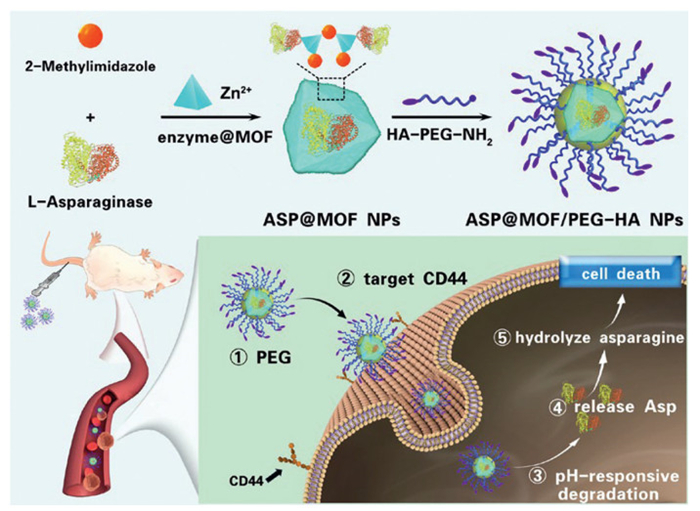
Scheme 1.
Schematic representation of the synthesis of ASP@MOF/PEG-HA NPs and the mechanism underlying anti-leukemia treatment.
Hyaluronic acid-modified MOF nanoparticles for encapsulating asparaginase in T-cell acute lymphoblastic leukemia treatment
Yeling Yuan , Lihua Du , Kejing Zeng , Yilu Zheng , Huaping Huang , Yu Shao , Wenqing Wang , Leping Yan , Jun Wu , Changhua Zhang , Hongman Xue , Haozhe He

Acute lymphoblastic leukemia (ALL) is a critical hematologic malignancy and the predominant form of pediatric cancer [1], contributing to about one-fourth of all pediatric cancer diagnoses [2,3]. ALL can be categorized into B-cell and T-cell types according to immunophenotype, with T-cell ALL constituting 15%–25% of new cases each year [4,5]. Patients with T-ALL typically exhibit a higher resistance to traditional chemotherapy, are associated with genetic subtypes indicative of poor outcomes, and have limited access to effective targeted treatments, immunotherapies, and T-cell therapies, leading to poorer survival rates compared to B-ALL patients [6].
L-Asparaginase (ASP) is an enzyme crucial for breaking down asparagine [7], an amino acid essential for the proliferation of lymphoblasts [8]. It has been a staple in multi-drug chemotherapy treatments for ALL since the 1960s [9]. Although effective, ASP can cause severe side effects such as allergic reactions, pancreatitis, liver damage, and thrombosis [10]. These adverse effects often lead many pediatric patients to stop the treatment [11], which negatively impacts the disease-free survival (DFS) among high-risk patients [12]. Thus, minimizing ASP's side effects while preserving its efficacy is a key research priority [13].
ASP immobilization is a strategy to lessen these side effects. One approach modifies the enzyme's structure either chemically or physically, which might expose it to environmental factors and reduce its functionality. Alternatively, embedding or encapsulating the enzyme within carrier materials preserves its activity and decreases toxicity, extends its in vivo half-life, enhances stability [14], and facilitates targeted delivery and controlled release [15–17]. Metal-organic frameworks (MOFs) are composed of organic ligands and metal ions/metal clusters linked through coordination bonds [18,19]. These frameworks boast large specific surface areas, customizable pore sizes, diverse compositions, and are easily functionalized on their surfaces [20–22]. By encapsulating enzymes within the cavities of MOFs (enzyme@MOFs), the enzymes are effectively shielded by a robust "MOF armor", which notably improves enzyme stability and prolongs drug half-life [23]. The porous architecture of MOFs also selectively enables substrates to access the active sites, boosting catalytic efficiency [24,25]. This dual enhancement renders enzyme@MOFs extremely promising for use in sensing, catalysis, and bioengineering [26].
The progression and recurrence of T-ALL are closely associated with the survival of leukemia-initiating cells (LIC) [27], identified by their high CD44 expression [28]. An early indicator in T-ALL progression is NOTCH activation, which dominates the oncogenic landscape in about 80% of T-ALL cases [29]. NOTCH1 signaling directly influences CD44 transcription in T-ALL cells, with NOTCH1 activation resulting in heightened CD44 expression [30]. Elevated CD44 levels serve as a biomarker for chemotherapy-resistant leukemia cells in T-ALL [31], and children with high CD44 levels often experience increased invasiveness and severe extramedullary infiltration. Thus, CD44 expression is strongly linked to high-risk T-ALL and is indicative of poor prognosis [32]. Targeting CD44 to reduce bone marrow infiltration and leukemia burden can subsequently enhance overall survival (OS) in children, suggesting that CD44 is a viable target for eradicating leukemia stem cells [32]. The interaction between CD44 and its main ligand, hyaluronic acid (HA), triggers several signaling pathways involved in cell proliferation, adhesion, migration, and invasion [33]. Utilizing this feature, we engineered nanoparticles with HA to promote their uptake via CD44-mediated endocytosis. Such an approach is already employed in nanomaterials for targeted treatment of acute myeloid leukemia (AML) [34].
Expanding on this foundation, as depicted in Scheme 1, we propose the development of a pH-responsive, targeted enzyme@MOFs nanoparticle using ASP-induced zeolitic imidazolate framework-8 (ZIF-8) nucleation for treating T-ALL. We altered the surface of ASP by acylation to add a negative charge, using a novel encapsulation technique that relies on rapid enzyme-triggered ZIF-8 nucleation. This method ensures that the encapsulated ASP retains high activity. The produced enzyme@MOF nanoparticles are further enhanced with polyethylene glycol (PEG) to improve their water solubility, stability, and biosafety, and to extend their half-life [35,36]. Additionally, surface modification with HA ensures targeted delivery to T-ALL cells overexpressing CD44 receptors. Overall, our designed nanodrug, ASP@MOF/PEG-HA NPs, aims to extend the drug's half-life, increase the targeting of ASP, and overcome drug resistance, offering a new approach to treat drug-resistant T-ALL characterized by high CD44 expression.
In this study, the anti-T-ALL chemotherapeutic agent ASP@MOF/PEG-HA was successfully synthesized. ZIF-8 NPs are widely acknowledged for their ability to carry molecules, enzymes, DNA, and proteins. Additionally, their pH-sensitive and biodegradable properties have been utilized to enable the pH-sensitive controlled release of nanoparticles [37]. A ZIF-8 enzyme-immobilized core was constructed through a one-step mineralization reaction using the "Zn2+−2-methylimidazole-ASP" ternary system. The coordination between the zinc and imidazolate ions dissociates at pH 5.0–6.0, which makes the drug release pH responsive and optimal for targeting cancer cells. Subsequently, HA-PEG-NH2 was employed to modify the surface of the drug-loaded ZIF-8 nanoparticles via coordination interactions between terminal amino groups and Zn2+, enhancing aqueous solubility and biocompatibility. The HA moiety, possessing CD44 receptor-targeting functionality, confers specific targeting and recognition capabilities toward leukemia cells. The resulting targeted nanomedicine is designated as ASP@MOF/PEG-HA. Measurements from the Malvern particle size analyzer revealed that the nanoparticles ASP@MOF/PEG and ASP@MOF/PEG-HA had average sizes of 97.95 and 98.05 nm, respectively (Fig. 1A). The polydispersity index (PDI) of these nanoparticles was 0.227 and 0.247 (Fig. 1B), suggesting a narrow particle size distribution. Furthermore, dynamic light scattering (DLS) analysis showed that both nanoparticles maintained a stable particle size distribution and PDI over a period of 7 days (Fig. 1B), and retained excellent stability in phosphate-buffered saline (PBS) with 10% fetal bovine serum (FBS) (Fig. S1 in Supporting information). Transmission electron microscopy (TEM) showed that ASP@MOF/PEG-HA in PBS) at pH 7.4 exhibited spherical nanoparticles with an average diameter of 100 nm, showing no degradation (Fig. 1C). However, when exposed to an acidic PBS solution for 24 h, ASP@MOF/PEG-HA underwent considerable degradation, leading to the loss of intact nanoparticles (Fig. 1D), confirming the pH-sensitive biodegradation and drug release properties of ASP@MOF/PEG-HA.

At pH 7.4, ASP exhibited its highest enzyme activity. However, a decrease in pH led to a reduction in enzyme activity. Despite this, ASP retained a significant portion of its activity even at pH 5. The activity of ASP@MOF/PEG and ASP@MOF/PEG-HA, was lowest at pH 7.4 but increased as the pH decreased, peaking at pH 5.0 (Fig. 1E). It is hypothesized that at pH 5.0, the MOF fully degraded, releasing the enzyme, which then interacted with the assay reagents. In contrast, at pH levels of 6.5 and 7.4, the MOF maintained its structural integrity, limiting the accessibility of the encapsulated ASP.
To verify the uptake of the nanodrugs by CCRF-CEM cells, MOF/PEG-ICG nanodrugs were synthesized. Indocyanine green (ICG), which fluoresces in the near-infrared spectrum, has been approved for tumor imaging, tracking, and treatment [38,39]. Confocal laser scanning microscopy (CLSM) showed that as the exposure time to the MOF/PEG-ICG nanodrug increased, cellular uptake significantly increased, with an increase in ICG fluorescence intensity (Fig. 1F). Additionally, flow cytometry confirmed time-dependent uptake (Fig. 1G).
To determine whether ASP@MOF/PEG-HA can specifically target CD44 on T-ALL cells, T-ALL cells were screened with a CD44 antibody, which identified CCRF-CEM cells as CD44-positive (Fig. 1H) and Jurkat cells as CD44-negative (Fig. 1I).
Using the cell counting kit-8 (CCK-8) assay, the effects of MOF/PEG, ASP@MOF/PEG, ASP@MOF/PEG-HA, and ASP were assessed. The results showed that ASP@MOF/PEG and ASP@MOF/PEG-HA induced dose-dependent cytotoxicity in CCRF-CEM cells, leading to a significant decrease in cell viability, with half-maximal inhibitory concentration (IC50) values of 0.907 ± 0.038 and 0.425 ± 0.035 mg/mL, respectively. These values were superior to those of ASP alone (IC50: 10.592 ± 0.858 mg/mL) and MOF/PEG (IC50: 3.878 ± 0.071 mg/mL) (Fig. 2A). ASP@MOF/PEG-HA achieved therapeutic effects with a lower concentration attributable to its specific targeting of CD44, Comparable results were observed in Jurkat cells (Fig. 2B).
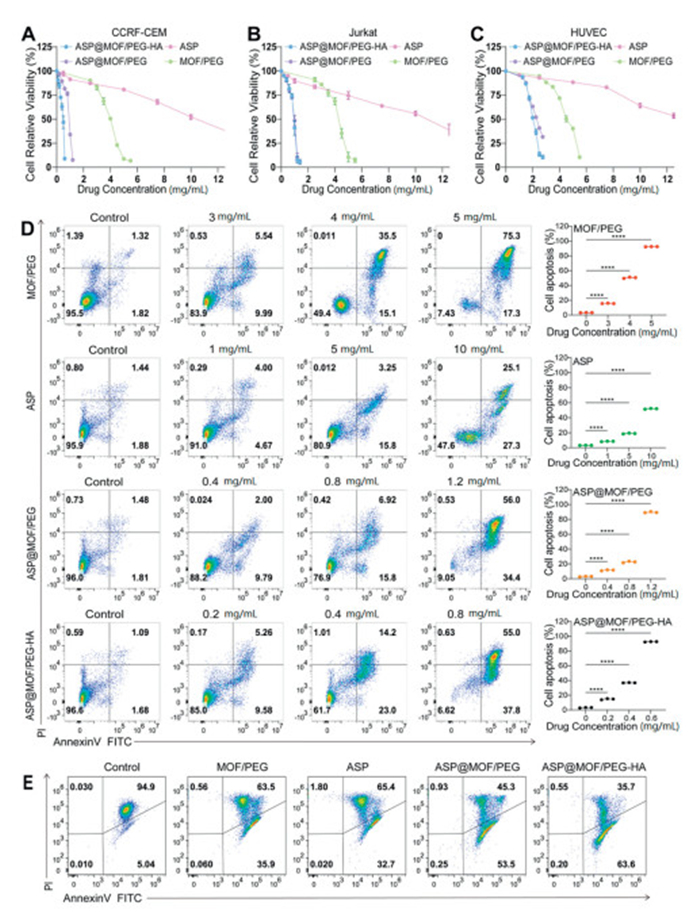
Safety experiments showed that at concentrations below 3.5 mg/mL of ASP or MOF/PEG, there was no significant impact on the cell viability of human umbilical vein endothelial cells (HUVEC). At concentrations below 1.25 mg/mL, ASP@MOF/PEG and ASP@MOF/PEG-HA preserved cell viability above 80%, with IC50 values (ASP@MOF/PEG: 2.28 ± 0.03 mg/mL; ASP@MOF/PEG-HA: 2.15 ± 0.04 mg/mL), significantly higher than those for T-ALL cells (Fig. 2C). This finding confirms the biosafety of the nanodrugs. Fluorescence microscope showed low toxicity of the nanodrugs to CCRF-CEM cells (Fig. S2 in Supporting information), Jurkat cells (Fig. S3 in Supporting information) and HUVEC (Fig. S4 in Supporting information).
Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) staining demonstrated that as the drug concentration increased, there was a corresponding rise in the apoptosis rate of CCRF-CEM (Fig. 2D) and Jurkat (Fig. S5 in Supporting information) cells. ASP@MOF-PEG and ASP@MOF-PEG-HA induced higher apoptosis rates than ASP and MOF/PEG alone, with similar results observed in HUVEC (Fig. S8 in Supporting information).
Mitochondrial depolarization was detected by JC-1 staining, revealing that ASP@MOF/PEG and ASP@MOF/PEG-HA significantly decreasedthe mitochondrial transmembrane potential (ΔΨm) in CCRF-CEM cells (Fig. 2E) and Jurkat cells (Fig. S6 in Supporting information), indicating activation of the intrinsic apoptotic pathway. ASP@MOF/PEG-HA, modified with HA for CD44 targeting, exhibited stronger apoptotic effect. Additionally, the apoptosis rate in HUVEC was notably lower than in T-ALL cells, suggesting that these nanoparticle drugs selectively target T-ALL cells and their high safety profile.
Propidium iodide staining combined with flow cytometry analysis showed that MOF/PEG, ASP@MOF/PEG and ASP@MOF/PEG-HA induced G2/M phase arrest in both CCRF-CEM and Jurkat cells (Fig. S7 in Supporting information). ASP primarily caused G2/M phase arrest in CCRF-CEM cells but induced G0/G1 phase arrest in Jurkat cells, suggesting differential response between the two cell lines, likely due to T-ALL heterogeneity.
To investigate the in vivo distribution of nanoparticles, CCRF-CEM cells were injected into the tail vein of NCG mice to create a cell-derived xenograft (CDX) model of T-ALL (Fig. 3A) [40,41]. All experimental procedures involving animals were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals under the approved protocol (No. SYSU-IACUC-2024-B0897). One week after transplantation, peripheral blood was collected for flow cytometry to measure the proportion of hCD45+ cells; a ratio exceeding 1% confirmed the successful establishment of the model. T-ALL mice were administered fluorescently labeled free ICG or MOF/PEG-ICG. In vivo imaging showed that free ICG fluorescence was detected within the first hour but rapidly decreased, with minimal retention after 24 h. In contrast, MOF/PEG-ICG fluorescence appeared within 1 h and declined slowly, with significant retention still observed after 24 h (Fig. 3B). These results confirm that MOF/PEG-ICG has a significantly higher in vivo uptake rate than free ICG. Imaging analyses of major organs revealed pronounced internalization of MOF/PEG-ICG in the bone marrow (an organ with high T-ALL cell concentration), while free ICG was mainly absorbed by the liver (Fig. 3C). These findings indicate that MOF/PEG-ICG may preferentially target leukemia cells and exhibits effective in vivo retention and organ targeting, laying the foundation for evaluating its anti-tumor efficacy in vivo.
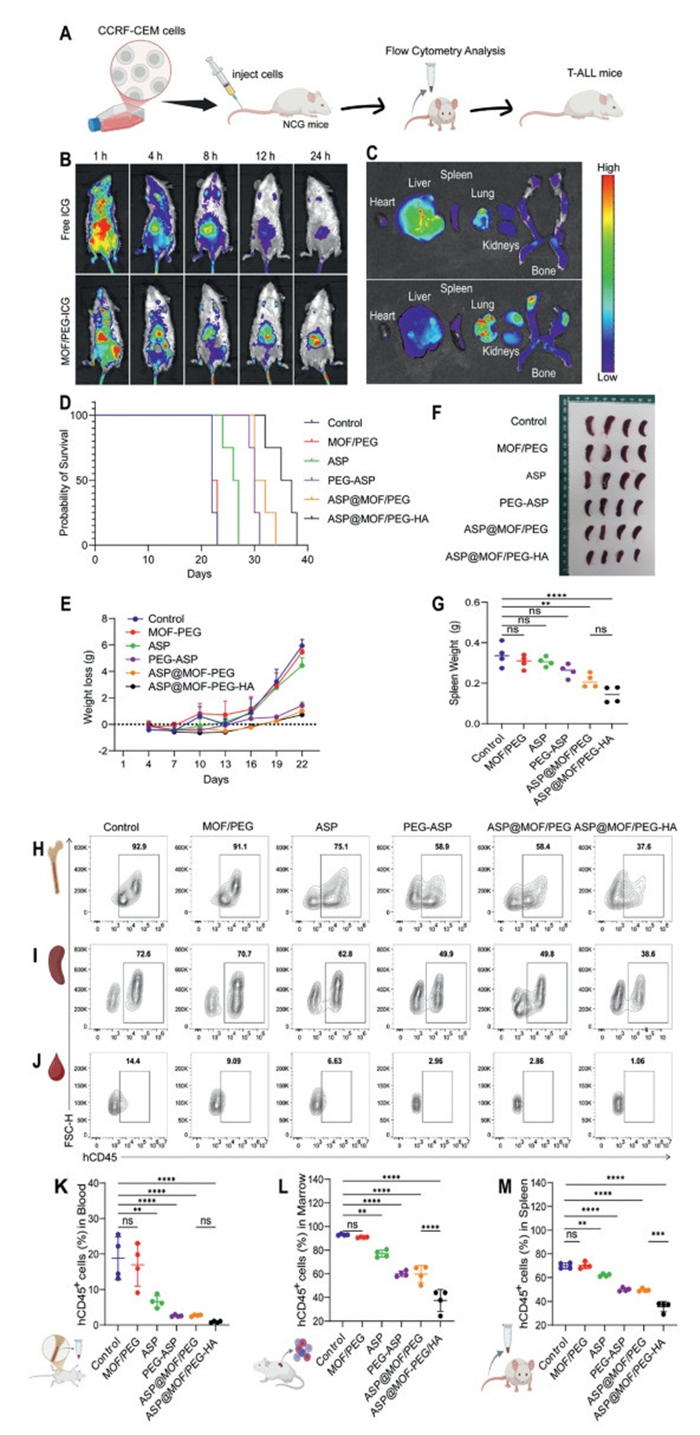
The T-ALL mice were randomly assigned to six treatment groups (8 mice per group) and treated with the following: PBS (control group), MOF/PEG, ASP, PEG-ASP, ASP@MOF/PEG, and ASP@MOF/PEG-HA. The in vivo anti-leukemic effects of the nanodrugs were evaluated by monitoring changes in body weight and survival rates, as well as by measuring the leukemia burden in blood, spleen, and bone marrow on day 22 post-cell injection. The survival analysis indicated that the ASP@MOF/PEG-HA group significantly prolonged the survival of the mice, while both the ASP@MOF/PEG and PEG-ASP groups also demonstrated improved survival rates (Fig. 3D).
The Control, MOF/PEG, and ASP groups showed significant weight loss, while the ASP@MOF/PEG and ASP@MOF/PEG-HA groups maintained stable body weight (Fig. 3E). Both the ASP@MOF/PEG and ASP@MOF/PEG-HA groups successfully inhibited splenic enlargement (Figs. 3F and G). Flow cytometry analysis of bone marrow (Fig. 3H) spleen (Fig. 3I), and peripheral blood (Fig. 3J) cells was conducted to determine the proportion of hCD45+ cells, followed by the quantification of leukemia burden (Figs. 3K–M). The data showed that the proportions of hCD45+ cells in the ASP@MOF/PEG, ASP@MOF/PEG-HA, and PEG-ASP groups were significantly lower than in the control group, with the ASP@MOF/PEG-HA group having the lowest proportion, likely due to the targeted inhibition of leukemia cells through CD44 binding. These findings suggest that both ASP@MOF/PEG-HA and ASP@MOF/PEG nanodrugs effectively reduce tumor burden in vivo and extend survival, with ASP@MOF/PEG-HA showing the highest efficacy. Additionally, no significant differences in body weight, survival, or other adverse reactions were observed between the groups treated with MOF/PEG and PBS, suggesting that the drug showed constrained therapeutic outcomes.
To assess the toxicity of the nanoparticles in tissues of T-ALL mice, various treatments including PBS, MOF/PEG, ASP, PEG-ASP, ASP@MOF/PEG, and ASP@MOF/PEG-HA were administered to NCG mice. Routine blood tests, such as white blood cells, red blood cells, platelets, and lymphocyte ratios, remained within normal ranges (Fig. 4A). This indicates that the nanodrugs ASP@MOF/PEG, and ASP@MOF/PEG-HA did not cause significant adverse effects on hematopoietic function. Liver function, measured by alanine aminotransferase (ALT) and aspartate aminotransferase (AST), showed no abnormalities, and kidney function markers, including blood urea nitrogen (BUN) and creatinine (CREA), were also within normal limits (Fig. 4B). Furthermore, histological analysis using hematoxylin and eosin (H&E) staining revealed no tissue damage in the major organs (heart, liver, spleen, lung, and kidney) across all treatment groups (Fig. 4C). This further supports the good biosafety profile of the synthesized nanodrugs. In conclusion, the data indicate that the synthesized nanodrugs, particularly ASP@MOF/PEG-HA, demonstrate superior therapeutic efficacy compared to ASP and PEG-ASP while exhibiting minimal toxicity and side effects.

ASP treatment for T-ALL is often associated with severe allergic reactions and other side effects, leading to discontinuation of therapy and a poor prognosis. In this study, we developed the nanoparticles ASP@MOF/PEG-HA by encapsulating ASP through a rapid enzyme-triggered MOF nucleation process. Effective uptake of these nanoparticles by T-ALL cells was demonstrated. The pH-responsive release properties of the MOF enabled targeted release of ASP within T-ALL cells, preserving high enzyme activity and significantly enhancing its cytotoxic effects. The PEG coating improved water solubility, extended the drug’s half-life, and increased biosafety, reducing toxicity to normal cells while maintaining strong cytotoxicity against T-ALL cells. Additionally, the HA coating, targeting CD44 overexpression on T-ALL cells, promoted increased apoptosis in CD44+ cells, overcoming resistance mechanisms. In vivo experiments revealed that the ASP@MOF/PEG-HA nanodrug prolonged survival in T-ALL model mice, reduced leukemia burden in the bone marrow and spleen, and exhibited potent anti-leukemia activity. This study emphasizes the importance of ASP@MOF/PEG-HA nanoparticles in treating T-ALL and presents a potential therapeutic approach for targeting CD44-expressing T-ALL cells that are resistant to conventional treatments. The results suggest that this nanodrug holds significant promise as a therapeutic agent for clinical applications.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Yeling Yuan: Writing – original draft, Formal analysis, Data curation. Lihua Du: Funding acquisition, Formal analysis, Data curation. Kejing Zeng: Funding acquisition, Formal analysis, Data curation. Yilu Zheng: Formal analysis, Data curation. Huaping Huang: Formal analysis, Data curation. Yu Shao: Visualization, Formal analysis, Data curation. Wenqing Wang: Formal analysis, Data curation. Leping Yan: Validation, Supervision, Funding acquisition. Jun Wu: Writing – review & editing, Funding acquisition, Conceptualization. Changhua Zhang: Writing – review & editing, Visualization, Project administration. Hongman Xue: Writing – review & editing, Project administration. Haozhe He: Writing – review & editing, Project administration, Funding acquisition, Conceptualization.
This research was supported by the National Natural Science Foundation of China (Nos. 52203206, 52173150, 81902004), Guangdong Basic and Applied Basic Research Foundation (Nos. 2023A1515012193, 2023A1515110025, 2023A1515012575, 2023A1515011962), Shenzhen Fundamental Research Program (Nos. JCYJ20220530144605012, JCYJ20240813150438050), China Postdoctoral Science Foundation (Nos. 2023M744090, GZC20233296), Research Start-up Fund of Post-doctoral of SAHSYSU (No. ZSQYRSFPD0071), Distinguished Young Scientists projects by Guangdong Second Provincial General Hospital (No. 2024E005), Guangdong International Science and Technology Cooperation Project (No. 2023A0505050120), Shenzhen International Cooperative Research Project (No. GJHZ20220913142401003), Open Fund Key Project of the Guangdong Provincial Key Laboratory of Digestive Cancer Research (No. GPKLDCR202201Z).
Supplementary material associated with this article can be found, in the online version, at doi:
L. Pagliaro, S.J. Chen, D. Herranz, et al., Nat. Rev. Dis. Primers 10 (2024) 41.
S. Essig, Q. Li, Y. Chen, et al., Lancet Oncol. 15 (2014) 841–851.
T. Moriyama, M.V. Relling, J.J. Yang, Blood 125 (2015) 3988–3995. doi: 10.1182/blood-2014-12-580001
S.P. Hunger, C.G. Mullighan, N. Engl. J. Med. 373 (2015) 1541–1552.
I. Iacobucci, C.G. Mullighan, J. Clin. Oncol. 35 (2017) 975–983.
D.T. Teachey, C.H. Pui, Lancet Oncol. 20 (2019) e142–e154.
A. Esen, A.A. Khan, J. Chan, et al., Blood 128 (2016) 4716.
S. Srivastava, J. Jiang, G. Seim, et al., Blood 140 (2022) 3040–3041. doi: 10.1182/blood-2022-170024
W.K. Chan, L. Tan, K. Harutyunyan, et al., Blood 132 (2018) 3959. doi: 10.1182/blood-2018-99-117818
I. Aldoss, D. Douer, Blood 135 (2020) 987–995.
K. Schmiegelow, A. Attarbaschi, S. Barzilai, et al., Lancet Oncol. 17 (2016) e231–e239.
S. Gupta, C. Wang, E.A. Raetz, et al., J. Clin. Oncol. 38 (2020) 1897–1905. doi: 10.1200/jco.19.03024
M.N. Offman, M. Krol, N. Patel, et al., Blood 117 (2011) 1614–1621. doi: 10.1182/blood-2010-07-298422
Y.S. Mao, C.F. Zou, Y.J. Jiang, et al., Chin. Chem. Lett. 32 (2021) 990–998.
V.P. Talluri, B. Mutaliyeva, A. Sharipova, et al., Adv. Colloid. Interface Sci. 316 (2023) 102915.
S. Zhang, Y. Sun, L. Zhang, et al., Adv. Sci. 10 (2023) e2300469.
Z. Yuan, B. Li, L. Niu, et al., Angew. Chem. Int. Ed. 59 (2020) 22378–22381. doi: 10.1002/anie.202004995
J.B. Zhang, Y.B. Tian, Z.G. Gu, et al., Nano-Micro Lett. 16 (2024) 253.
Z. Ye, Y. Jiang, L. Li, et al., Nano-Micro Lett. 13 (2021) 203.
M. Ding, R.W. Flaig, H.L. Jiang, et al., Chem. Soc. Rev. 48 (2019) 2783–2828. doi: 10.1039/c8cs00829a
C. Yang, J. Xu, D.D. Yang, et al., Chin. Chem. Lett. 29 (2018) 1421–1424.
J. Xu, J. Ma, Y. Peng, et al., Chin. Chem. Lett. 34 (2023) 107527.
S. Huang, X. Kou, J. Shen, et al., Angew. Chem. Int. Ed. 59 (2020) 8786–8798. doi: 10.1002/anie.201916474
G. Chen, X. Kou, S. Huang, et al., Angew. Chem. Int. Ed. 59 (2020) 2867–2874. doi: 10.1002/anie.201913231
X. Lian, Y. Fang, E. Joseph, et al., Chem. Soc. Rev. 46 (2017) 3386–3401.
G. Chen, S. Huang, X. Kou, et al., Angew. Chem. Int. Ed. 58 (2019) 1463–1467. doi: 10.1002/anie.201813060
J. Tatarek, K. Cullion, T. Ashworth, et al., Blood 118 (2011) 1579–1590. doi: 10.1182/blood-2010-08-300343
S. Piya, Y. Yang, S. Bhattacharya, et al., Leukemia 36 (2022) 1261–1273. doi: 10.1038/s41375-022-01516-1
F. Malard, M. Mohty, Lancet 395 (2020) 1146–1162.
M. García-Peydró, P. Fuentes, M. Mosquera, et al., J. Clin. Invest. 128 (2018) 2802–2818. doi: 10.1172/jci92981
J. Calvo, I. Naguibneva, A. Kypraios, et al., Leukemia 39 (2025) 323–336. doi: 10.1038/s41375-024-02473-7
R.La Starza, C. Borga, G. Barba, et al., Blood 124 (2014) 3577–3582.
C. Chen, S. Zhao, A. Karnad, et al., J. Hematol. Oncol. 11 (2018) 64. doi: 10.1145/3210284.3210287
J. Li, H. Wu, Z. Yu, et al., Nat. Commun. 15 (2024) 5689.
C. Guo, H. Yuan, Y. Wang, et al., Adv. Drug. Deliv. Rev. 200 (2023) 115044.
M. Zoulikha, Z.J. Chen, J. Wu, et al., Chin. Chem. Lett. 36 (2025) 110225.
X. Fu, G.Q. Zhang, Y.L. Zhang, et al., Chin. Chem. Lett. 32 (2021) 1559–1562.
H. Wang, X. Li, B.W. Tse, et al., Theranostics 8 (2018) 1227–1242.
E.P. Porcu, A. Salis, E. Gavini, et al., Biotechnol. Adv. 34 (2016) 768–789.
M. Zhang, D. Chen, X. Fu, et al., Clin. Cancer. Res. 28 (2022) 2830–2843. doi: 10.1158/1078-0432.ccr-21-4097
J. Shi, Z. Zhang, H. Cen, et al., J. Hematol. Oncol. 14 (2021) 162.

Scheme 1 Schematic representation of the synthesis of ASP@MOF/PEG-HA NPs and the mechanism underlying anti-leukemia treatment.

Figure 1 Characterization, enzyme activity, cellular uptake of nanodrugs, and cellular CD44 expression. (A) DLS analysis of the particle size distribution of ASP@MOF/PEG-HA and ASP@MOF/PEG NPs in PBS (pH 7.4). (B) Stability evaluation of ASP@MOF/PEG-HA and ASP@MOF/PEG NPs dissolved in PBS buffer over 7 days using DLS. Representative TEM images of ASP@MOF/PEG-HA in PBS solution at pH 7.4 (C) and pH 5.0 (D). (E) Enzyme activity of free ASP, ASP@MOF/PEG-HA, and ASP@MOF/PEG at pH values of 7.4, 6.5, and 5. Data are presented as mean ± standard deviation (SD) (n = 3). P < 0.05, **P < 0.01, ****P < 0.0001. Two-way ANOVA, post hoc comparisons, Tukey's test. Fluorescence intensity images (F) and flow cytometry analysis (G) of ICG uptake in CCRF-CEM cell after incubation of MOF/PEG-ICG for specified times. CD44 expression on the surface of CCRF-CEM (H) and Jurkat (I) cells.

Figure 2 Anti-tumor effects of nanodrugs in vitro. The percentage of cell viability in CCRF-CEM (A), Jurkat (B), and HUVEC (C) cells after treatment with nanodrugs MOF/PEG, ASP@MOF/PEG, ASP@MOF/PEG-HA, and ASP. Following 24-h treatment of CCRF-CEM cells with specific concentrations of nanoparticle drugs (MOF/PEG, ASP@MOF/PEG, ASP@MOF/PEG-HA, and ASP). Cell apoptosis was measured by flow cytometry using the Annexin V/PI method. Dot plot: mean ± SD from 3 separate studies (right). ns, not significant. P < 0.05, ***P < 0.001. One-way ANOVA, post hoc comparisons, Tukey's test. (D) Changes in mitochondrial potential in CCRF-CEM cells post-treatment with various drugs were assessed by JC-1 staining (E).

Figure 3 In vivo anti-leukemic efficacy and biosafety of nanodrugs. (A) Establishment of CDX T-ALL mouse model. (B) Bioluminescence imaging of T-ALL mice at different time points after injection of free ICG or MOF/PEG-ICG. (C) Fluorescence imaging of major organs in T-ALL mice was performed 24 h post-injection of free ICG or MOF/PEG-ICG nanodrugs. (D) Survival curves of T-ALL mice across treatment groups. (E) Body weight change curve of T-ALL mice in different treatment group. (F) Spleen morphology and (G) weight of T-ALL mice in each treatment group at day 22 post-modeling. (H–J) Flow cytometry analysis of leukemia cell proportions in bone marrow (H), spleen (I), and peripheral blood (J) of T-ALL mice from each treatment group at day 22 post-modeling, with quantitative analysis for bone marrow (K), spleen (L), and peripheral blood (M). Dot plot: mean ± SD from 4 mice. P < 0.05, ***P < 0.001. One-way ANOVA, post hoc comparisons, Tukey's test.

Figure 4 In vivo biosafety assessment. (A) Analysis of routine blood tests and (B) liver and kidney functionin T-ALL mice under different treatment regimens Dot plot: mean ± SD from 4 mice. P < 0.05, ***P < 0.001. One-way ANOVA, post hoc comparisons, Tukey's test. (C) Representative histopathological images of major organs in T-ALL mice after various treatment. Scale bar: 100 µm.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
 下载:
下载: