-IS-Co. (g) The wavelet transform of ZrT-1-NH2-IS-Co.
Figure 2
(a) Tauc plots of ZrT-1-NH2, ZrT-1-NH2-IS and ZrT-1-NH2-IS-Co. Mott-Schottky plots of ZrT-1-NH2 (b), ZrT-1-NH2-IS (c) and ZrT-1-NH2-IS-Co (d). (e) EIS Nyquist plots of ZrT-1-NH2, ZrT-1-NH2-IS and ZrT-1-NH2-IS-Co. (f) Transient photocurrent response of ZrT-1-NH2, ZrT-1-NH2-IS and ZrT-1-NH2-IS-Co.
Figure 3
(a) Photocatalytic performances under different reaction conditions. (b) Trend of CO2 reduction of ZrT-1-NH2-IS-Co over time. (c) The result of the isotopic experiment. (d) Reproducibility experiment of ZrT-1-NH2-IS-Co. (e) Trend in CO2 reduction of monodisperse ZrT-1-NH2-IS-Co over time. (f) The syngas yields compared with other reported literature.
Figure 4
(a) Post-catalytic TEM and mapping images of ZrT-1-NH2-IS-Co. (b) Pre-catalytic and post-catalytic FT-IR spectra of ZrT-1-NH2-IS-Co. Post-catalytic XPS of ZrT-1-NH2-IS-Co (c) C 1s, (d) O 1s, (e) Zr 3d, (f) Co 2p.
Figure 5
(a) In situ infrared spectrum of CO2 reduction on ZrT-1-NH2-IS-Co. (b) Emission spectra of [Ru(bpy)3]2+ in the varying amounts of ZrT-1-NH2-IS-Co and TEOA. (c) Gibbs free energy diagram for CO2 reduction of ZrT-1-NH2-IS-Co. (d) Possible mechanisms of ZrT-1-NH2-IS-Co for photocatalytic CO2 reduction.

 DownLoad:
DownLoad:

 下载:
下载:






 下载:
下载:
