Scheme 1.
Research background and this work.
Single-electron transfer enables enantioselective synthesis of spirooxindoles via dual copper and phosphoric acid catalysis
Jun Shi , Xueting Zhou , Bing-Qing Yang , Biaobiao Jiang , Wei Wu , Hai Ren

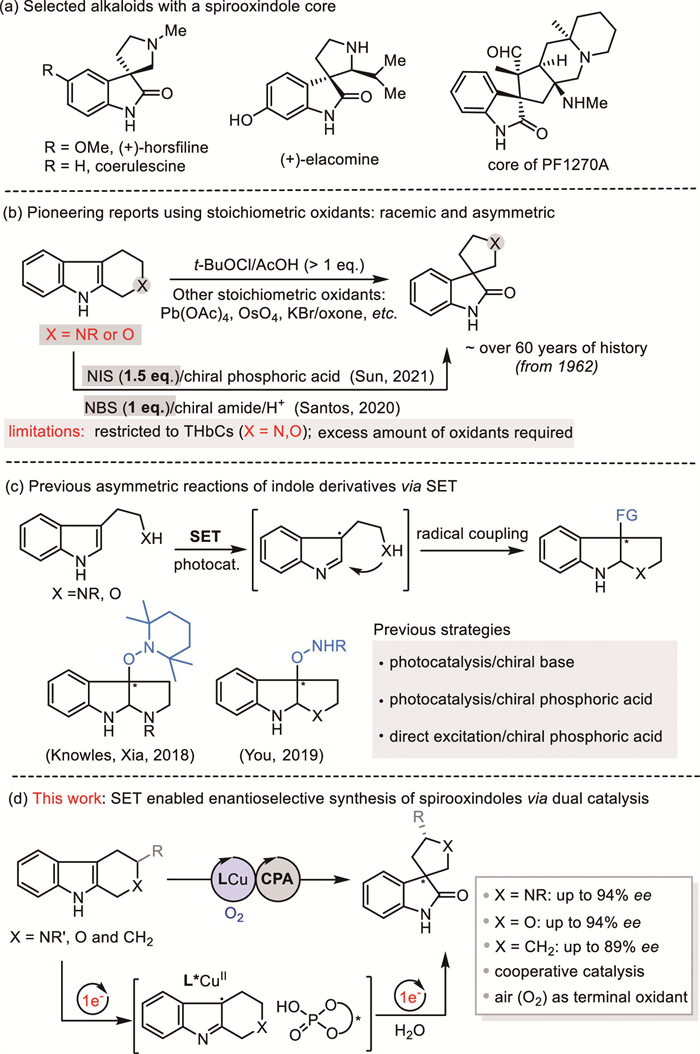
The spirooxindole moiety is a ubiquitous substructure in natural products and bioactive molecules. For examples, pyrrolidinyl- and cyclopentyl-bearing spiroindolinone units represent two common privileged cores present in a number of alkaloids and therapeutic agents (Scheme 1a) [1-5]. Accordingly, it has received substantial attention from the scientific community due to its appealing molecular architecture and versatility as a pharmacophore [6-11].
Owing to their biomimetic potential, syntheses of these types of framework have been intensively investigated through direct oxidative rearrangement of cyclic indoles since the 1960s [12,13]. Many classic oxidants, such as Pb(OAc)2, OsO4, NBS, and peroxide reagents have been successfully applied to these transformations (Scheme 1b) [14-20]. However, the development of asymmetric catalytic methods has been demonstrated to be highly challenging. Recently, the Santos [21] and Sun [22] groups reported breakthroughs in this field by the combination of classical halide oxidation with chiral organocatalysts (Scheme 1b). However, an excess of NBS or NIS as the oxidant was necessary, and only active tetrahydro-β-carbolines (THβCs) and tetrahydropyrano[3,4-b]indoles (THPIs) were explored in both these works. Therefore, new synthetic protocols that allow the expansion of this strategy’s applications via catalytic enantioselective oxidation remain highly desirable.
During the past decade, single-electron transfer (SET)-induced oxidation has emerged as a powerful and straightforward strategy for indole-based transformations. Many oxidative strategies including transition-metal-[23-27], photoredox-[28-32], and electrochemistry-based protocols [33-36] have been extensively explored in this field. Accordingly, a variety of elegant and useful reactions have been developed, some of which have found widespread application in the total synthesis of natural products and biologically active compounds. In sharp contrast, only a few examples of enantioselective catalytic variants of such reactions have been reported. For example, in 2018, Knowles et al. [37] reported a pioneering photocatalytic enantioselective cyclization of tryptamine catalyzed by an iridium photoredox/chiral-phosphate-base catalyst in the presence of TEMPO. The same reaction was also achieved in a photocatalyst-free fashion with excited-state TEMPO as a hydrogen-atom-transfer catalyst by Xia et al. [38]. Further work by You et al. [39] demonstrated a catalytic asymmetric dearomatization cyclization of indole derivatives using dual photoredox and chiral phosphoric acid (CPA)-based catalysis (Scheme 1c). Notably, these examples elegantly demonstrated that indole derivatives can serve as radical species in photocatalytic asymmetric dearomative cyclization reactions. However, to the best of our knowledge, further reaction modes that provide enantiocontrol over in-situ generated open-shell indole radical cations are scarce.
Recently, our research group has successfully demonstrated the exceptional regulatory ability of a copper-complex-based catalytic system in the SET-induced oxidative transformation of indole derivatives [40-45], including the synthesis of spiroindolinones [44,45]. However, high enantioselective control only by using chiral copper complex in this field was always found to fail. Herein, we report the first catalytic enantioselective oxidative rearrangement of cyclic indoles through two sequential SETs using a dual chiral copper/CPA catalyst (Scheme 1d).
We commenced our investigation with N-protected THβC 1a as the model substrate. Inspired by the successful racemic reaction enabled by copper catalysis in our previous work [44], we reasoned that an enantioselective reaction mode might be realized by the addition of a chiral organocatalyst, exploiting its hydrogen-bonding interactions with the substrate. When racemic bisbenzoxazoline L1/CuBr2 and chiral phosphoric acid A1 were used as co-catalysts in chlorobenzene under open air conditions, the oxidative rearrangement product 2a was obtained in 12% yield with 22% ee (Table 1, entry 1). This preliminary result indicated that our dual catalysis strategy could work, encouraging us to optimize the reaction conditions with other chiral oxazoline ligands (Table 1, entries 2–4). When bisphenyl BOX L4 was used, the enantioselectivity was improved to 41% ee with a better yield of 18% (Table 1, entry 4). Attempts to enhance the yield and enantioselectivity using other copper salts were unsuccessful (Table 1, entries 5−8). Various chiral phosphoric acids with different sterically hindering substituents were then evaluated (Table 1, entries 9–12; see Supporting information for more details), and better enantioselectivity was achieved using 2,4,6-triisopropylphenyl modified phosphoric acid A5 (65% ee). Furthermore, it was found that the skeleton of the CPA catalyst also plays an important role in the enantioselectivity of this protocol. The use of spirophosphoric acid (A6 or A7) further improved the enantioselectivity (Table 1, entries 13−15). However, the yield was still low. Gratifyingly, when benzyloxycarbonyl (Cbz)-substituted THβC 1b was employed, a good yield and excellent enantioselectivity were achieved (74% yield, 94% ee) (Table 1, entry 16).
 DownLoad:
CSV
DownLoad:
CSV
 |
|||||
| Entry | L | Metal salt | CPA | 2a yield (%) | ee (%)b |
| 1 | L1 | CuBr2 | (R)-A1 | 12 | 22 |
| 2 | L2 | CuBr2 | (R)-A1 | 22 | 38 |
| 3 | L3 | CuBr2 | (R)-A1 | 15 | 41 |
| 4 | L4 | CuBr2 | (R)-A1 | 18 | 41 |
| 5 | L4 | CuCl2 | (R)-A1 | 41 | 10 |
| 6 | L4 | CuBr | (R)-A1 | 0 | – |
| 7 | L4 | Cu(OTf)2 | (R)-A1 | 0 | – |
| 8 | L4 | Cu(OAc)2 | (R)-A1 | trace | – |
| 9 | L4 | CuBr2 | (R)-A2 | 15 | 21 |
| 10 | L4 | CuBr2 | (R)-A3 | 24 | 16 |
| 11 | L4 | CuBr2 | (R)-A4 | 37 | 16 |
| 12 | L4 | CuBr2 | (R)-A5 | 35 | 65 |
| 13 | L4 | CuBr2 | (R)-A6 | 22 | 77 |
| 14 | L4 | CuBr2 | (R)-A7 | 21 | 91 |
| 15c | L4 | CuBr2 | (R)-A7 | 54 | 93 |
| 16c,d | L4 | CuBr2 | (R)-A7 | 74 (2b) | 94 |
| a 1 (0.2 mmol), L (0.024 mmol, 12 mol%), CuBr2 (0.02 mmol, 10 mol%), CPA (0.02 mmol, 10 mol%), PhCl (2.0 mL), H2O (1 mmol, 5 equiv.), 50 ℃. b ee value determined by HPLC with a chiral stationary phase. c 1 (0.1 mmol), L (0.012 mmol, 12 mol%), CuBr2 (0.01 mmol, 10 mol%), CPA (0.01 mmol, 10 mol%), PhCl (2.0 mL), H2O (1 mmol, 10 equiv.), 40 ℃. d 1b used as substrate. |
|||||
The substrate scope of the current protocol was then investigated under the optimized conditions. As shown in Scheme 2, a wide range of THβCs bearing different Nβ-esterified substituents reacted smoothly to give the desired pyrrolidinyl-spirooxindoles enantioselectively in good to excellent yields (2b–2f). Methylsulfonyl-substituted THβC was also found to be compatible, providing spirooxindole 2g in 50% yield with 91% ee. The use of various THβCs containing different substituents at the indole phenyl moiety with different electronic characteristics, such as fluoro (2h), methyl (2i, 2k), methoxy (2j), and chloro (2l) groups, afforded the corresponding products in moderate yields with high enantioselectivities. The C3-ester-substituted substrate was also suitable substrate, producing the product 2m in 79% yield with high diastereoselectivity.
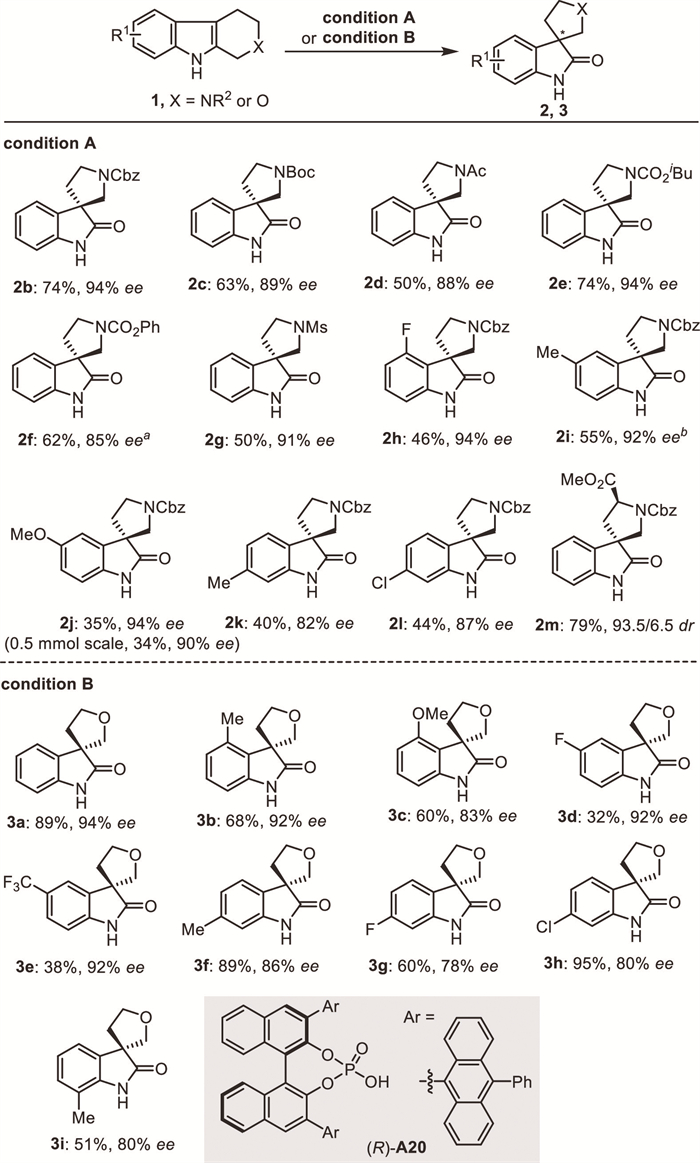
This protocol was also investigated for the THPI substrates. Further condition optimization indicated that the use of CPA A20 as co-catalyst induced high enantioselectivities for this reaction (Section 2.2 in Supporting information for more details). Under the optimal conditions, THPIs that bear either an electron-rich or electron-poor group at different positions of the indole ring reacted smoothly to deliver the tetrahydrofuranyl-spirooxindoles 3a−3i in moderate to excellent yields with good to excellent enantioselectivities. The absolute configuration of 2a and 3a were confirmed by comparison with Sun′s work [22].
Encouraged from these results, we conceived that the tetrahydrocarbazoles could be extended for the preparation of enantioenriched cyclopentyl-spirooxindoles, another important skeletons found in a range of natural and bioactive molecules. Notably, due to the absence of electron-donating group at β-position, the oxidative rearrangement of tetrahydrocarbazoles to cyclopentyl-spirooxindoles remains challenging and its enantioselective variant has not yet been reported. After reaction condition optimization, we found that the isopropyl BOX L14/CuBr2 with CPA A21 was the effective dual catalyst, producing the desired product 5a in 43% yield with 89% ee and >20/1 diastereoselectivity (dr) (see Section 2.3 in Supporting information for more details of optimization). The substrate scope was further investigated. As showed in Scheme 3, m-fluoro-phenyl and p-chloro-phenyl substituted tetrahydrocarbazoles proceeded smoothly to give the products 5b and 5c in moderate yields with high enantioselectivities. The γ-methyl- and -ethyl-substituted tetrahydrocarbazoles were also tolerated, producing the corresponding products 5d−5f in 30%−47% yields with 80%−84% ee and >20/1 dr. Notably, along with the generation of product 5d, the hydroxyl-imine indoline 6 was isolated in 22% yield with 52% ee and >99/1 dr, indicating that the γ-methyl group has a great influence for the chemoselectivity of this reaction. The relative configuration of 5d was confirmed by comparison with our previous work [45], and the absolute configuration was deduced from the formation of 3a.

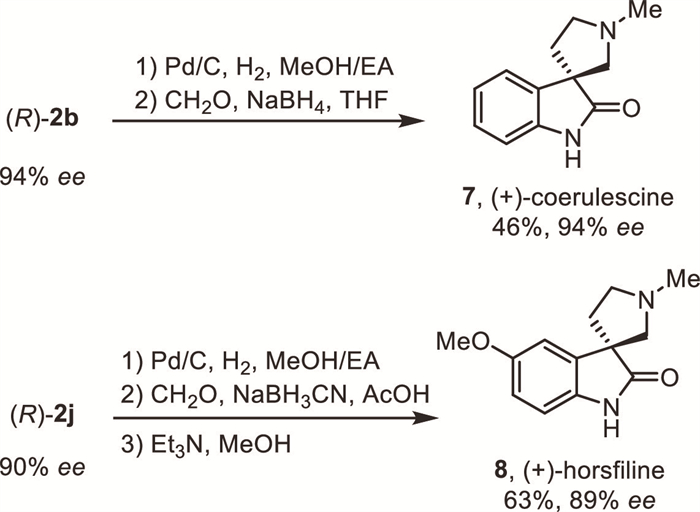
The synthetic utility of this method for the preparation of alkaloids was demonstrated by a concise catalytic enantioselective synthesis of natural product (+)-coerulescine 7 and (+)-horsfiline 8 via simple transformations of oxidative rearrangement products 2b and 2j, respecitively (Scheme 4). Notably, this is the first catalytic enantioselective synthetic route avoiding the use of excess amount of oxidant.
For the mechanism of the reaction, several control experiments were carried out. When the H218O was used instead of H2O, the heavy oxygen atom labeled product [18O]−2b was identified by high-resolution mass spectrometry (HRMS) analysis (Scheme 5, Eq. 1). This evidence indicates that the oxygen of the carbonyl group in product 2a mainly comes from H2O. When the reaction was conducted under Ar atmosphere, only a trace amount of desired product 2b was detected. The radical inhibitor TEMPO greatly suppressed the reaction (Scheme 5, Eq. 2). In addition, the 2-hydroxyl, 3-hydroperooxide indoline 9 was detected by HRMS analysis in situ under the standard conditions (Scheme 5, Eq. 3). These results suggested that the reaction may proceed through an SET-induced aerobic oxidation process.
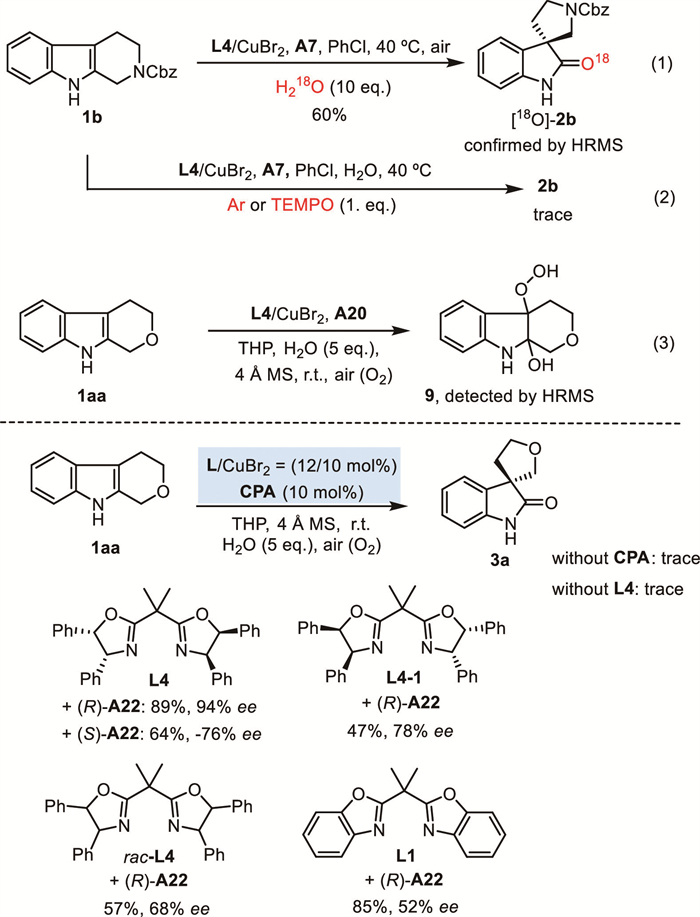
Some control experiments were designed to understand the functions of co-catalysts (Scheme 5). For the reaction of THPI 1aa, when the opposite enantiomer ligand L4–1 or racemic ligand rac-L4 was used instead of L4, the enantioselection of the reaction decreased. When racemic bisbenzoxazoline L1 was employed, only 52% ee was obtained. When the opposite enantiomer CPA (S)-A22 was used, the reaction proceeded smoothly, but with opposite enantioinduction (−76% ee). Notably, only a trace amount of product 4a was detected in the absence of CPA A22 or ligand L4. These results suggested that the enantioselection of reaction was originally induced by the CPA catalyst, but the structure of ligand has significant influences for the enantioselectivity control.
Based on the results described above and previous studies, a plausible reaction pathway is proposed in Fig. 1a. Initialy, the SET-induced oxidation of cyclic indole by CuⅡ species proceeds to generate the indole radical imine A. The C3 radical is further oxidized by the CuⅡ species to form the organo-copper complex B [46], which could be confirmed by HRMS analysis in situ. The CPA assisted nucleophilic addition of H2O to the imine motif to generate the cis-substituted indoline C. As showed, the CPA serves as a bifunctional catalyst to activate both imine and water, and the selectivity is controlled due to recognition by the two dual catalysts. The reductive elimination of intermediate C gave cis-2,3-dibydroxyl indoline D, which could also be deteced by HRMS analysis in situ. Then, the 3-OH is protonated by the CPA and the H2O is concomitantly released to form an intact ion pair including a tertiary carbocation and a chiral phosphate anion (E). Finally, the chiral phosphate anion induced 1, 2-rearrangement of hemiaminal to afford the optically enriched product and complete the CPA catalytic cycle. Simultaneously, the CuⅠ species is oxidized to CuⅡ species (LCuⅡBrOH) by O2 in the catalytic cycle [47,48].
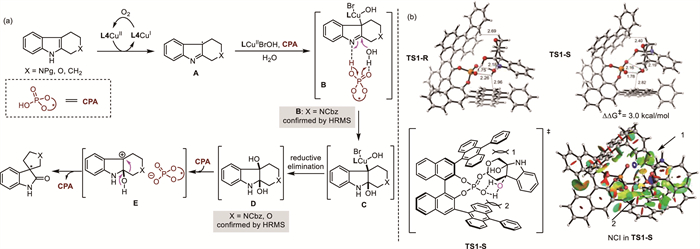
Furthermore, theoretical calculations were performed to investigate the key steps and the origin of enantioselectivity of the THPI (see Section 4 in Supporting information for details). Calculations reveals that the 2,3-cis-dihydroxylated indoline is the most stable intermediate, which is favored over the trans-species by 6.5 kcal/mol. The protonation of 3-OH and dehydration enabled by CPA has the highest energy barrier and serves as the enantioselectivity-determining step. The transition state that leads to R product (TS1-R) is favored over TS1-S by 3.0 kcal/mol, coinciding with the experimental results. The enantioselectivity-determining model suggested that the steric repulsion between the methylene of substrate and the phenyl group of catalyst, as well as that between the newly-formed water and the anthracenyl scaffold of catalyst destabilize TS1-S (Fig. 1b).
In summary, a general, unified copper/CPA co-catalyzed enantioselective SET-induced oxidative rearrangement of cyclic indoles has been developed. With atmospheric oxygen (O2) as the terminal oxidant, THβCs, THPIs and tetrahydrocarbazoles are all suitable substrates, providing highly streamlined access to optically enriched pyrrolidinyl-, tetrahydrofuranyl-, and cyclopentyl-bearing spiroindolinones. The synthetic utility of this protocol was demonstrated in a concise catalytic synthesis of (+)-coerulescine and (+)-horsfiline. Mechanistic studies suggest that this process involves SET-induced aerobic oxygenation, and which is followed by a pinacol-like rearrangement. Further detailed mechanistic studies and the extension of the present catalytic protocol to other asymetric reactions are underway in our laboratory.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Jun Shi: Investigation. Xueting Zhou: Investigation. Bing-Qing Yang: Investigation. Biaobiao Jiang: Data curation. Wei Wu: Data curation. Hai Ren: Writing – review & editing, Project administration.
We thank the National Natural Science Foundation of China (Nos. 22261010, 22201053, 22461012), Guizhou Provincial Major Scientific and Technological Program, China (No. QKHZDZX [2024]015) for financial support.
Supplementary material associated with this article can be found, in the online version, at doi:
C.V. Galliford, K.A. Scheidt, Angew. Chem. Int. Ed. 46 (2007) 8748–8758. doi: 10.1002/anie.200701342
G.S. Singh, Z.Y. Desta, Chem. Rev. 112 (2012) 6104–6155. doi: 10.1021/cr300135y
L. Hong, R. Wang, Adv. Synth. Catal. 355 (2013) 1023–1053. doi: 10.1002/adsc.201200808
D. Cheng, Y. Ishihara, B. Tan, et al., ACS Catal. 4 (2014) 743–762. doi: 10.1021/cs401172r
A. Váradi, G.F. Marrone, T.C. Palmer, et al., J. Med. Chem. 59 (2016) 8381–8397. doi: 10.1021/acs.jmedchem.6b00748
Z. Cao, F. Zhou, J. Zhou, Acc. Chem. Res. 51 (2018) 1443–1454. doi: 10.1021/acs.accounts.8b00097
F. Zhou, Y.L. Liu, J. Zhou, Adv. Synth. Catal. 352 (2010) 1381–1407. doi: 10.1002/adsc.201000161
Y. Liu, S.J. Han, W.B. Liu, et al., Acc. Chem. Res. 48 (2015) 740–751. doi: 10.1021/ar5004658
K.R. Holman, A.M. Stanko, S.E. Reisman, Chem. Soc. Rev. 50 (2021) 7891–7908. doi: 10.1039/d0cs01385d
A. Wetzel, F. Gagosz, Angew. Chem. Int. Ed. 50 (2011) 7354–7358. doi: 10.1002/anie.201102707
H. Leng, Q. Zhao, Q. Mao, et al., Chin. Chem. Lett. 32 (2021) 2567–2571. doi: 10.1016/j.cclet.2021.03.009
N. Finch, W.I. Taylor, J. Am. Chem. Soc. 84 (1962) 1318–1320. doi: 10.1021/ja00866a062
J. Shavel, H. Zinnes, J. Am. Chem. Soc. 84 (1962) 1320–1321. doi: 10.1021/ja00866a063
S.D. Edmondson, S.J. Danishefsky, Angew. Chem. Int. Ed. 37 (1998) 1138–1140. doi: 10.1002/(SICI)1521-3773(19980504)37:8<1138::AID-ANIE1138>3.0.CO;2-N
S. Edmondson, S.J. Danishefsky, L. Sepp-Lorenzino, et al., J. Am. Chem. Soc. 121 (1999) 2147–2155. doi: 10.1021/ja983788i
M. Ito, C.W. Clark, M. Mortimore, et al., J. Am. Chem. Soc. 123 (2001) 8003–8010. doi: 10.1021/ja010935v
P.S. Baran, J.M. Richter, J. Am. Chem. Soc. 127 (2005) 15394–15396. doi: 10.1021/ja056171r
S. Han, M. Movassaghi, J. Am. Chem. Soc. 133 (2011) 10768–10771. doi: 10.1021/ja204597k
J. Xu, L. Liang, H. Zheng, et al., Nat. Commun. 10 (2019) 4754. doi: 10.1038/s41467-019-12768-4
G. Zhao, L. Liang, E. Wang, et al., Green Chem. 23 (2021) 2300–2307. doi: 10.1039/d1gc00297j
M. Sathish, F.M. Nachtigall, L.S. Santos, RSC Adv. 10 (2020) 38672–38677. doi: 10.1039/d0ra07705d
C. Qian, P. Li, J. Sun, Angew. Chem. Int. Ed. 60 (2021) 5871–5875. doi: 10.1002/anie.202015175
P.S. Baran, J.M. Richter, J. Am. Chem. Soc. 126 (2004) 7450–7451. doi: 10.1021/ja047874w
S. Tadano, Y. Mukaeda, H. Ishikawa, Angew. Chem. Int. Ed. 52 (2013) 7990–7994. doi: 10.1002/anie.201303143
K. Liang, X. Deng, X. Tong, et al., Org. Lett. 17 (2015) 206–209. doi: 10.1021/ol5032365
L. Li, X.M. Chen, Z.S. Wang, et al., ACS Catal. 7 (2017) 4004–4010. doi: 10.1021/acscatal.7b01038
A.Y. Ji, A. Thirupathi, J.Y. Hwang, et al., Org. Lett. 25 (2023) 1541–1546. doi: 10.1021/acs.orglett.3c00330
Y. Chen, L.Q. Lu, D.G. Yu, et al., Sci. China Chem. 62 (2019) 24–57. doi: 10.1007/s11426-018-9399-2
C. Zhang, S. Li, F. Bureš, et al., ACS Catal. 6 (2016) 6853–6860. doi: 10.1021/acscatal.6b01969
J. Ye, J. Wu, T. Lv, et al., Angew. Chem. Int. Ed. 56 (2017) 14968–14972. doi: 10.1002/anie.201708893
M. Frahm, T. von Drathen, L.M. Gronbach, et al., Angew. Chem. Int. Ed. 59 (2020) 12450–12454. doi: 10.1002/anie.202007549
L.M. Gronbach, A. Voss, M. Frahm, et al., Org. Lett. 23 (2021) 7834–7838. doi: 10.1021/acs.orglett.1c02857
W. Zhou, X. Chen, L. Lu, et al., Chin. Chem. Lett. 35 (2024) 108902. doi: 10.1016/j.cclet.2023.108902
J. Wu, Y. Dou, R. Guillot, et al., J. Am. Chem. Soc. 141 (2019) 2832–2837. doi: 10.1021/jacs.8b13371
C.J. Long, H. Cao, B.K. Zhao, et al., Angew. Chem. Int. Ed. 61 (2022) e202203666. doi: 10.1002/anie.202203666
X. Liu, D. Yang, Z. Liu, et al., J. Am. Chem. Soc. 145 (2023) 3175–3186. doi: 10.1021/jacs.2c12902
E.C. Gentry, L.J. Rono, M.E. Hale, et al., J. Am. Chem. Soc. 140 (2018) 3394–3402. doi: 10.1021/jacs.7b13616
K. Liang, X. Tong, T. Li, et al., J. Org. Chem. 83 (2018) 10948–10958. doi: 10.1021/acs.joc.8b01597
Y.Z. Cheng, Q.R. Zhao, X. Zhang, et al., Angew. Chem. Int. Ed. 58 (2019) 18069–18074. doi: 10.1002/anie.201911144
Y.S. Peng, W. Wang, J. Shi, et al., Angew. Chem. Int. Ed. 62 (2023) e202313687. doi: 10.1002/anie.202313687
Y.Z. Sun, H.C. Yang, J.R. Song, et al., ACS Catal. 14 (2024) 17025–17032.
J. Shi, X.J. Li, S.Y. Jiang, et al., ACS Catal. 14 (2024) 5605–5611. doi: 10.1021/acscatal.4c00637
S.Y. Jiang, J. Shi, W. Wang, et al., ACS Catal. 13 (2023) 3085–3092. doi: 10.1021/acscatal.2c05881
J. Shi, R.A. Wang, W. Wu, et al., Org. Lett. 24 (2022) 3358–3362. doi: 10.1021/acs.orglett.2c01059
H. Ren, B.Q. Yang, J. Shi, et al., Chem. Eur. J. 30 (2024) e202401293. doi: 10.1002/chem.202401293
G. Zhang, S. Zhou, L. Fu, et al., Angew. Chem. Int. Ed. 59 (2020) 20439–20444. doi: 10.1002/anie.202008338
A.E. King, L.M. Huffman, A. Casitas, et al., J. Am. Chem. Soc. 132 (2010) 12068–12073. doi: 10.1021/ja1045378
C. Zhang, N. Jiao, Angew. Chem. Int. Ed. 49 (2010) 6174–6177. doi: 10.1002/anie.201001651

Scheme 2 Substrate scope. Reactions carried out under air atmosphere. Condition A: 1 (0.1 mmol), L4 (12 mol%), CuBr2 (10 mol%), (R)-A7 (10 mol%), PhCl (2 mL), H2O (1.0 mmol, 10 equiv.), 40 ℃. Isolated yields. a (R)-A7 (5 mol%). b H2O (55 equiv.). Condition B: 1 (0.1 mmol), L4 (0.012 mmol, 12 mol%), CuBr2 (0.01 mmol, 10 mol%), (R)-A20 (0.01 mmol, 10 mol%), THP (1 mL), H2O (0.5 mmol, 5 equiv.), 4 Å MS (50 mg), room temperature. All yields are isolated yield.

Scheme 3 Substrate scope. Reactions carried out under air atmosphere: 4 (0.1 mmol), L14 (0.015 mmol, 15 mol%), CuBr2 (0.01 mmol, 10 mol%), (R)-A21 (0.01 mmol, 10 mol%), THF (1 mL), room temperature. All yields are isolated yield. The dr values were confirmed by NMR analysis.
Table 1. Optimization of reaction conditions.a
|
|||||
| Entry | L | Metal salt | CPA | 2a yield (%) | ee (%)b |
| 1 | L1 | CuBr2 | (R)-A1 | 12 | 22 |
| 2 | L2 | CuBr2 | (R)-A1 | 22 | 38 |
| 3 | L3 | CuBr2 | (R)-A1 | 15 | 41 |
| 4 | L4 | CuBr2 | (R)-A1 | 18 | 41 |
| 5 | L4 | CuCl2 | (R)-A1 | 41 | 10 |
| 6 | L4 | CuBr | (R)-A1 | 0 | – |
| 7 | L4 | Cu(OTf)2 | (R)-A1 | 0 | – |
| 8 | L4 | Cu(OAc)2 | (R)-A1 | trace | – |
| 9 | L4 | CuBr2 | (R)-A2 | 15 | 21 |
| 10 | L4 | CuBr2 | (R)-A3 | 24 | 16 |
| 11 | L4 | CuBr2 | (R)-A4 | 37 | 16 |
| 12 | L4 | CuBr2 | (R)-A5 | 35 | 65 |
| 13 | L4 | CuBr2 | (R)-A6 | 22 | 77 |
| 14 | L4 | CuBr2 | (R)-A7 | 21 | 91 |
| 15c | L4 | CuBr2 | (R)-A7 | 54 | 93 |
| 16c,d | L4 | CuBr2 | (R)-A7 | 74 (2b) | 94 |
| a 1 (0.2 mmol), L (0.024 mmol, 12 mol%), CuBr2 (0.02 mmol, 10 mol%), CPA (0.02 mmol, 10 mol%), PhCl (2.0 mL), H2O (1 mmol, 5 equiv.), 50 ℃. b ee value determined by HPLC with a chiral stationary phase. c 1 (0.1 mmol), L (0.012 mmol, 12 mol%), CuBr2 (0.01 mmol, 10 mol%), CPA (0.01 mmol, 10 mol%), PhCl (2.0 mL), H2O (1 mmol, 10 equiv.), 40 ℃. d 1b used as substrate. |
|||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们




 下载:
下载: