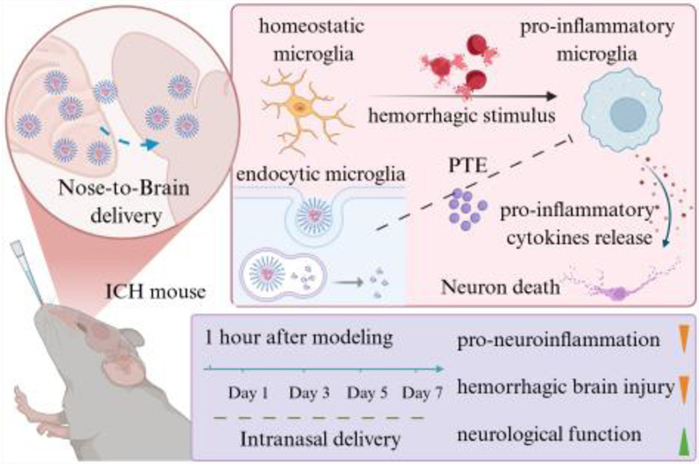
Scheme 1.
PTE-NPs intranasal administration alleviated neuroinflammation after ICH via suppressing pro-inflammatory microglia activation and pro-inflammatory cytokine release.
Intranasal pterostilbene nanoparticles delivery alleviates neuroinflammation and brain injury after intracerebral hemorrhage
Zhongxin Duan , Yue Wang , Yunchu Zhang , Xia Liu , Wanyu Wang , Hua Li , Qingyang Lu , Chao You , Yongzhong Cheng , Cong Wu , Xiang Gao

Intracerebral hemorrhage (ICH) is a form of spontaneous, non-traumatic bleeding resulting from vascular rupture, leading to hematoma formation within the brain parenchyma [1,2]. ICH is the second most prevalent stroke type after ischemic stroke, comprising about 10%–15% of all stroke incidents. The global incidence of ICH is approximately 24.6 cases per 100,000 person-years, with a higher prevalence in the elderly population [3]. Notably, individuals aged 85 years and older have a 9.6-fold higher incidence compared to middle-aged adults [4]. ICH is associated with a high mortality rate; studies indicated that the 1-month case fatality rate was approximately 40%, and the 1-year mortality rate reached around 54% [5]. Other reports [4,6,7] showed that the 30-day mortality ranged between 34% and 50%, with half of the deaths occurring within the first 2 days after onset. ICH posed a significant global health burden. In 2021, an estimated 3.44 million new cases of ICH were reported worldwide, with an age-standardized prevalence of 40.8 cases per 100,000 people. Despite a decrease in incidence, mortality, prevalence, and disability-adjusted life years (DALYs) related to ICH from 1990 to 2021, the total number of cases has increased. Moreover, just 12%–39% of ICH survivors achieve long-term functional independence, while many endure severe disabilities, imposing significant personal and societal burdens [8]. Taken together, ICH remains a severe medical condition with high morbidity and mortality, representing a major challenge to global health. Therefore, exploring effective therapeutic strategies to reduce mortality and improve patient outcomes is important.
The disruption of homeostasis in the brain microenvironment after ICH is driven by a cascade of primary and secondary brain injuries [9,10], which are major contributors to the high mortality and poor prognosis associated with ICH [11,12]. Neuroinflammation is crucial in brain injury and repair, making its modulation a potential therapeutic target [13–15]. In the initial phase of ICH, blood component leakage and erythrocyte metabolic products activate microglia, inducing their pro-inflammatory polarization and the release of cytokines like tumor necrosis factor-alpha (TNF-α) and interleukin-1 beta (IL-1β), which worsen neuroinflammation and lead to secondary neuronal damage near the hematoma [16,17]. Inhibiting early pro-inflammatory microglia activation and cytokine release may offer a therapeutic approach to mitigate ICH-induced brain injury.
Pterostilbene (PTE), a natural polyphenol primarily found in blueberries, grapes, and the heartwood of Pterocarpus species, is a dimethylated derivative of resveratrol, a classical anti-inflammation and antioxidation compound. The substitution of two hydroxyl groups with methoxy groups enhances its bioavailability compared to resveratrol, leading to superior anti-inflammatory and antioxidant properties [18,19]. Previous studies demonstrated that PTE exerts neuroprotective effects in ischemic stroke models by alleviating neuronal injury through its anti-inflammatory and antioxidative mechanisms [20,21]. Furthermore, PTE demonstrated therapeutic efficacy in neurodegenerative diseases such as Parkinson's, Alzheimer's, and Huntington's diseases [22–24]. Two clinical studies on non-alcoholic fatty liver disease and muscle injury indicated that oral PTE administration is biosafe [25,26]. However, the poor water solubility of PTE limits its oral bioavailability and blood-brain barrier (BBB) permeability, thereby inhibiting its therapeutic efficacy in neuroinflammatory diseases [27]. Nanotechnology-based drug delivery strategies offer a promising approach to enhance the solubility and bioavailability of hydrophobic drugs [15,28]. Intranasal (IN) administration offers a direct, non-invasive method for delivering drugs to the central nervous system, effectively crossing the BBB and increasing brain drug accumulation while reducing systemic side effects [29]. The combination of nanotechnology and intranasal delivery presents a potential solution for enhancing the therapeutic efficacy of PTE in neurological diseases.
In this study, we encapsulated PTE in a methoxy poly(ethylene glycol)-poly(ε-caprolactone) (mPEG-PCL) amphiphilic block copolymer to improve its bioavailability. IN administration was performed to facilitate BBB penetration and enhance PTE accumulation at the nidus of ICH. When PTE-NPs were delivered into the hemorrhagic brain microenvironment and internalized by microglia surrounding the hematoma, the endocytosed PTE could inhibit microglia pro-inflammatory polarization, reduce the release of pro-inflammatory cytokines, subsequently alleviate inflammation-mediated neuronal injury in the peri–hematomal region (Scheme 1). In two ICH mouse models, PTE-NPs demonstrated significant therapeutic efficacy, reducing brain injury and neurological deficits while exhibiting good biosafety.
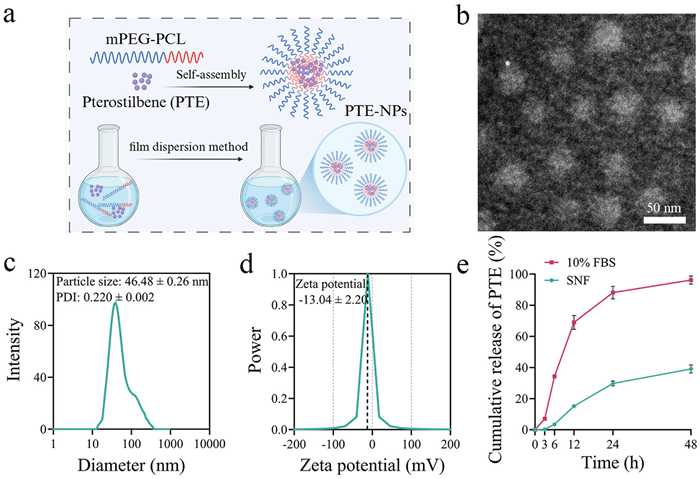
To resolve the poor water solubility of PTE, we synthesized mPEG-PCL via ring-opening polymerization (Fig. S1 in Supporting information). Proton nuclear magnetic resonance (1H NMR) spectroscopy confirmed the polymer's molecular structure (Fig. S2 in Supporting information). PTE-NPs were synthesized via a thin-film hydration technique (Fig. 1a). Dynamic light scattering (DLS) analysis revealed that PTE-NPs had a hydrodynamic diameter of 46.48 ± 0.26 nm and a polydispersity index (PDI) of 0.220 ± 0.002 (Fig. 1c). The zeta potential was measured at −13.04 ± 2.20 mV (Fig. 1d). Transmission electron microscopy (TEM) imaging confirmed that PTE-NPs exhibited a uniform spherical morphology (Fig. 1b). The encapsulation efficiency and drug loading capacity of PTE-NPs were determined to be 96.3% ± 0.8% and 15.6% ± 0.2%, respectively. Nanoparticles have emerged as a highly promising strategy for brain-targeted drug delivery [30]. Typically, nanoparticles ranging from 20 nm to 100 nm enhance drug selectivity, improving efficacy while minimizing off-target effects and systemic toxicity [31]. For IN administration, an optimal nanocarrier size should be below 200 nm, with a PDI under 0.3 [32]. Given these parameters, our PTE-NPs are well-suited for intranasal delivery. To evaluate the stability of PTE-NPs, we measured their hydrodynamic diameter and PDI over time. As shown in Fig. S3 (Supporting information), both parameters remained stable for up to 7 days. In vitro release studies were conducted in simulated nasal fluid (SNF) and simulated blood (phosphate-buffered saline containing 10% fetal bovine serum, 10% FBS-PBS) to assess the drug release behavior of PTE-NPs. As shown in Fig. 1e, PTE-NPs exhibited a slow and sustained release of PTE in SNF over 48 h. In contrast, a more rapid release of PTE-NPs was observed in simulated blood. This release behavior of PTE-NPs in nasal fluid indicates its potential to reduce dosing frequency and enhance therapeutic convenience.
Intranasal drug delivery is a non-invasive method that enables direct crossing of the BBB via several pathways, including the olfactory pathway, the trigeminal nerve pathway, and the paracellular pathway. Olfactory epithelial cells transport nanoparticles to the olfactory bulb via the olfactory nerve, subsequently delivering them to the brain. This is considered the primary brain-targeting mechanism for IN administration. Additionally, the trigeminal nerve, which is widely distributed, enables nanoparticles transport from the nasal cavity to the brainstem. Moreover, nanoparticles pass through the nasal epithelium via the paracellular space, then move through the perineural space to reach the subarachnoid space and cerebrospinal fluid in the brain [33]. The initial step in this process is the drug crossing the nasal mucosal epithelial cells and mucus layer [34]. We developed an in vitro transwell co-culture model simulating the nasal mucosal epithelial barrier with a mucus layer to assess the permeability of PTE-NPs (Fig. 2a). Firstly, we evaluated the cytotoxicity of mPEG-PCL for RPMI 2650, a human nasal mucosa epithelial cell. As shown in Fig. S4 (Supporting information), RPMI 2650 cell viability remained unchanged across increasing concentrations of mPEG-PCL compared to the control group. To measure the nasal barrier permeability of the mPEG-PCL nanocarrier, chlorin e6 (Ce6), a fluorescent tracer, was encapsulated into mPEG-PCL nanoparticles (Ce6-NPs) to track nanoparticle penetration. Compared to free-Ce6, Ce6-NPs exhibited significantly enhanced cumulative permeability across the epithelial barrier (Fig. 2b). Microglia, as the main phagocytic cells in the brain and essential mediators of neuroimmune responses, are vital for drug uptake and efficacy. We assessed Ce6-NPs uptake in BV2 microglia cells through the nasal mucosal epithelial barrier, including a mucus layer. As shown in Figs. 2c and d, BV2 cells internalized Ce6-NPs significantly more than free Ce6. These findings indicated that nanoencapsulation enhanced the permeability of hydrophobic drugs across the nasal mucosal barrier and facilitated uptake by microglia. To further investigate the biodistribution of PTE-NPs following different administration routes, we performed in vivo imaging system (IVIS) fluorescence imaging of ICH mice at 3, 6, 9, 12, and 24 h after administration. All experimental procedures adhered to the Guide for the Care and Use of Laboratory Animals and received approval from the Animal Ethics Committee of the State Key Laboratory of Biotherapy, Sichuan University (Chengdu, China). As shown in Figs. 2e and f, no detectable fluorescence signals were observed in the brain after IN administration of free Ce6. After intravenous injection, Ce6-NPs rapidly entered the systemic circulation at a relatively high concentration. Due to BBB disruption following ICH, passive targeting became the primary mechanism for Ce6-NPs to reach brain tissue. Consequently, within the first 9 h post-administration, the brain drug concentration in the intravenous (IV) group was significantly higher than in the IN group. However, intravenous administration also results in systemic distribution, with metabolism primarily occurring in organs like the liver, leading to a rapid decline in brain concentration over time [35]. In contrast, the fluorescence intensity in the brain of the intranasal Ce6-NPs group remained stable for up to 9 h post-administration before gradually decreasing. This process occurs through two mechanisms: the paracellular pathway, which allows drugs to reach the brain within approximately 30 min, and the transcellular pathway, which involves receptor-mediated endocytosis or passive diffusion and requires more time for brain distribution. The use of nanomaterials further prolongs Ce6-NPs retention in the nasal cavity. By bypassing systemic circulation, intranasal delivery reduces drug metabolism and clearance, resulting in a more stable fluorescence intensity of Ce6-NPs in the brain over the 9 h observation period.
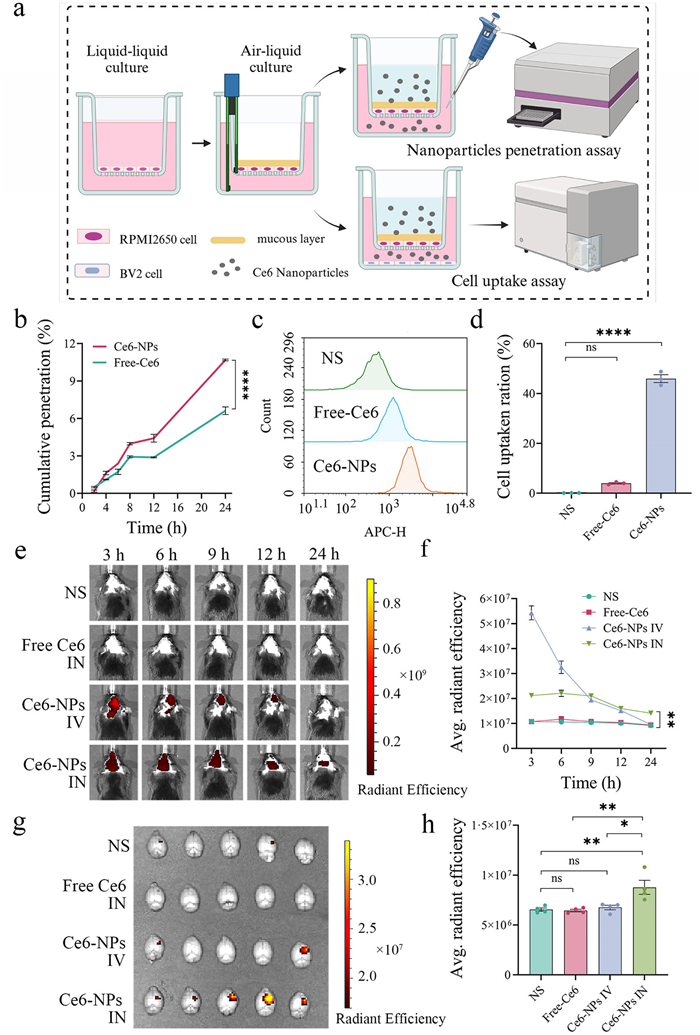
Ex vivo fluorescence analysis at 24 h post-administration revealed significantly higher brain fluorescence in the intranasal Ce6-NPs group compared to other groups. On the contrary, the fluorescence intensity of major organs in the intranasal Ce6-NPs group was lower than in the intravenous group (Figs. 2g and h, Fig. S5 in Supporting information). These findings demonstrated that PTE-NPs had sustained drug retention in the brain following IN administration, reduced peripheral drug exposure, and potentially minimized systemic side effects.
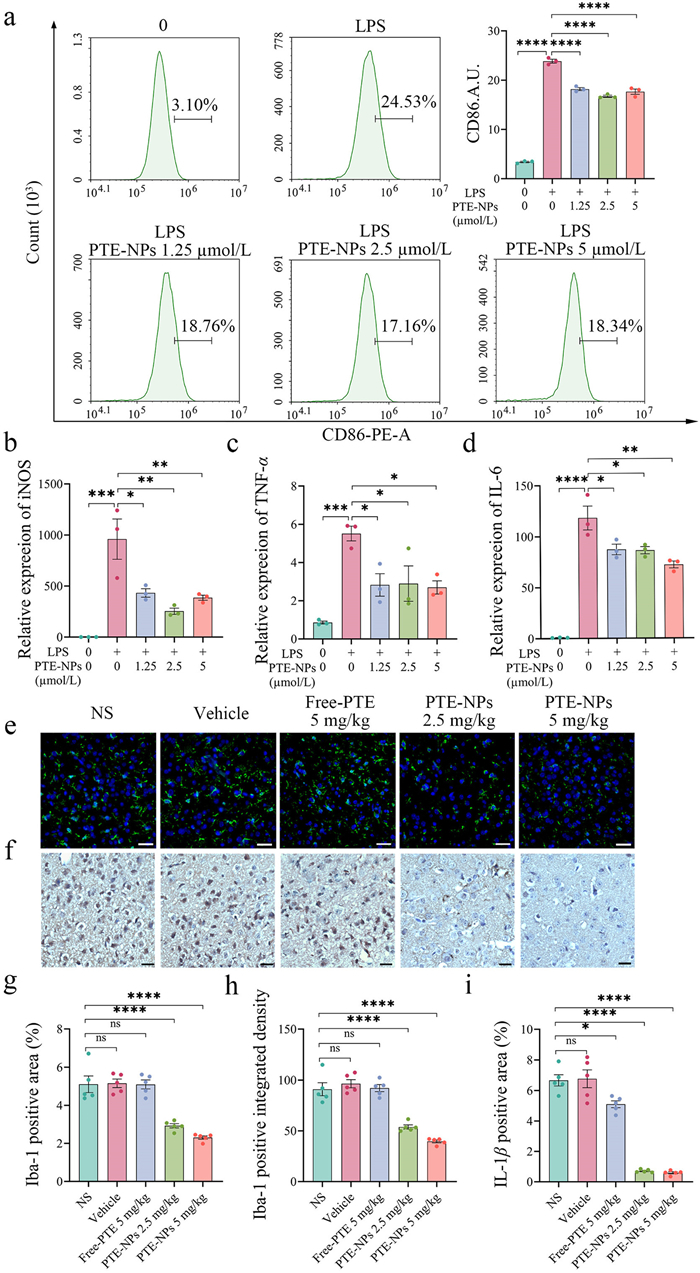
In the early stage of ICH, microglia-mediated pro-neuroinflammation is a key contributor to brain injury [36–38]. To assess the anti-neuroinflammatory effects of PTE-NPs, we established a BV2 microglia inflammation model in vitro. The gradient concentrations of PTE-NPs were selected based on BV2 cell viability (Fig. S6 in Supporting information). Flow cytometry analysis revealed that PTE-NPs pre-treatment significantly reduced CD86 expression in BV2 cells following lipopolysaccharide (LPS) stimulation (Fig. 3a), indicating inhibition of pro-inflammatory polarization. Real-time quantitative polymerase chain reaction (RT-qPCR) analysis demonstrated that PTE-NPs notably reduced the expression of pro-inflammatory cytokines such as inducible nitric oxide synthase (iNOS), TNF-α, and IL-6 following LPS stimulation (Figs. 3b–d). We further evaluated the anti-neuroinflammatory effects of PTE-NPs in vivo using a collagenase-induced ICH mouse model. Three days after IN administration of PTE-NPs, immunofluorescence and immunohistochemical staining were performed to assess microglia activation and IL-1β expression around the hematoma. Ionized calcium-binding adapter molecule 1 (Iba-1), an indicator of microglial activation, was measured in the peri–hematomal area. As shown in Figs. 3e, g and h, IN administration of free PTE failed to suppress microglia activation, whereas PTE-NPs significantly reduced Iba-1 expression at an equivalent PTE dose of 2.5 mg/kg. Additionally, while a free PTE dose of 5 mg/kg slightly reduced IL-1β expression, a PTE-NPs dose of 2.5 mg/kg exhibited a more significant reduction in IL-1β levels (Figs. 3f and i).
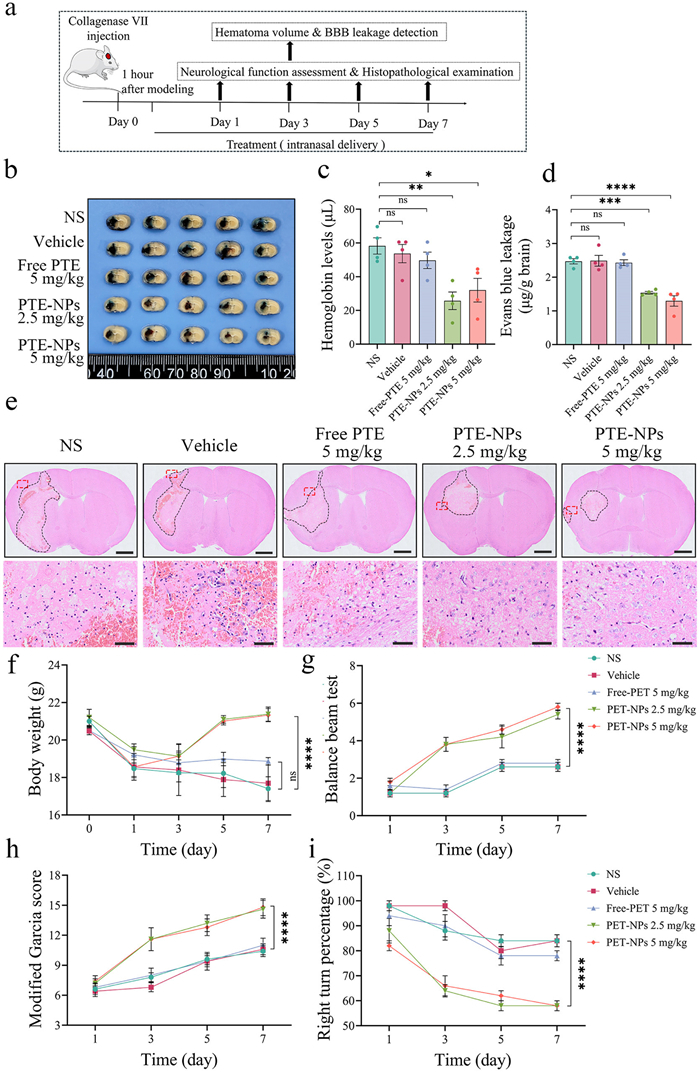
To comprehensively evaluate the therapeutic efficacy of PTE-NPs, we used two ICH mouse models, a collagenase-induced model and an autologous blood injection model. As shown in Fig. 4a, treatment was initiated 1 h after collagenase injection, with IN administration performed once daily for 7 days. On day 3 post-ICH induction, we measured hemoglobin levels in the ipsilateral brain tissue and assessed BBB integrity using Evans blue leakage assay. As shown in Figs. 4b–d, free PTE (5 mg/kg) failed to reduce hematoma size or BBB disruption, whereas PTE-NPs at 2.5 mg/kg significantly decreased both hematoma volume and Evans blue leakage. Neurological deficits were assessed using body weight changes, the balance beam test, the modified Garcia score, and the right-turn test. As shown in Fig. 4f, PTE-NPs significantly attenuated ICH-induced weight loss compared to the normal saline (NS), vehicle, and free-PTE groups. Moreover, mice treated with PTE-NPs exhibited better performance in the balance beam test and modified Garcia score (Figs. 4g and h), along with a significant reduction in right-turn percentage (Fig. 4i). Correlations between hematoma size, BBB disruption, and neurological deficits were further analyzed. As shown in Fig. S7 (Supporting information), hemoglobin content and Evans blue leakage on day 3 correlated positively with the balance beam and modified Garcia scores but negatively with the right turn percentage. Histopathological analysis revealed that PTE-NPs reduced monocyte infiltration around the hematoma (Fig. 4e). Nissl staining showed that PTE-NPs significantly increased neuronal survival around the hematoma, whereas free PTE failed to do so (Figs. S8a and b in Supporting information). The basal ganglia is a common site of ICH onset, and neuronal survival in this region is closely associated with functional recovery. Correlation analysis confirmed a significant association between striatal neuron density on day 3 post-ICH and neurological performance (Figs. S8c–e in Supporting information). After day 7 of ICH, the brain histopathological evaluation revealed that PTE-NPs reduced the glial scar formation, protected myelin integrity, and alleviated iron deposition around the hematoma (Fig. S9 in Supporting information). Similarly, the therapeutic efficacy of PTE-NPs was assessed in the autologous blood injection ICH mouse model (Fig. S10 in Supporting information). PTE-NPs IN administration significantly reduced hematoma size, BBB damage, and neurological deficits of ICH mice. Correlations between hematoma volume, BBB leakage, and neurological function outcomes were consistent with findings from the collagenase-induced ICH mouse model (Fig. S11 in Supporting information).
Finally, we assessed the biosafety of PTE-NPs. On day 7 after treatment, major organs and peripheral blood were collected. Hematoxylin and eosin (HE) staining of major organs revealed no histopathological abnormalities after PTE-NPs treatment (Fig. S12 in Supporting information). The blood biochemical parameters in the PTE-NPs treatment group showed no significant differences compared to the NS group (Fig. S13 in Supporting information). These findings confirm that IN administration of PTE-NPs is well biosafe and holds promise for clinical translation.
In conclusion, we developed a nanoparticle-based drug delivery system encapsulating PTE and demonstrated its efficient, non-invasive delivery to the ICH microenvironment via IN administration. PTE-NPs exhibited uniform particle size and sustained-release properties in SNF. Compared to free PTE, PTE-NPs efficiently crossed the nasal mucosal epithelial barrier and mucus layer, facilitating enhanced uptake by microglia. Intranasally administered PTE-NPs demonstrated slow and stable penetration across the nasal mucosal barrier, enabling targeted accumulation at the ICH nidus. This approach offered easy administration and prolonged therapeutic effects. Once internalized by microglia surrounding the hematoma, PTE-NPs effectively inhibited pro-inflammatory polarization of microglia and the release of pro-inflammatory cytokines, thereby reducing inflammation-induced neuronal damage in the peri–hematomal region. In two ICH mouse models, IN administration of free PTE failed to show therapeutic effects. In contrast, PTE-NPs, even at a lower equivalent free PTE dose, significantly reduced hematoma volume and BBB leakage, alleviated pathological brain damage, and markedly improved neurological function in ICH mice. Moreover, PTE-NPs demonstrated excellent biosafety. This research offers novel perspectives on optimizing brain-targeted drug delivery and increasing therapeutic effectiveness for ICH treatment.
The authors declare no known competing interests or personal relationships that could have influenced the work reported in this paper.
Zhongxin Duan: Resources, Methodology, Investigation. Yue Wang: Methodology, Investigation, Formal analysis. Yunchu Zhang: Resources, Methodology. Xia Liu: Project administration, Methodology. Wanyu Wang: Software, Methodology. Hua Li: Methodology, Investigation. Qingyang Lu: Investigation. Chao You: Supervision, Methodology. Yongzhong Cheng: Formal analysis. Cong Wu: Writing – review & editing, Writing – original draft, Investigation, Formal analysis. Xiang Gao: Writing – review & editing, Writing – original draft, Funding acquisition.
This work was supported by the National Natural Science Foundation of China (Nos. 32222046, 82172630), the National Key Research and Development Program of China (No. 2024YFA1210202), the Sichuan Science and Technology Program (No. 2023NSFSC1931), and 1·3·5 Project for Disciplines of Excellent, West China Hospital, Sichuan University (No. ZYYC25008). We would also like to thank BioRender (
Supplementary material associated with this article can be found, in the online version, at doi:
L. Puy, A.R. Parry-Jones, E.C. Sandset, et al., Nat. Rev. Dis. Primers 9 (2023) 14. doi: 10.1038/s41572-023-00424-7
J. Magid-Bernstein, R. Girard, S. Polster, et al., Circ. Res. 130 (2022) 1204–1229. doi: 10.1161/circresaha.121.319949
K.N. Sheth, N. Engl. J. Med. 387 (2022) 1589–1596. doi: 10.1056/nejmra2201449
C.J. van Asch, M.J. Luitse, G.J. Rinkel, et al., Lancet Neurol. 9 (2010) 167–176. doi: 10.1016/S1474-4422(09)70340-0
S.J. An, T.J. Kim, B.W. Yoon, J. Stroke 19 (2017) 3–10. doi: 10.5853/jos.2016.00864
B.M. Hansen, O.G. Nilsson, H. Anderson, et al., J. Neurol. Neurosurg. Psychiatry 84 (2013) 1150–1155. doi: 10.1136/jnnp-2013-305200
S.M. Greenberg, W.C. Ziai, C. Cordonnier, et al., Stroke 53 (2022) e282–e361.
V.L. Feigin, B.A. Stark, C.O. Johnson, et al., Lancet Neurol. 20 (2021) 795–820. doi: 10.1016/S1474-4422(21)00252-0
H. Ren, R. Han, X. Chen, et al., J. Cereb. Blood Flow Metab. 40 (2020) 1752–1768. doi: 10.1177/0271678x20923551
M. Xue, V.W. Yong, Lancet Neurol. 19 (2020) 1023–1032. doi: 10.1016/S1474-4422(20)30364-1
H. Chu, C. Huang, Z. Zhou, et al., Int. J. Surg. 109 (2023) 266–276. doi: 10.1097/js9.0000000000000191
E.R. Bader, T.A. Pana, R.S. Barlas, et al., J. Neurol. 269 (2022) 6330–6341. doi: 10.1007/s00415-022-11284-8
T. Shichita, H. Ooboshi, A. Yoshimura, Nat. Rev. Neurosci. 24 (2023) 299–312. doi: 10.1038/s41583-023-00690-0
Q. Li, X. Lan, X. Han, et al., Brain Behav. Immun. 94 (2021) 437–457. doi: 10.1016/j.bbi.2021.02.001
C. Xie, J. Liao, N. Zhang, et al., Chin. Chem. Lett. 35 (2024) 109149. doi: 10.1016/j.cclet.2023.109149
X. Li, X. Gao, W. Zhang, et al., Cell Death Dis. 13 (2022) 33. doi: 10.1038/s41419-021-04424-x
W. Chen, Y. Zhang, X. Zhai, et al., J. Cereb. Blood Flow Metab. (2022) 1579–1596. doi: 10.1177/0271678x221098841
H. Kim, K.H. Seo, W. Yokoyama, J. Agric. Food Chem. 68 (2020) 12836–12841. doi: 10.1021/acs.jafc.0c00070
B.J. Dutta, P.S. Rakshe, N. Maurya, et al., Ageing Res. Rev. 92 (2023) 102125. doi: 10.1016/j.arr.2023.102125
Z.H. Yang, Y.J. Liu, W.K. Ban, et al., Food Funct. 14 (2023) 8291–8308. doi: 10.1039/d3fo02639f
Y. Chen, W. He, J. Qiu, et al., Cell Mol. Biol. Lett. 29 (2024) 114. doi: 10.1186/s11658-024-00634-1
J. Liu, J. Xu, L. Jia, et al., Int. J. Pharm. 655 (2024) 124002. doi: 10.1016/j.ijpharm.2024.124002
J. Xu, J. Liu, Q. Li, et al., Phytomedicine 119 (2023) 155011. doi: 10.1016/j.phymed.2023.155011
F. Pacifici, C. Salimei, D. Pastore, et al., Int. J. Mol. Sci. 23 (2022) 3110. doi: 10.3390/ijms23063110
R.W. Dellinger, H.E. Holmes, T. Hu-Seliger, et al., Hepatology 78 (2023) 863–877. doi: 10.1002/hep.32778
J.B. Jensen, O.L. Dollerup, A.B. Moller, et al., JCI Insight 7 (2022) e158314. doi: 10.1172/jci.insight.158314
M. Waszczuk, S.E. Bianchi, V. Pittol, et al., Int. J. Pharm. 635 (2023) 122736. doi: 10.1016/j.ijpharm.2023.122736
B. Wang, S. Hu, Y. Teng, et al., Signal Transduct. Target. Ther. 9 (2024) 200. doi: 10.1201/9781003483755-20
H. Wu, Y. Wang, Z. Ren, et al., Chin. Chem. Lett. 36 (2025) 109996. doi: 10.1016/j.cclet.2024.109996
J.K. Patra, G. Das, L.F. Fraceto, et al., J. Nanobiotechnology 16 (2018) 71.
J. Li, M. Zheng, O. Shimoni, et al., Adv. Sci. 8 (2021) e2101090. doi: 10.1002/advs.202101090
C.P. Costa, J.N. Moreira, J.M. Sousa Lobo, et al., Acta Pharm. Sin. B 11 (2021) 925–940. doi: 10.1016/j.apsb.2021.02.012
H. Shen, N. Aggarwal, B. Cui, et al., Cell 188 (2025) 1545–1562. doi: 10.1016/j.cell.2025.01.017
D. Wu, Q. Chen, X. Chen, et al., Signal Transduct. Target. Ther. 8 (2023) 217.
A.A. Yakubova, K.A. Mitusova, A. Darwish, et al., J. Control. Release 359 (2023) 400–414. doi: 10.1016/j.jconrel.2023.06.012
Y. Wang, P. Tang, W. Tu, et al., Chin. Chem. Lett. 36 (2025) 109955. doi: 10.1016/j.cclet.2024.109955
C. Wei, C. Chen, S. Li, et al., Redox Biol. 80 (2025) 103487. doi: 10.1016/j.redox.2024.103487
X. Lan, X. Han, Q. Li, et al., Nat. Rev. Neurol. 13 (2017) 420–433. doi: 10.1038/nrneurol.2017.69

Scheme 1 PTE-NPs intranasal administration alleviated neuroinflammation after ICH via suppressing pro-inflammatory microglia activation and pro-inflammatory cytokine release.

Figure 1 The characteristics of PTE-NPs. (a) illustration of PTE-NPs preparation. (b) TEM image of PTE-NPs (scale bar: 50 nm). (c) Particle size of PTE-NPs. (d) Zeta potential of PTE-NPs. (e) Cumulation release of PTE from PTE-NPs in 10% FBS-PBS and SNF (n = 3). Data are presented as mean ± standard error of the mean (SEM).

Figure 2 The nasal Ce6-NPs permeability assay in vitro and brain targeting of Ce6-NPs via different administration routes in vivo. (a) illustration of nasal mucosal epithelium model, PTE-NPs permeability assay, and BV2 cell uptake assay in vitro. (b) Ce6-NPs had a significant uptrend of penetration with incubation time compared to free Ce6 (n = 3). (c, d) Ce6-NPs were significantly taken in by BV2 compared to free Ce6 in the nasal mucosal epithelium model (n = 3). (e, f) Brain fluorescence intensity changed over time in ICH mice after Ce6-NPs intranasal or intravenous administration (n = 5). (g, h) Ex vivo brain fluorescence intensity was measured 24 h post-administration of Ce6-NPs via intranasal or intravenous routes (n = 4). ns, no significance. *P < 0.05, *P < 0.01, ****P < 0.0001. Data are presented as mean ± SEM.

Figure 3 The anti-neuroinflammatory effect evaluation of PTE-NPs in vitro and in vivo. (a) Flow cytometry analysis revealed that 12 h pre-treatment with PTE-NPs significantly decreased CD86 fluorescence intensity on BV2 cell surfaces following 24 h LPS stimulation (n = 3). (b–d) Real-time quantitative PCR analysis revealed that pre-treatment with PTE-NPs for 12 h, followed by 24 h of LPS stimulation, significantly decreased the mRNA expression of pro-inflammatory cytokines iNOS, TNF-α, and IL-6 in BV2 cells (n = 3). (e, g, h) immunofluorescence assay of Iba-1 expression around hematoma of ICH mice after 3 days PTE-NPs IN administration, green fluorescence was defined as Iba-1 positive signal, blue fluorescence was defined as 4′, 6-diamidino-2-phenylindole (DAPI) positive signal. PTE-NPs administration significantly reduced Iba-1 expression around hematoma of ICH mice (n = 5). (f, i) immunohistochemistry assay of IL-1β expression around hematoma of ICH mice after 3 days PTE-NPs IN administration, the brown area was defined as IL-1β positive signal, blue area as defined as cell nucleus positive signal. The administration of PTE-NPs notably decreased IL-1β expression around the hematoma in ICH mice (n = 5). Scale bar: 20 µm. *P < 0.05, *P < 0.01, ***P < 0.001, ****P < 0.0001. Data are presented as mean ± SEM.

Figure 4 The therapeutic efficacy of PTE-NPs IN administration in ICH mice induced by collagenase injection. (a) Schedule of experimental design for the therapeutic efficacy of PTE-NPs IN administration in ICH mice. (b–d) IN administration of PTE-NPs significantly decreased hematoma volume and BBB disruption in ICH mice 3 days post-administration (n = 4). (e) The representative brain section of ICH mice after 3 days of different treatments. PTE-NPs IN administration reduced monocyte infiltration around the hematoma. Scale bar: 1 mm (up), 50 µm (down). (f) PTE-NPs IN administration significantly increased the body weight of ICH mice after 3 days of administration (n = 5). (g, h) PTE-NPs IN administration significantly increased balance beam test score and modified Garcia score of ICH mice after 3 days of administration (n = 5). (i) PTE-NPs IN administration significantly reduced the right turn percentage of ICH mice after 3 days of administration (n = 5). *P < 0.05, *P < 0.01, ***P < 0.001, ****P < 0.0001. Data are presented as mean ± SEM.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
 下载:
下载: