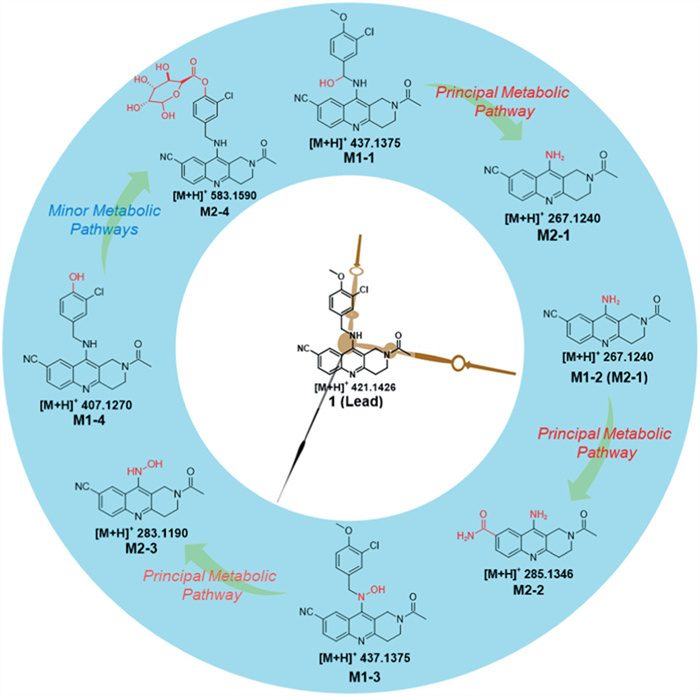
Figure 1.
Metabolite analysis of the hit compound reveals that the "N-(3-chloro-4-methoxybenzyl)" group was the metabolic soft spots.
Plasma metabolites-based drug design: Discovery of novel and highly selective phosphodiesterase 5 inhibitors
Deyan Wu , Qingjiang Ma , Yanquan Chen , Guofeng Yang , Fengcai Zhang , Meiyan Jiang , Xue Wang , Xingfu Liu , Qian Zhou , Yi-You Huang , Zhe Li , Hai-Bin Luo

One of the biggest challenges in the drug discovery is to identify the high-quality hit or lead compounds. In general, early drug discovery and development usually starts with accidental discovery and natural product [1]. With the improvements in drug development strategies the new drug screening methods have emerged and always played an important role [2,3], such as structure-based drug design (SBDD), fragment-based drug discovery (FBDD), high-throughput screening, scaffold hopping, biocatalysis, structural biology, and artificial Intelligence techniques [4-6]. However, the fate of a drug depends on each other administration in the body involves absorption, distribution, metabolism, and excretion (ADME), which is a complex biotransformation process [7,8]. Therefore, the metabolization characteristics of the drug on pharmacokinetics (PK), pharmacodynamics (PD), and safety should be carefully considered.
There are generally accepted rules were effectively applied to improving PK and PD properties, such as structural modification on or around the metabolic soft spots [9,10], metabolic switching and deuterium replacement [11,12], prodrugs or active metabolites as new drug candidates [4,13,14], minimizing toxicity potential associated with bioactivation [13,14], and the machine learning techniques improve the decision-making in drug discovery in recent years [15,16], but the structure-metabolism relationships of the drug is complex and still not completely understood [7]. Therefore, new strategies for drug discovery still need to be discovered.
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive and irreversible interstitial lung disease and effective therapy is lacking that resulting in a median survival of only 2.5–3.5 years after diagnosis [17-21]. However, effective therapy for IPF is still lacking, and only two anti-fibrotic drugs (pirfenidone and nintedanib) are available to delay the disease progression [22-25]. Sildenafil, a phosphodiesterase 5 (PDE5) inhibitor, has already used in anti-IPF study [26,27], but it has poor selectivity of 10-fold over PDE6 and leading to several side effects [28,29]. Moreover, almost all of the currently marketed PDE5 inhibitors (such as sildenafil, vardenafil, and tadalafil) existing of these common side effects [28]. Sildenafil, vardenafil, and avanafil results in the visual side effects by inhibiting PDE6 [28,30]. Tadalafil, on the other hand, is selective versus PDE6, but potently inhibits PDE11 which result in the back and muscle pain [28]. Therefore, the discovery of novel selective PDE5 inhibitors with improved safety profiles would be a welcome contribution to drugs study.
Herein, we reported a metabolites-based scaffold hopping strategy with the absolute binding free energy calculation for the discovery of 2,3,3a,4,5,6-hexahydro-1H-benzo[b]pyrido[2,3,4-de][1,6]naphthyridines as novel and highly selective PDE5 inhibitors with remarkable metabolic stability for the treatment of IPF. In this study, the hit compound (1) with a 1,2,3,4-tetrahydro benzo[b][1,6]naphthyridine scaffold and exhibited potent inhibitory activity as well as excellent selectivity, whereas it was unstable in rat liver microsomes with the t1/2 of 3.2 min [31]. And the subsequent metabolic studies shows that the "N-(3-chloro-4-methoxybenzyl)" group was the metabolic soft spots (Fig. 1 and Table S1 in Supporting information).
In order to achieve the goal of discovering highly potent and selective PDE5 inhibitors with reasonable drug-like properties, a "2,3,3a,4,5,6-hexahydro-1H-benzo[b]pyrido[2,3,4-de][1,6]naphthyridine" scaffold was designed to blocking the metabolic sites of 1 via cyclizing the N-(3-chloro-4-methoxybenzyl) motif to the 1,2,3,4-tetrahydrobenzo[b][1,6]naphthyridines (Scheme 1A). Following the liver microsomal stability study show that the novel compound L1 have a dramatic improvement on the metabolic stability with t1/2 of 139 min. It is worth mentioning that we have developed an innovative drug design method, a Gaussian algorithm-enhanced free energy perturbation (GA-FEP) protocol [32-34], which has been used in this study to discover highly potent PDE5 inhibitors (Scheme 1B). In addition, their predicted binding patterns were further verified by determining a cocrystal structure of the PDE5-inhibitor complex (Scheme 1C), and the predicted binding free energies correlate well with the experimental activity data (Scheme 1B).
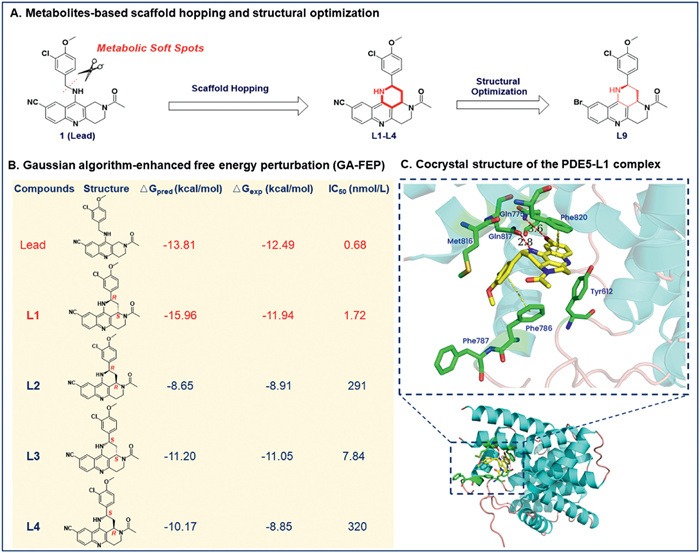
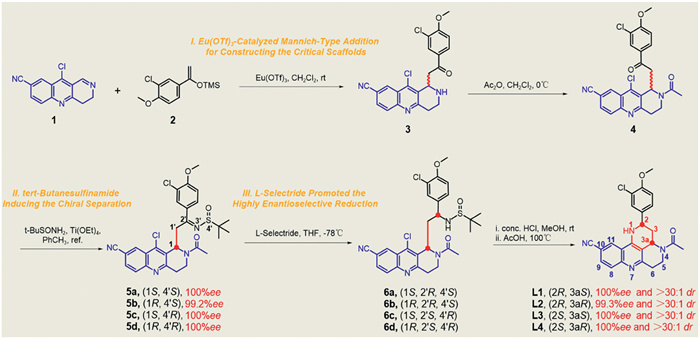
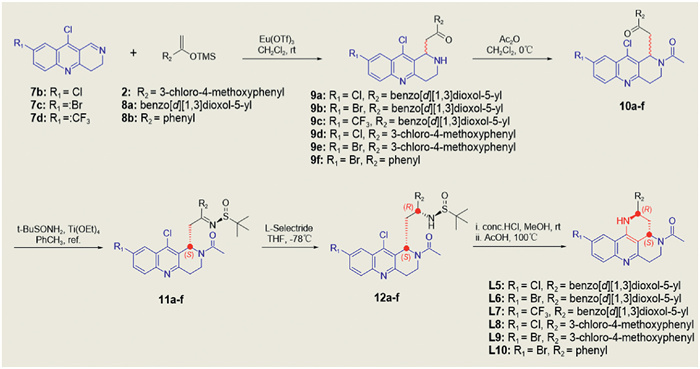
However, these chiral 1,2,3,4-tetrahydrobenzo[b][1,6]naphthyridines has not been explored. In this study, we wish to disclosure a concise and efficient method for the preparation of chiral 2,3,3a,4,5,6-hexahydro-1H-benzo[b]pyrido[2,3,4-de][1,6] naphthyridines as novel PDE5 inhibitors with high enantioselectivity (> 99% ee and > 30:1 dr) and good yields which highlighted the utility of a Eu(OTf)3-catalyzed Mannich-type reaction for constructing the key scaffolds (Scheme 2). Specifically, by use 10-chloro-3,4-dihydrobenzo[b][1,6]naphthyridine-8-carbonitrile (1) and ((1-(3-chloro-4-methoxyphenyl)vinyl)oxy)trimethylsilane (2) as the starting materials and treated with Eu(OTf)3 or Sc(OTf)3 in dichloromethane to get the key compound 3. Then acetylation of the amine 3 to afford the ketone 4. This intermediate is a reactive electrophilic reagent that readily react with the chiral tert-butanesulfinamide [35] resulting in afforded sulfonamides (5) and make the separation of the diastereoisomers easier by column chromatography, respectively. After separation of the diastereomers, an l-selectride catalyzed highly enantioselective reduction reaction [36] was conducted to afford the functionalized chiral amines (6), respectively. Finally, an intramolecular cyclization of the amines 6 giving the target compounds L1–4 with high enantioselectivities (> 99% ee and > 30:1 dr) and high yields, respectively. Under optimized reaction conditions and for the purpose of further explore the structure−activity relationships around the novel scaffold, derivatives L5–10 were synthesized (Scheme 3). The results of exploratory studies have demonstrated that the Eu(OTf)3-catalyzed Mannich-type reaction followed by l-selectride catalyzed reduction serves as an effective method for the preparation of useful chiral 2,3,3a,4,5,6-hexahydro-1H-benzo[b]pyrido[2,3,4-de][1,6] naphthyridines.
As a result, ten 2,3,3a,4,5,6-hexahydro-1H-benzo[b]pyrido[2,3,4-de][1,6]naphthyridines were successfully synthesized with excell ent enantioselectivity (> 99% ee and > 30:1 dr) and potent inhibitory activities against PDE5 (Table 1). It is worth mentioning that the hit compound has a half maximal inhibitory concentration (IC50) of 0.68 nmol/L under identical assay conditions, and sildenafil was used as the positive control compound. The results showed that compound L1 based on scaffold hopping has the equivalent inhibitory potency as the hit compound. However, other configurations of L1 exhibited the decreased inhibitory activities, especially for the compounds with an 2S-configuration (L3 and L4). Further investigation revealed that the -CN group may be replaced with a -Br or -Cl group (L1, L8, and L9), while the -Br group exhibited the better inhibitory activities. However, other modifications, such as replacement of a 3-chloro-4-methoxyphenyl group with a benzo[d][1,3]dioxol-5-ylmethyl group (L6) or a phenyl group (L10) exhibited weaker PDE5 inhibitory activities than compound L9. Finally, L9 exhibited highly potent inhibitory activity against PDE5 with the IC50 of 1.03 ± 0.03 nmol/L was selected as the potent inhibitor for subsequent study.
 DownLoad:
CSV
DownLoad:
CSV
| Compound | R1 | R2 | Stereochemistry | ee (%)a | dra | PDE5, IC50 (nmol/L) |
| Hit | − | − | − | − | − | 0.68 |
| Sildenafilb | − | − | − | − | − | 5.0 |
| L1 | -CN |  |
2R, 3aS | 100 | > 30:1 | 1.72 ± 0.34 |
| L2 | -CN |  |
2R, 3aR | 99.3 | > 30:1 | 7.84 ± 0.98 |
| L3 | -CN |  |
2S, 3aS | 100 | > 30:1 | 291 ± 16 |
| L4 | -CN |  |
2S, 3aR | 100 | > 30:1 | 320 ± 27 |
| L5 | -Cl |  |
2R, 3aS | 100 | > 30:1 | 16.1 ± 1.1 |
| L6 | -Br |  |
2R, 3aS | 99.2 | > 30:1 | 19.9 ± 1.5 |
| L7 | -CF3 |  |
2R, 3aS | 100 | > 30:1 | 276 ± 26 |
| L8 | -Cl |  |
2R, 3aS | 100 | > 30:1 | 16.4 ± 0.8 |
| L9 | -Br |  |
2R, 3aS | 100 | > 30:1 | 1.03 ± 0.03 |
| L10 | -Br |  |
2R, 3aS | 100 | > 30:1 | 47.2 ± 6.2 |
| a Determined by chiral HPLC analysis (Opti-Chiral A1–5). b Sildenafil, the reference compound. |
||||||
Given the highly potent activities of compound L9, we measured the selectivity index (SI) across phosphodiesterase (PDE) subtypes. Under identical assay conditions, L9 have a SI of 898-fold against PDE6, and the SI values against PDE1, PDE2, PDE3, PDE4, PDE7, PDE8, PDE9, PDE10, PDE11 were uniformly greater than 1000-fold (Table 2). These results suggest that L9 exhibited at least 898-fold selectivity over other PDE families (Table 2). It is worth mentioned is sildenafil only has a SI of 8~10-fold against PDE6 [28,37-41], while tadalafil only has a SI of 5~9-fold against PDE11 (Table 2) [28,37]. Therefore, the study discovered 2,3,3a,4,5,6-hexahydro-1H-benzo[b]pyrido[2,3,4-de][1,6]naphthyridines as novel PDE5 inhibitors with higher selectivity over other PDE families, especially for PDE6 and PDE11, which is of great significance in the potential relief of several side effects.
DownLoad:
CSV
| PDE isozyme | L9 | Sildenafila | Tadalafila | |||||
| IC50 (nmol/L) | SI | IC50 (nmol/L) | SI | IC50 (nmol/L) | SI | |||
| PDE5 | 1.03 ± 0.03 | − | 4.31 ± 0.46 | − | 2.35 ± 0.28 | − | ||
| PDE1 | > 10,300 | > 10,000 | 819 ± 89 | 190 | > 10,000 | > 4255 | ||
| PDE2 | > 10,300 | > 10,000 | > 10,000 | > 2320 | > 10,000 | > 4255 | ||
| PDE3 | > 10,300 | > 10,000 | > 10,000 | > 2320 | > 10,000 | > 4255 | ||
| PDE4 | > 1030 | > 10,000 | > 10,000 | > 2320 | > 10,000 | > 4255 | ||
| PDE6 | 924.5 | 898 | 36.4 ± 1.8 | 8 | 402 ± 56 | 171 | ||
| PDE7 | > 10,300 | > 10,000 | > 10,000 | > 2320 | > 10,000 | > 4255 | ||
| PDE8, | > 10,300 | > 10,000 | > 10,000 | > 2320 | > 10,000 | > 4255 | ||
| PDE9 | > 10,300 | > 10,000 | > 10,000 | > 2320 | > 10,000 | > 4255 | ||
| PDE10 | > 10,300 | > 10,000 | > 10,000 | > 2320 | > 10,000 | > 4255 | ||
| PDE11 | > 1030 | > 1000 | 4930 ± 1140 | 1144 | 22.1 ± 5.9 | 9 | ||
| aThe PDE selectivity data of the reference drugs from Ref. [35]. | ||||||||
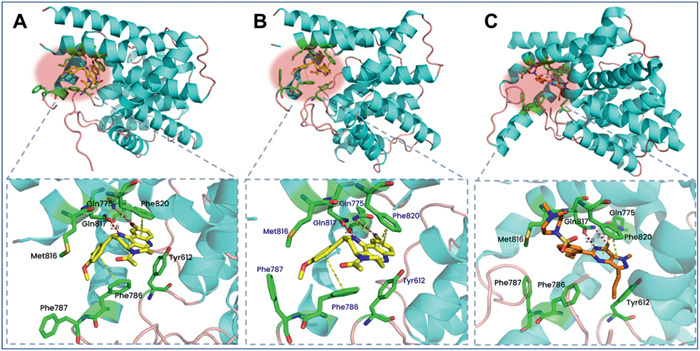
The co-crystals further revealed that 2,3,3a,4,5,6-hexahydro-1H-benzo[b]pyrido[2,3,4-de][1,6]naphthyridines had a specific binding pattern. In this study, the cocrystal structures of PDE5 with L1 and L9 were successfully determined (PDB ID: 9LSL and 9LSM), respectively. As shown in Fig. 2, the structures indicated that L1 (Fig. 2A) and L9 (Fig. 2B) occupies the active pocket with a unique binding pattern. Specifically, each of the NH group formed H-bond with residue Gln817 and the tetracyclic ring formed π−π stacking interaction with residue Phe820, which are two characteristic interactions of the reported PDE5 inhibitors (e.g., sildenafil, Fig. 3C). However, the -CN group of L1 or bromine of L9 formed another H-bond with residue Gln775, and the 3-chloro-4-methoxyphenyl group occupies the small pocket lined by Phe786, Phe787, Leu804, and Met816. These interactions were not observed in the binding pattern of PDE5/sildenafil (Fig. 3C), interpreting that 2,3,3a,4,5,6-hexahydro-1H-benzo[b]pyrido[2,3,4-de][1,6]naphthyridines exhibited higher selectivity.
Since compounds exhibited potent activities against PDE5 and highly selectivity over other PDEs, its drug-like profile evaluations were performed. We examined the metabolic stability of designed compounds using a standard microsomal stability assay. The results indicate that L1 and L9 made a dramatic improvement on the metabolic stability with the t1/2 of 139 and 79 min than the hit 1 of 3.2 min, respectively. In this study, the inhibitory activities of compound L9 against human hepatic CYP enzymes (Table S3 in Supporting information) were tested. And the results shows that L9 has an IC50 of 2.2 µmol/L against CYP1A2, and its IC50 values for the other CYPs (CYP2C9, CYP2C19, CYP2D6, and CYP3A4) were uniformly > 25 µmol/L, suggesting that L9 exhibited a very weak inhibitory effect on these CYP isoenzymes. Besides, compound L9 inhibited the human ether-à-go-go-related gene (hERG) with an IC50 of 0.18 µmol/L using an automated patch clamp electrophysiology measurement in CHO-hERG cells (Fig. S1 in Supporting information). The results suggest that L9 have a moderate inhibitory effect on hERG.
In terms of the reasonable druglike properties and higher selectivity of L9, its pharmacokinetic profile assessment in Sprague Dawley rats after oral administration (po) and intravenous administration (iv) of L9·HCl were performed. Note that all animal care and experimental programs were in line with the "Guide of Laboratory Animals" (National Institutes of Health Publication, revised 1996, No. 86–23, Bethesda, MD) and approved by the Institutional Ethical Committee for Animal Research of Sun Yat-sen University (No. SYSU-IACUC-2021–000184). As a result, compound L9·HCl exhibited favorable pharmacokinetic properties, and its pharmacokinetic data are summarized in Table 3. After oral administration of a 5.0 mg/kg dose of L9·HCl to rats, pharmacokinetic analysis revealed it have a t1/2 of 3.79 ± 0.41 h, Cmax of 119 ± 11.5 ng/mL, area under curve (AUC(0-t)) of 1227 ± 161 ng h mL−1 and oral bioavailability of 47.2%, indicating that it could give a reasonable blood exposure (Table 3). On the basis of its pharmacokinetic profile, compound L9·HCl was further subjected to pharmacodynamics studies in vivo.
DownLoad:
CSV
| Route | t1/2 (h) | tmax (h) | Cmax (ng/mL) | AUC(0-t) (ng h mL−1) | AUC(0-∞) (ng h mL−1) | MRT(0-t) (h)a | F (%) |
| po (5.0 mg/kg) | 3.79 ± 0.41 | 4.67 ± 2.31 | 119 ± 11.5 | 1227 ± 161 | 1249 ± 170 | 6.54 ± 0.34 | 47.2 ± 0.01 |
| iv (2.5 mg/kg) | 4.56 ± 0.34 | 0.083 ± 0.00 | 363 ± 56.5 | 1295 ± 130 | 1323 ± 138 | 4.67 ± 0.02 | |
| a MRT: mean residence time. | |||||||
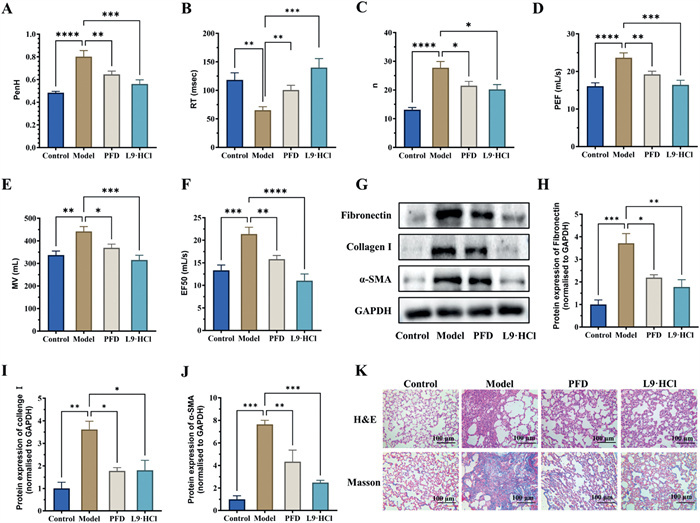
The therapeutic results of L9·HCl as anti-IPF agents in vivo are shown in Fig. 3, the pulmonary function parameters of rats in the blcomycin (BLM)-induced model group, including enhanced pause (penH), respiratory rate (n), peak expiratory flow (PEF), and mid-expiratory flow (EF50) remarkably increased compared with those in the control group, indicating that the BLM-induced IPF model was successfully established. After 28 consecutive days of oral administration, compound L9·HCl had remarkable efficacy in improving the lung function of IPF rats in terms of those parameters such as EF50, PEF, and penH reverted to resemble those in the control group (Figs. 3A–F). As a result, L9·HCl at an oral dose of 5.0 mg/kg had better anti-IPF effect than the marketed drug pirfenidone (PFD) administered at an oral dose of 150 mg/kg. Usually, collagen deposition was one of key pathological features in IPF, both L9·HCl and PFD almost completely inhibited collagen deposition to a normal level in this study. The expression levels of the fibronectin (FN), collagen I, and α-smooth muscle actin protein (α-SMA) in the lung tissue, the marker proteins in IPF, were upregulated in the model group relative to those in the control group as determined by Western blot (Figs. 3G–J). And L9·HCl and PFD significantly decreased the levels of FN, collagen I, and α-SMA. Hematoxylin and eosin (H & E) staining showed disordered alveoli, ruptured alveolar walls, and apparent inflammatory cell infiltration in the lung tissues of the model group (Fig. 3K). L9·HCl (5.0 mg/kg) and PFD (150 mg/kg) could both effectively alleviate this structural damage to the lungs with fewer fibrotic lesions. Besides, blue staining indicated that the expression of collagen deposition was greatly increased with large areas of collagen deposition around the trachea and cell hyperplasia in the model group (Fig. 3K). These results demonstrated that the PDE5 inhibitor L9·HCl ameliorated the BLM induced pulmonary fibrosis.
In conclusion, one of the biggest challenges in the drug discovery is to identify the highly potent hit or lead compounds with remarkable safety and pharmacokinetic profiles. Until now, almost all of the currently marketed PDE5 inhibitors (such as sildenafil, vardenafil, and tadalafil) have poor selectivity over PDE6 or PDE11, which leads to several side effects. In the study, a metabolites-based scaffold hopping strategy with the absolute binding free energy calculation was used to discover highly selective PDE5 inhibitors with remarkable metabolic stability from the starting compound 1 with a poor t1/2 of 3.2 min. It is worth noting that the Eu(OTf)3-catalyzed Mannich-type reaction followed by l-selectride catalyzed reduction serves as an efficient method for preparation of useful chiral 2,3,3a,4,5,6-hexahydro-1H-benzo[b]pyrido[2,3,4-de][1,6]naphthyridines as novel PDE5 inhibitors with high enantioselectivity (> 99% ee and > 30:1 dr) and high selectivity over other PDE subtypes. Further investigation of the scope of the cascade reaction and 10 derivatives of 2,3,3a,4,5,6-hexahydro-1H-benzo[b]pyrido[2,3,4-de][1,6]naphthyridines were discovered, resulting in 7 compounds with an IC50 values ranging from 1 nmol/L to 100 nmol/L and 3 compounds with the IC50 < 10 nmol/L. The most potent inhibitor L9 has an IC50 of 1.03 ± 0.03 nmol/L with high selectivity (≥898-fold) over other PDEs and oral bioavailability of 47.2%. And oral administration of L9·HCl (5.0 mg/kg) exhibited better anti-pulmonary fibrotic effects than the reference compound pirfenidone (150 mg/kg) in a bleomycin-induced IPF rat model, indicate that L9·HCl is a potential lead for the treatment of IPF.
In short, the strategies for improving both inhibitor selectivity against other isoforms and metabolic stability discussed above may be applicable for other drug discovery programs.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Deyan Wu: Writing – review & editing, Writing – original draft, Validation, Software, Resources, Formal analysis. Qingjiang Ma: Methodology, Investigation, Conceptualization. Yanquan Chen: Methodology, Investigation, Formal analysis, Data curation. Guofeng Yang: Methodology, Investigation, Formal analysis, Data curation. Fengcai Zhang: Methodology, Investigation, Formal analysis. Meiyan Jiang: Methodology, Investigation. Xue Wang: Investigation, Formal analysis. Xingfu Liu: Resources, Methodology. Qian Zhou: Resources, Methodology. Yi-You Huang: Methodology, Investigation. Zhe Li: Writing – review & editing, Writing – original draft, Visualization, Resources, Project administration. Hai-Bin Luo: Writing – review & editing, Supervision, Resources.
We acknowledge financial support from the Natural Science Foundation of China (Nos. 82373732, 22377023, 22307031, 81872727), Excellent Talent Team Project in Hainan Province (No. HNYT20250004), Hainan Provincial Natural Science Foundation of China (Nos. KJRC2023B10, 222RC556, 823CXTD375, 324MS018), Fundamental Research Funds for Hainan University (Nos. XTCX2022JKA01, KYQD(ZR)-21126, KYQD(ZR)-23003).
Supplementary material associated with this article can be found, in the online version, at doi:
J.W.H. Li, J.C. Vederas, Science 325 (2009) 161–165. doi: 10.1126/science.1168243
K.F. Zhu, C. Yuan, Y.M. Du, et al., Military Med. Res. 10 (2023) 10. doi: 10.1109/bigdatasecurity-hpsc-ids58521.2023.00013
Q. Gong, J. Song, Y. Song, et al., Chin. Chem. Lett. 36 (2025) 110456. doi: 10.1016/j.cclet.2024.110456
L.J. Gershell, J.H. Atkins, Nat. Rev. Drug Dis. 2 (2003) 321–327. doi: 10.1038/nrd1064
M. Bissaro, M. Sturlese, S. Moro, Drug Discov. Today 25 (2020) 1693–1701. doi: 10.1016/j.drudis.2020.06.023
A. Fryszkowska, P.N. Devine, Curr. Opin. Chem. Biol. 55 (2020) 151–160. doi: 10.1016/j.cbpa.2020.01.012
Z. Zhang, W. Tang, Acta Pharm. Sin. B 8 (2018) 721–732. doi: 10.1016/j.apsb.2018.04.003
Z. Zhang, M. Zhu, W. Tang, Curr. Pharm. Design 15 (2009) 2220–2235. doi: 10.2174/138161209788682460
D. Zhang, G. Luo, X. Ding, C. Lu, Acta Pharm. Sin. B 2 (2012) 549–561. doi: 10.1016/j.apsb.2012.10.004
M. Trunzer, B. Faller, A. Zimmerlin, J. Med. Chem. 52 (2009) 329–335. doi: 10.1021/jm8008663
N. Harada, G.T. Miwa, J.S. Walsh, A.Y. Lu, J. Biol. Chem. 259 (1984) 3005–3010. doi: 10.1016/S0021-9258(17)43249-2
C.J. Wenthur, R. Morrison, A.S. Felts, et al., J. Med. Chem. 56 (2013) 5208–5212. doi: 10.1021/jm400439t
J.E. Gallant, S. Deresinski, Clin. Infect. Dis. 37 (2003) 944–950. doi: 10.1086/378068
T.M. Chapman, J.K. McGavin, S. Noble, Drugs 63 (2003) 1597–1608. doi: 10.2165/00003495-200363150-00006
J. Vamathevan, D. Clark, P. Czodrowski, et al., Nat. Rev. Drug Dis. 18 (2019) 463–477. doi: 10.1038/s41573-019-0024-5
N. Pillai, A. Dasgupta, S. Sudsakorn, J. Fretland, P.D. Mavroudis, Drug Dis. Today 27 (2022) 2209–2215. doi: 10.1016/j.drudis.2022.03.017
T.E. King, A. Pardo, M. Selman, Lancet 378 (2011) 1949–1961. doi: 10.1016/S0140-6736(11)60052-4
F.J. Martinez, H.R. Collard, A. Pardo, et al., Nat. Rev. Dis. Primers 3 (2017) 17014. doi: 10.1038/nrdp.2017.14
B. Ley, H.R. Collard, T.E. King Jr, Am. J. Resp. Crit. Care 183 (2011) 431–440. doi: 10.1164/rccm.201006-0894CI
G. Raghu, S.Y. Chen, Q. Hou, W.S. Yeh, H.R. Collard, Eur. Respir. J. 48 (2016) 179–186. doi: 10.1183/13993003.01653-2015
T. Zhang, C. Sun, S. Yang, et al., Chin. Chem. Lett. 35 (2024) 109248. doi: 10.1016/j.cclet.2023.109248
G. Sgalla, B. Iovene, M. Calvello, et al., Resp. Res. 19 (2018) 32.
A. Azuma, T. Nukiwa, E. Tsuboi, et al., Am. J. Resp. Crit. Care 171 (2005) 1040–1047. doi: 10.1164/rccm.200404-571OC
O. Distler, K.B. Highland, M. Gahlemann, et al., New Engl. J. Med. 380 (2019) 2518–2528. doi: 10.1056/nejmoa1903076
P. Spagnolo, J.A. Kropski, M.G. Jones, et al., Pharmacol Therapeut 222 (2021) 107798.
The Idiopathic Pulmonary Fibrosis Clinical Research Network, New Engl. J. Med. 363 (2010) 620–628.
M.K. Han, D.S. Bach, P.G. Hagan et al., Chest 143 (2013) 1699–1708. doi: 10.1378/chest.12-1594
D. Pissarnitski, Med. Res. Rev. 26 (2006) 369–395. doi: 10.1002/med.20053
D. Wu, T. Zhang, Y. Chen, et al., J. Med. Chem. 60 (2017) 6622–6637. doi: 10.1021/acs.jmedchem.7b00523
A. Yamazaki, O. Moskvin, R.K. Yamazaki, Phosphorylation by cyclin-dependent protein kinase 5 of the regulatory subunit (Pγ) of retinal cGMP phosphodiesterase (PDE6): its implications in phototransduction, in: W. Baehr, K. Palczewski (Eds.), Advances in Experimental Medicine and Biology Photoreceptors and Calcium, Springer, Boston, 2002, pp. 131–153.
J. Fiorito, J. Vendome, F. Saeed, et al., J. Med. Chem. 60 (2017) 8858–8875. doi: 10.1021/acs.jmedchem.7b00979
Z. Li, X. Li, Y. Huang, et al., Proc. Natl. Acad. Sci. U. S. A. 17 (2020) 27381–27387. doi: 10.1073/pnas.2010470117
Z. Li, Y. Huang, Y. Wu, et al., J. Med. Chem. 62 (2019) 2099–2111. doi: 10.1021/acs.jmedchem.8b01763
D. Wu, X. Zheng, R. Liu, et al., Acta Pharm. Sin. B 12 (2022) 1351–1362.
J.L. Garcia Ruano, J. Alemán, C. Fajardo, A. Parra, Org. Lett. 7 (2005) 5493–5496. doi: 10.1021/ol052250w
Z.J. Liu, J.T. Liu, Chem. Commun. (2008) 5233–5235. doi: 10.1039/b810459j
Z. Wang, X. Jiang, X. Zhang, et al., J. Med. Chem. 62 (2019) 4979–4990. doi: 10.1021/acs.jmedchem.9b00123
D.P. Rotella, Nat. Rev. Drug Dis. 1 (2002) 674–682. doi: 10.1038/nrd893
T. Ukita, Y. Nakamura, A. Kubo, et al., Bioorg. Med. Chem. Lett. 13 (2003) 2341–2345.
H. Mochida, M. Takagi, H. Inoue, et al., Eur. J. Pharm. 456 (2002) 91–98.
N.K. Terrett, A.S. Bell, D. Brown, P. Ellis, Bioorg. Med. Chem. Lett. 6 (1996) 1819–1824.

Figure 1 Metabolite analysis of the hit compound reveals that the "N-(3-chloro-4-methoxybenzyl)" group was the metabolic soft spots.

Scheme 1 Schematic diagram for the metabolites-based scaffold hopping strategy combined with the absolute binding free energy calculation for the discovery of novel selective PDE5 inhibitors, and cocrystal structure of PDE5-L1 revealed that the novel scaffold has a suitable binding pattern.

Scheme 2 Synthetic pathway for the production of 2,3,3a,4,5,6-hexahydro-1H-benzo[b]pyrido[2,3,4-de][1,6]naphthyridines as novel and highly selective PDE5 inhibitors.

Scheme 3 Synthesis of 2,3,3a,4,5,6-hexahydro-1H-benzo[b]pyrido[2,3,4-de][1,6]naphthyridine derivatives.

Figure 2 Cocrystal structures of the PDE5-L1 and PDE5-L9 complexes (PDB codes 9LSL and 9LSM): (A) surface model of L1 with PDE5; (B) surface model of L9 with PDE5; (C) surface model of sildenafil with PDE5. The dotted lines refer to H-bonds (red) or π−π stacking interactions (yellow).

Figure 3 Anti-IPF effect of L9·HCl in a BLM-induced rat model. (A–F) Indicators of the pulmonary respiratory function in different groups of rats. Rats in each group: n = 12. (G–J) Western blot analysis of FN, collagen I, and α-SMA protein expression levels [relative to glyceraldehyde 3-phosphate dehydrogenase (GAPDH)] (n = 3). (K) The representative image of H & E strain and Masson strain. Scale bar: 100 µm. Data are shown as means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Table 1. 2,3,3a,4,5,6-Hexahydro-1H-benzo[b]pyrido[2,3,4-de][1,6]naphthyridines with high enantioselectivity and remarkable inhibitory activities against PDE5.a
| Compound | R1 | R2 | Stereochemistry | ee (%)a | dra | PDE5, IC50 (nmol/L) |
| Hit | − | − | − | − | − | 0.68 |
| Sildenafilb | − | − | − | − | − | 5.0 |
| L1 | -CN | |
2R, 3aS | 100 | > 30:1 | 1.72 ± 0.34 |
| L2 | -CN | |
2R, 3aR | 99.3 | > 30:1 | 7.84 ± 0.98 |
| L3 | -CN | |
2S, 3aS | 100 | > 30:1 | 291 ± 16 |
| L4 | -CN | |
2S, 3aR | 100 | > 30:1 | 320 ± 27 |
| L5 | -Cl | |
2R, 3aS | 100 | > 30:1 | 16.1 ± 1.1 |
| L6 | -Br | |
2R, 3aS | 99.2 | > 30:1 | 19.9 ± 1.5 |
| L7 | -CF3 | |
2R, 3aS | 100 | > 30:1 | 276 ± 26 |
| L8 | -Cl | |
2R, 3aS | 100 | > 30:1 | 16.4 ± 0.8 |
| L9 | -Br | |
2R, 3aS | 100 | > 30:1 | 1.03 ± 0.03 |
| L10 | -Br | |
2R, 3aS | 100 | > 30:1 | 47.2 ± 6.2 |
| a Determined by chiral HPLC analysis (Opti-Chiral A1–5). b Sildenafil, the reference compound. |
||||||
 下载: 导出CSV
下载: 导出CSV
Table 2. 2,3,3a,4,5,6-Hexahydro-1H-benzo[b]pyrido[2,3,4-de][1,6]naphthyridines with high enantioselectivity and remarkable inhibitory activities against PDE5.a
| PDE isozyme | L9 | Sildenafila | Tadalafila | |||||
| IC50 (nmol/L) | SI | IC50 (nmol/L) | SI | IC50 (nmol/L) | SI | |||
| PDE5 | 1.03 ± 0.03 | − | 4.31 ± 0.46 | − | 2.35 ± 0.28 | − | ||
| PDE1 | > 10,300 | > 10,000 | 819 ± 89 | 190 | > 10,000 | > 4255 | ||
| PDE2 | > 10,300 | > 10,000 | > 10,000 | > 2320 | > 10,000 | > 4255 | ||
| PDE3 | > 10,300 | > 10,000 | > 10,000 | > 2320 | > 10,000 | > 4255 | ||
| PDE4 | > 1030 | > 10,000 | > 10,000 | > 2320 | > 10,000 | > 4255 | ||
| PDE6 | 924.5 | 898 | 36.4 ± 1.8 | 8 | 402 ± 56 | 171 | ||
| PDE7 | > 10,300 | > 10,000 | > 10,000 | > 2320 | > 10,000 | > 4255 | ||
| PDE8, | > 10,300 | > 10,000 | > 10,000 | > 2320 | > 10,000 | > 4255 | ||
| PDE9 | > 10,300 | > 10,000 | > 10,000 | > 2320 | > 10,000 | > 4255 | ||
| PDE10 | > 10,300 | > 10,000 | > 10,000 | > 2320 | > 10,000 | > 4255 | ||
| PDE11 | > 1030 | > 1000 | 4930 ± 1140 | 1144 | 22.1 ± 5.9 | 9 | ||
| aThe PDE selectivity data of the reference drugs from Ref. [35]. | ||||||||
下载: 导出CSV
Table 3. Pharmacokinetic profile of L9·HCl in rats.
| Route | t1/2 (h) | tmax (h) | Cmax (ng/mL) | AUC(0-t) (ng h mL−1) | AUC(0-∞) (ng h mL−1) | MRT(0-t) (h)a | F (%) |
| po (5.0 mg/kg) | 3.79 ± 0.41 | 4.67 ± 2.31 | 119 ± 11.5 | 1227 ± 161 | 1249 ± 170 | 6.54 ± 0.34 | 47.2 ± 0.01 |
| iv (2.5 mg/kg) | 4.56 ± 0.34 | 0.083 ± 0.00 | 363 ± 56.5 | 1295 ± 130 | 1323 ± 138 | 4.67 ± 0.02 | |
| a MRT: mean residence time. | |||||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载: