
Figure 1.
Phosphorodithioate derivatives and asymmetric synthesis of axially chiral organophosphorus derivatives from anhydride derivatives.
Organocatalytic enantioselective construction of axially chiral phosphorodithiolated styrenes
Fuxing Yang , Mengjie Gong , Yifei Zhang , Bangchi Wei , Nan Huang , Jun Jiang

Organophosphorodithioates have a wide variety of applications in pharmacy [1-4], agrochemistry [5-8], materials chemistry [9,10] and synthetic chemistry [11,12]. Consequently, great effort has been devoted to the development of efficient methods for the construction of organophosphorodithioates and related derivatives in recent years (Fig. 1a) [13-18]. Enantioselective preparation of organophosphorodithioates mainly relied on asymmetric induction approaches began with stoichiometric amounts of chiral auxiliaries [19-28]. The emerging P(Ⅴ)-based phosphorylation protocol developed by Baran et al. provided a robust synthetic strategy for stereocontrolled synthesis of chiral phosphorodithioate derivatives with P-stereogenic centers by using limonene-derived chiral oxathiophospholane reagents [19,22-28]. Nevertheless, the catalytic asymmetric reaction for accessing chiral organophosphorodithioates was exclusively explored via (salen)Ti(Ⅳ)-catalyzed asymmetric ring opening of epoxide with dithiophosphorus acid, but unfortunately the enantioselective control of carbon stereocenter remains challenging (up to 73% ee) [29]. Furthermore, the direct catalytic asymmetric approach toward axially chiral phosphorodithioates has never been reported.
Anhydride was an important synthon in synthetic chemistry to provide electrophilic acyl (C═O) moiety in various reactions such as Friedel-Crafts acylations [30,31], esterifications [32-34], and Allan-Robinson reactions [35,36]. Phosphinic anhydrides, a type of highly reactive anhydrides, were recently utilized as electrophilic reagents to afford phosphoryl group (P═O) for the construction of phosphoryl compounds [37,38]. In 2021, the Wang group demonstrated a highly enantiodivergent Atherton-Todd reaction of biaryldiol and phosphinic anhydride generated in situ from phosphine oxide for the synthesis of atropisomeric O-phosphorylated diaryl diols (Fig. 1b) [38].
Recently, the catalytic asymmetric nucleophilic addition reactions of vinylidene ortho-quinone methide (VQM) intermediates with various nucleophiles provided an efficient approach to the construction of axially chiral styrenes [39-50]. In light of the challenge and importance of catalytic enantioselective construction of axially chiral phosphorodithioates, we hypothesize that phosphinothioic thioanhydride reacts as the nucleophilic reagent (S—P═S moiety), as phosphinothioic thioanhydride could exhibit stronger nucleophilicity compared to phosphinic anhydride, which enables the development of an asymmetric nucleophilic addition of VQM to directly afford axially chiral phosphorodithiolated styrenes (Fig. 1c). The reaction suffers from two major challenges: (1) divergent potential reactive sites of phosphinothioic thioanhydride as both nucleophile and electrophile; (2) stereoselective control of axially chiral styrenes due to their low rotational barrier leading to racemization. Herein, we describe a method for the in situ generation of phosphinodithioic acid anion as nucleophiles for the successful synthesis of axially chiral phosphorodithiolated styrenes with excellent enantioselectivity. The reaction was achieved through the activation of phosphinothioic thioanhydride using 4-dimethylaminopyridine (DMAP), coupled with the stereoselective control guided by chiral catalyst.
Initially, 1-(phenylethynyl)naphthalen-2-ol 1a and diphenylphosphinothioic thioanhydride 2a were chosen as the model substrates to optimize the conditions (Table 1). The reaction was first investigated in the presence of commercially available quinine in toluene at 50 ℃. Fortunately, the desired axially chiral product 3a could be obtained in 58% yield with −24% ee (entry 1). Inspired by this success, we next screened chiral organocatalysts. Cinchona alkaloid-derived urea, thioureas and squaramides (C-1~5) could efficiently improve the enantioselectivity (entries 2−6), and C-5 gave the product 3a with 77% ee (entry 6). The reaction at 30 ℃ gave higher yield and ee than that at 50 ℃ and 0 ℃ (entries 7 vs. 6 and 8). Further investigation on various solvents including dichloromethane (DCM), tetrahydrofuran (THF), mesitylene and ethyl acetate (EA) showed that toluene was the best solvent (entries 7, 9−12). Gratifyingly, pyridine derivatives as the additive were found to improve the reaction yield and activation of anhydride 2a (entries 13−15) [51,52]. Notably, DMAP as the additive boosted the enantioselectivity to 96% ee together with improved yield (entry 15). In addition, the catalyst loading of C-5 and DMAP could be reduced to 10 mol% while keeping almost the same yield and ee (entry 16). Considering that phosphonic anhydride has a structure similar to that of phosphinothioic thioanhydride, we also tested phosphonic anhydride 2a’ as a nucleophile in this reaction. However, only a trace amount of product 3a’ was obtained (entry 17). This result highlighted the crucial role of phosphinothioic thioanhydride in the nucleophilic addition of VQM.
 DownLoad:
CSV
DownLoad:
CSV
 |
||||||
| Entry | Catalyst | T (℃) | Time (h) | Solvent | Yield (%)b | ee (%)c |
| 1 | QN | 50 | 72 | Toluene | 58 | −24 |
| 2 | C-1 | 50 | 72 | Toluene | 56 | 65 |
| 3 | C-2 | 50 | 72 | Toluene | 72 | 74 |
| 4 | C-3 | 50 | 72 | Toluene | 73 | 63 |
| 5 | C-4 | 50 | 72 | Toluene | 70 | 71 |
| 6 | C-5 | 50 | 72 | Toluene | 76 | 77 |
| 7 | C-5 | 30 | 120 | Toluene | 78 | 84 |
| 8 | C-5 | 0 | >168 | Toluene | 43 | 79 |
| 9 | C-5 | 30 | 72 | DCM | 78 | 55 |
| 10 | C-5 | 30 | 72 | THF | 20 | 26 |
| 11 | C-5 | 30 | 120 | Mesitylene | 58 | 80 |
| 12 | C-5 | 30 | 168 | EA | 80 | 72 |
| 13d | C-5 | 30 | 24 | Toluene | 82 | 76 |
| 14e | C-5 | 30 | 24 | Toluene | 80 | 81 |
| 15f | C-5 | 30 | 12 | Toluene | 86 | 96 |
| 16g | C-5 | 30 | 12 | Toluene | 84 | 96 |
| 17h | C-5 | 30 | 12 | Toluene | Trace | – |
| a Reaction condition: 1a (36.6 mg, 0.15 mmol), 2a (46.6 mg, 0.1 mmol), catalyst (20 mol%), toluene (2 mL), 50 ℃. DCM = Dichloromethane; THF = Tetrahydrofuran; EA = Ethyl acetate; DMAP = 4-Dimethylaminopyridine. b Isolated yield. c Determined by HPLC on a chiral stationary phase. d 20 mol% pyridine. e 20 mol% 2,6-lutidine. f 20 mol% DMAP. g 10 mol% C-5 and 10 mol% DMAP. h Phosphonic anhydride 2a’ instead of phosphinothioic thioanhydride 2a, 10 mol% C-5 and 10 mol% DMAP. |
||||||
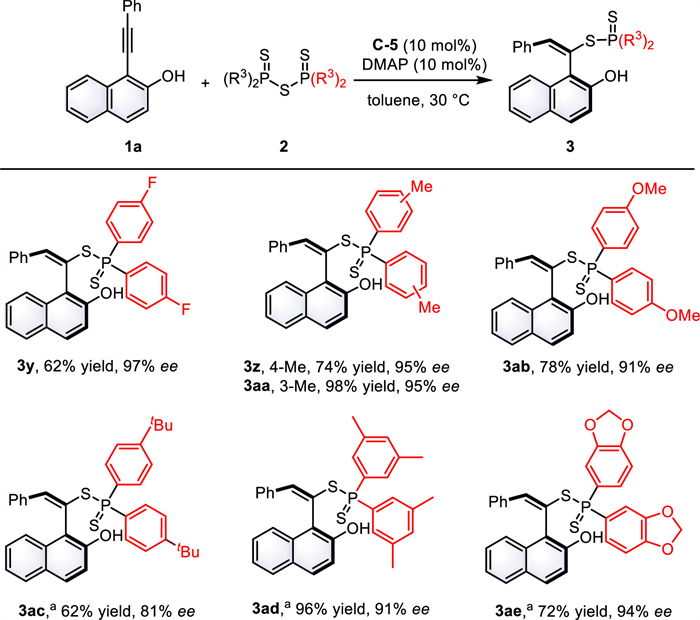
With the optimized conditions in hand, we first evaluated the substrate scope of o-alkynylnaphthols (Fig. 2). The reactions with o-alkynylnaphthols containing various electron-deficient and electron-rich groups at para-position of the aryl ring proceeded smoothly to afford the corresponding products 3b−k in 54%−95% yields with 88%−97% ee. o-Alkynylnaphthol bearing 1-naphthyl group generated the desired product 3l in 63% yield with 82% ee. The meta- and ortho-substituent on aryl ring gave good enantioselectivity, ranging from 84% to 95% ee (3m−r). In addition, 3,5-dimethyl-substituted product 3s could be obtained in 78% yield with 90% ee, while 3,5-bis(trifluoromethyl) substituent decreased the reactivity and enantioselectivity. It is notable that heteroaromatic substituted o-alkynylnaphthols also reacted smoothly with 2a to furnish the corresponding 3u−w with good results (76%−78% yields, 85%−95% ee). Furthermore, naphthalene ring bearing 6-bromo-substituted group was also tolerated in this transformation and afforded product 3x in 81% yield with 96% ee.

Subsequently, we further investigated the scope of phosphinothioic thioanhydrides 2 (Fig. 3). The reaction with 2 bearing halogen and electrondonating groups, such as p-fluoro, p-methyl, m-methyl, or p-methoxy yielded the corresponding products 3y-3ab with excellent enantioselectivity ranging from 91% to 97% ee. Notably, the bulky tert-butyl group at the para position of phenyl ring gave 3ac in 62% yield with 81% ee. Moreover, phosphinothioic thioanhydride with 3,5-dimethyl substituent generated product 3ad in 96% yield with 91% ee. Furthermore, bis(benzo[d][1,3]dioxol-4-yl)thiophosphoric anhydride was also feasible for this reaction, producing 3ae with 94% ee.
In order to explore the configurational stability of axially chiral phosphorodithiolated styrenes, the racemization experiment of 3a was carried out in toluene at 50 ℃. The rotation barrier of 3a was determined as 29.0 kcal/mol, which is just slightly higher than the required racemization barrier (24 kcal/mol) for isolating the individual atropisomers [53]. Besides, we also calculated the rotation barrier of 3a (Figs. S1−S3 in Supporting information), and the calculated value (∆G‡ = 29.86 kcal/mol at 50 ℃) was in good accordance with the experimental data. These results demonstrated the challenges in constructing the axial chirality of phosphorodithiolated styrenes due to the low racemization barrier and the weak configurational stability.
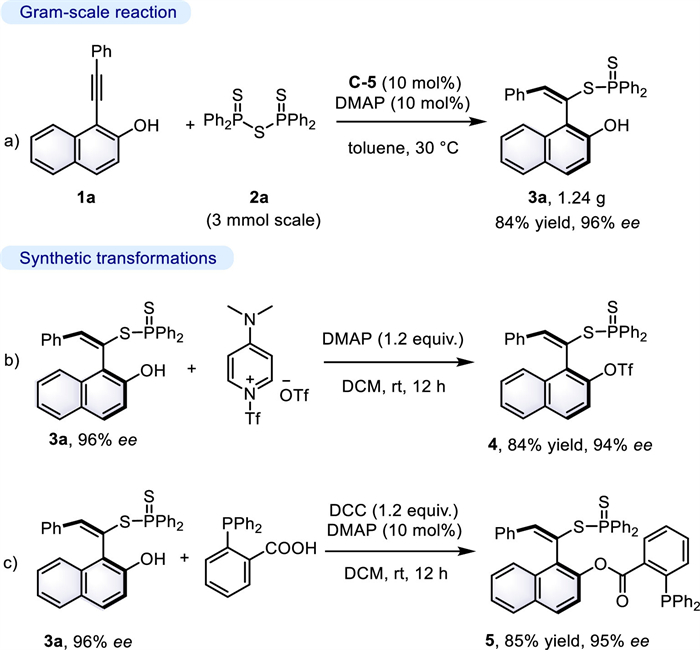
To demonstrate the utility of this protocol, a gram-scale reaction was performed on a 3 mmol scale under the standard reaction conditions (Scheme 1a), furnishing the desired product 3a maintaining the same level of yield and enantioselectivity (84% yield, 96% ee). Moreover, the trifluoromethanesulfonation of 3a could easily produce the synthetically useful triflate 4 in 84% yield with 94% ee (Scheme 1b). Furthermore, esterification of 3a with 2-(diphenylphosphaneyl)benzoic acid afforded axially chiral phosphine-containing styrene 5 (Scheme 1c).
To gain further insight into the mechanism of this reaction, a series of control experiments were conducted (Scheme 2). O-Acyled substrate 1y generated no desired product 3af under the standard reaction conditions, indicating that OH group of o-alkynylnaphthol could play a crucial role in forming VQM intermediate (Scheme 2a). Interestingly, the reaction of 1a and benzoic anhydride 6 under the standard conditions only generated product 7, and no desired product 8 was generated from nucleophilic addition of VQM with the anhydride 6 as a nucleophilic reagent (Scheme 2b). To further explore the influence of nucleophiles, we conducted a series of experiments using various S-nucleophiles. The ee values of the resulting products exhibited significant variation, indicating that the nucleophilic attack on VQM may represent the enantioselectivity-determining step (Scheme 2c). The recovery of 93% of racemic 3a in the presence of catalyst C-5 excluded the possibility that the reaction initially produced racemic 3a, which was subsequently isomerized to one enantiomer under the influence of C-5 (Scheme 2d). Moreover, a deuterium labeling experiment with D2O in the reaction system gave the corresponding product with a 61% D-labelled ratio, suggesting that external trace amounts of water might provide proton for the generation of product (Scheme 2e). Besides the desired product 3a, this reaction between 1a (1.5 equiv.) and 2a (1.0 equiv.) generated byproducts including O-(1-(phenylethynyl)naphthalen-2yl)diphenylphosphinothioate 11 and pyridinium 12 which were identified by NMR, mass spectrometric and X-ray crystallographic analysis, indicating that excess phosphinothioic thioanhydride 1a could react as the electrophilic reagent to give 11 (Scheme 2f). Interestingly, the reaction with pyridinium 12 instead of DMAP under the standard reaction conditions also proceeded smoothly to afford product 3a in 81% yield with 97% ee, maintaining almost the same level of enantioselectivity, and treatment of 1a with pyridinium 12 (1.0 equiv.) in toluene at 30 ℃ produced 3a in 77% yield with 80% ee. This observation suggested that pyridinium 12 may serve as the true nucleophiles in the addition reaction. Moreover, the reaction without 1a under standard conditions also afforded pyridinium 12 (for details see Supporting information). The Hammett relationship was established for para-pyridine, allowing for a Hammett analysis to be performed on the nature of these substituents and their effect on asymmetric induction in the formation of 3a [54]. A linear plot of log(er) for 3a versus σp for para-pyridine was obtained, revealing a negative slope in the relationship between enantiomeric ratios and σp constants for 3a (Fig. S5 in Supporting information). This result indicates that the reaction might undergo an ionic transition state and the electron-donating effect of pyridine enhances the ionization of pyridinium salt.

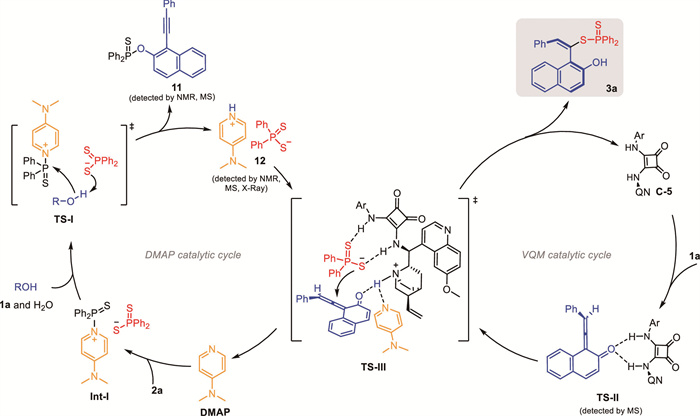
Based on the above experimental observations, a plausible mechanism was elucidated (Scheme 3). First, DMAP as a nucleophilic catalyst reacted with phosphinothioic thioanhydride 2a to produce the activated thiophosphoryl donor intermediate Int-I. Subsequently, 1a and H2O interacted with Int-I to generate pyridinium 12 and byproduct 11 through TS-I [55]. Meanwhile, the hydrogen-bonding interactions of 1a and cinchona alkaloid-derived squaramide catalyst C-5 could generate VQM intermediate. Concurrently, the proton of pyridinium 12 was neutralized by the quinine moiety of C-5. Then, C-5 efficiently guided nucleophilic addition with enantioselective control to the formation of TS-Ⅲ by the hydrogen-bonding interactions [56-59]. Finally, the nucleophilic addition and protonation of TS-Ⅲ led to axially chiral styrene 3a, together with catalyst C-5 and DMAP released into the catalytic cycle respectively.
In conclusion, we have developed an efficient catalytic asymmetric nucleophilic addition of in situ generated VQMs with phosphinothioic thioanhydride as nucleophilic reagent by the dual catalysis of cinchona alkaloid-derived squaramide and DMAP. The reaction rate could be significantly enhanced through the in situ generation of pyridinium, facilitated by the interaction of DMAP with phosphinothioic thioanhydride. Chiral squaramide catalyst controlled the stereoselectivity of the reaction by hydrogen-bonding interactions. This protocol provides an efficient and straightforward route for the construction of axially chiral phosphorodithiolated styrenes in good yields with high enantioselectivities. Our work not only successfully solved the unprecedented catalytic asymmetric synthesis of axially chiral phorodithioates but also showcased the potential of combining chiral organocatalysis with achiral organocatalysis to achieve new asymmetric transformations that are otherwise unreachable.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Fuxing Yang: Writing – review & editing, Writing – original draft, Visualization, Validation, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Mengjie Gong: Writing – review & editing, Visualization, Validation, Methodology, Investigation, Formal analysis, Data curation. Yifei Zhang: Validation, Software, Formal analysis, Data curation. Bangchi Wei: Validation, Data curation. Nan Huang: Writing – review & editing, Visualization, Validation, Supervision, Project administration, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Jun Jiang: Writing – review & editing, Visualization, Supervision, Resources, Project administration, Methodology, Investigation, Funding acquisition, Formal analysis, Conceptualization.
We are grateful for financial support from Natural Science Foundation of China (No. 22161005) and Guangxi Natural Science Foundation (Nos. 2021GXNSFDA075005, 2024GXNSFFA010001). We thank the Center for Instrumental Analysis at the Guangxi University for running the high-resolution mass spectra (HRMS) for the products.
Supplementary material associated with this article can be found, in the online version, at doi:
W.S. Marshall, M.H. Caruthers, Science 259 (1993) 1564–1570. doi: 10.1126/science.7681216
T.S. Kumar, T. Yang, S. Mishra, et al., J. Med. Chem. 56 (2013) 902–914. doi: 10.1021/jm301372c
S.Y. Wu, X. Yang, K.M. Gharpure, et al., Nat. Commun. 5 (2014) 3459. doi: 10.1038/ncomms4459
V. Zelikman, J. Pelletier, L. Simhaev, et al., J. Med. Chem. 61 (2018) 3939–3951. doi: 10.1021/acs.jmedchem.7b01906
P.J. Skouras, J.T. Margaritopoulos, N.A. Seraphides, et al., Pest Manag. Sci. 63 (2007) 42–48. doi: 10.1002/ps.1306
C. Magaña, P. Hernández-Crespo, A. Brun-Barale, et al., Insect Biochem. Mol. Biol. 38 (2008) 756–762. doi: 10.1016/j.ibmb.2008.05.001
J. Soleño, L. Anguiano, A.P. de D’Angelo, et al., Pest Manag. Sci. 64 (2008) 964–970. doi: 10.1002/ps.1582
M. Kim, G. An, J. Park, G. Song, W. Lim, J. Hazard. Mater. 455 (2023) 131577. doi: 10.1016/j.jhazmat.2023.131577
N.J. Mosey, T.K. Woo, J. Phys. Chem. A 107 (2003) 5058–5070.
J. Bao, K.S. Kang, J. Molineux, et al., Angew. Chem. Int. Ed. 63 (2024) e202315963. doi: 10.1002/anie.202315963
T. Ozturk, E. Ertas, O. Mert, Chem. Rev. 110 (2010) 3419–3478. doi: 10.1021/cr900243d
N.D. Shapiro, V. Rauniyar, G.L. Hamilton, J. Wu, F.D. Toste, Nature 470 (2011) 245–249. doi: 10.1038/nature09723
G.R. Norman, W. LeSuer, T. Mastin, J. Am. Chem. Soc. 74 (1952) 161–163. doi: 10.1021/ja01121a040
Y. Zhu, T. Chen, S. Li, S. Shimada, L.B. Han, J. Am. Chem. Soc. 138 (2016) 5825–5828. doi: 10.1021/jacs.6b03112
H.F. Piedra, V. Gebler, C. Valdés, M. Plaza, Chem. Sci. 14 (2023) 12767–12773. doi: 10.1039/d3sc05263j
C. Qu, Y. Lv, J. Huang, et al., Green Chem. 25 (2023) 10678–10683. doi: 10.1039/d3gc02665e
X.M. Chen, L. Song, J. Pan, et al., Chin. Chem. Lett. 35 (2024) 110112. doi: 10.1016/j.cclet.2024.110112
J. Hao, Y. Lv, S. Tian, et al., Chin. Chem. Lett. 35 (2024) 109513. doi: 10.1016/j.cclet.2024.109513
K.W. Knouse, D.T. Flood, J.C. Vantourout, et al., ACS Cent. Sci. 7 (2021) 1473–1485. doi: 10.1021/acscentsci.1c00487
W.J. Stec, A. Grajkowski, A. Kobylanska, et al., J. Am. Chem. Soc. 117 (1995) 12019–12029. doi: 10.1021/ja00154a001
P. Guga, K. Domanski, W.J. Stec, Angew. Chem. Int. Ed. 40 (2001) 610–613. ´ doi: 10.1002/1521-3773(20010202)40:3<610::AID-ANIE610>3.0.CO;2-W
K.W. Knouse, J.N. deGruyter, M.A. Schmidt, et al., Science 361 (2018) 1234–1238. doi: 10.1126/science.aau3369
C. He, H. Chu, T.P. Stratton, et al., J. Am. Chem. Soc. 142 (2020) 13683–13688. doi: 10.1021/jacs.0c06641
J.C. Vantourout, S.R. Adusumalli, K.W. Knouse, et al., J. Am. Chem. Soc. 142 (2020) 17236–17242. doi: 10.1021/jacs.0c05595
D. Xu, N. Rivas-Bascón, N.M. Padial, et al., J. Am. Chem. Soc. 142 (2020) 5785–5792. doi: 10.1021/jacs.9b13898
Y. Huang, K.W. Knouse, S. Qiu, et al., Science 373 (2021) 1265–1270. doi: 10.1126/science.abi9727
M. Nassir, M. Ociepa, H.J. Zhang, et al., J. Am. Chem. Soc. 145 (2023) 15088–15093. doi: 10.1021/jacs.3c05655
H.J. Zhang, M. Ociepa, M. Nassir, et al., Nat. Chem. 16 (2024) 249–258. doi: 10.1038/s41557-023-01347-2
Z. Zhou, Z. Li, W. Quanyong, et al., J. Organomet. Chem. 691 (2006) 5790–5797. doi: 10.1016/j.jorganchem.2006.09.049
P.H. Gore, Chem. Rev. 55 (1955) 229–281. doi: 10.1021/cr50002a001
P. Ji, X. Feng, P. Oliveres, et al., J. Am. Chem. Soc. 141 (2019) 14878–14888. doi: 10.1021/jacs.9b07891
F.J. Bristow, Nature 162 (1948) 1004-1004.
M. Stacey, E.J. Bourne, J.C. Tatlow, J.M. Tedder, Nature 164 (1949) 705-705.
I. Shiina, K. Nakata, K. Ono, Y.S. Onda, M. Itagaki, J. Am. Chem. Soc. 132 (2010) 11629–11641. doi: 10.1021/ja103490h
J. Allan, R. Robinson, J. Chem. Soc. 125 (1924) 2192–2195. doi: 10.1039/CT9242502192
T. Horie, M. Tsukayama, Y. Kawamura, M. Seno, J. Org. Chem. 52 (1987) 4702–4709. doi: 10.1021/jo00230a009
S. Fang, Z. Liu, H. Zhang, et al., ACS Catal. 11 (2021) 13902–13912. doi: 10.1021/acscatal.1c03966
S. Fang, J.P. Tan, J. Pan, et al., Angew. Chem. Int. Ed. 60 (2021) 14921–14930. doi: 10.1002/anie.202102352
W. Qin, Y. Liu, H. Yan, Acc. Chem. Res. 55 (2022) 2780–2795. doi: 10.1021/acs.accounts.2c00486
S.C. Zheng, S. Wu, Q. Zhou, et al., Nat. Commun. 8 (2017) 15238. doi: 10.1038/ncomms15238
Y. Tan, S. Jia, F. Hu, et al., J. Am. Chem. Soc. 140 (2018) 16893–16898. doi: 10.1021/jacs.8b09893
S. Jia, Z. Chen, N. Zhang, et al., J. Am. Chem. Soc. 140 (2018) 7056–7060. doi: 10.1021/jacs.8b03211
Y.B. Wang, P. Yu, Z.P. Zhou, et al., Nat. Catal. 2 (2019) 504–513. doi: 10.1038/s41929-019-0278-7
S. Jia, S. Li, Y. Liu, W. Qin, H. Yan, Angew. Chem. Int. Ed. 58 (2019) 18496–18501. doi: 10.1002/anie.201909214
Y. Liang, J. Ji, X. Zhang, et al., Angew. Chem. Int. Ed. 59 (2020) 4959–4964. doi: 10.1002/anie.201915470
Y. Chang, C. Xie, H. Liu, et al., Nat. Commun. 13 (2022) 1933. doi: 10.1038/s41467-022-29557-1
D. Xu, S. Huang, F. Hu, et al., CCS Chem. 4 (2022) 2686–2697. doi: 10.31635/ccschem.021.202101154
B. Cai, Y. Cui, J. Zhou, et al., Angew. Chem. Int. Ed. 62 (2023) e202215820. doi: 10.1002/anie.202215820
T.D. Tan, G.L. Qian, H.Z. Su, et al., Sci. Adv. 9 (2023) eadg4648. doi: 10.1126/sciadv.adg4648
Y. Cui, Y.B. Wang, H.H. Liu, S.H. Xiang, B. Tan, J. Am. Chem. Soc. 147 (2025) 3450–3458. doi: 10.1021/jacs.4c14589
G. Höfle, W. Steglich, H. Vorbrüggen, Angew. Chem. Int. Ed. 17 (1978) 569–583. doi: 10.1002/anie.197805691
H. Huang, J. Denne, C.H. Yang, H. Wang, J.Y. Kang, Angew. Chem. Int. Ed. 57 (2018) 6624–6628. doi: 10.1002/anie.201802082
E. Kumarasamy, R. Raghunathan, M.P. Sibi, J. Sivaguru, Chem. Rev. 115 (2015) 11239–11300. doi: 10.1021/acs.chemrev.5b00136
W.E.G. Osminski, Z. Lu, W. Zhao, et al., J. Org. Chem. 86 (2021) 17762–17773. doi: 10.1021/acs.joc.1c01769
A.C. Spivey, S. Arseniyadis, Angew. Chem. Int. Ed. 43 (2004) 5436–5441. doi: 10.1002/anie.200460373
C.K. De, E.G. Klauber, D. Seidel, J. Am. Chem. Soc. 131 (2009) 17060–17061. doi: 10.1021/ja9079435
J.A. Birrell, J.N. Desrosiers, E.N. Jacobsen, J. Am. Chem. Soc. 133 (2011) 13872–13875. doi: 10.1021/ja205602j
N. Mittal, K.M. Lippert, C.K. De, et al., J. Am. Chem. Soc. 137 (2015) 5748–5758. doi: 10.1021/jacs.5b00190
L.M. Entgelmeier, O.G. Mancheño, Synthesis 54 (2022) 3907–3927. doi: 10.1055/a-1846-6139

Figure 1 Phosphorodithioate derivatives and asymmetric synthesis of axially chiral organophosphorus derivatives from anhydride derivatives.

Figure 2 Scope of o-alkynylnaphthols. Reaction condition: 1 (0.15 mmol), 2a (46.6 mg, 0.1 mmol), C-5 (6.3 mg, 10 mol%), DMAP (1.2 mg, 10 mol%), toluene (2 mL), 30 ℃. The enantiomeric excesses were determined by HPLC on a chiral stationary phase. a 20 mol% C-5, 20 mol% DMAP.

Figure 3 Scope of phosphinothioic thioanhydrides. Reaction condition: 1a (36.6 mg, 0.15 mmol), 2a (0.1 mmol), C-5 (6.3 mg, 10 mol%), DMAP (1.2 mg, 10 mol%), toluene (2 mL), 30 ℃, 12 h. The enantiomeric excesses were determined by HPLC on a chiral stationary phase. a 20 mol% C-5, 20 mol% DMAP.
Table 1. Reaction optimization.a
|
||||||
| Entry | Catalyst | T (℃) | Time (h) | Solvent | Yield (%)b | ee (%)c |
| 1 | QN | 50 | 72 | Toluene | 58 | −24 |
| 2 | C-1 | 50 | 72 | Toluene | 56 | 65 |
| 3 | C-2 | 50 | 72 | Toluene | 72 | 74 |
| 4 | C-3 | 50 | 72 | Toluene | 73 | 63 |
| 5 | C-4 | 50 | 72 | Toluene | 70 | 71 |
| 6 | C-5 | 50 | 72 | Toluene | 76 | 77 |
| 7 | C-5 | 30 | 120 | Toluene | 78 | 84 |
| 8 | C-5 | 0 | >168 | Toluene | 43 | 79 |
| 9 | C-5 | 30 | 72 | DCM | 78 | 55 |
| 10 | C-5 | 30 | 72 | THF | 20 | 26 |
| 11 | C-5 | 30 | 120 | Mesitylene | 58 | 80 |
| 12 | C-5 | 30 | 168 | EA | 80 | 72 |
| 13d | C-5 | 30 | 24 | Toluene | 82 | 76 |
| 14e | C-5 | 30 | 24 | Toluene | 80 | 81 |
| 15f | C-5 | 30 | 12 | Toluene | 86 | 96 |
| 16g | C-5 | 30 | 12 | Toluene | 84 | 96 |
| 17h | C-5 | 30 | 12 | Toluene | Trace | – |
| a Reaction condition: 1a (36.6 mg, 0.15 mmol), 2a (46.6 mg, 0.1 mmol), catalyst (20 mol%), toluene (2 mL), 50 ℃. DCM = Dichloromethane; THF = Tetrahydrofuran; EA = Ethyl acetate; DMAP = 4-Dimethylaminopyridine. b Isolated yield. c Determined by HPLC on a chiral stationary phase. d 20 mol% pyridine. e 20 mol% 2,6-lutidine. f 20 mol% DMAP. g 10 mol% C-5 and 10 mol% DMAP. h Phosphonic anhydride 2a’ instead of phosphinothioic thioanhydride 2a, 10 mol% C-5 and 10 mol% DMAP. |
||||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们



 下载:
下载: