Scheme 1.
(a) Synthesis of imides from amides. (b) Synthesis of imides as minor products from N2. (c) Synthesis of N-methylimides as sole products from N2.
Titanium-promoted conversion of N2 into N-methylimides
Rui Hu , Yidan Qi , Xingyu Wang , Yunhui Yang , Congyang Wang

As the predominant component of the Earth’s atmosphere, dinitrogen (N2) has garnered substantial research interest in its activation and utilization [1-10]. The industrial synthesis of ammonia via the Haber-Bosch process remains the sole method for converting dinitrogen into usable forms of nitrogen and serves as the foundation for producing nitrogen-containing organic compounds. However, its requirements for elevated temperatures (350–550 ℃) and high pressure (150–350 atm.) result in considerable energy consumption [11]. As such, development of mild and efficient approaches to directly synthesize nitrogen-containing organic compounds from N2 emerges as an urgent and significant challenge.
Imides and their derivatives are widely found in natural products and bioactive compounds, exhibiting significant biological functions [12]. Additionally, imides are extensively used in the modification of carbon materials, thereby enhancing their performance in fields such as photoconversion and chiral luminescence [13]. Traditional methods for synthesizing imides typically involve reactions of amides with acylating reagents such as acyl chlorides [14] and aldehydes [15], or oxidation of nitrogen-substituted amides [16] (Scheme 1a). In these methods, the starting amides are invariably derived from multi-step reactions of ammonia. Consequently, the direct synthesis of imides from N2, bypassing the use of ammonia, holds significant economic and environmental potentials. Mori and co-workers reported synthesis of diphenyl imide using a structure-undefined titanium-nitrogen complex obtained through reduction of TiCl₃ with magnesium [17]. However, the amide was the primary product in this reaction with the yield of imide being only 24%, and the substrate scope was rather limited (Scheme 1b). Of note, reactions involving transition metal nitrides prepared from N2 mostly tend to favor the formation of nitriles [18-24].
Herein, we reported a method for the preparation of N-methyl imides from N2, involving sequential N-methylation and N-acylation reactions of structurally well-defined dinitrogen dititanium complexes. Remarkably, the formation of amides could be circumvented, affording imides as the sole products (Scheme 1c).
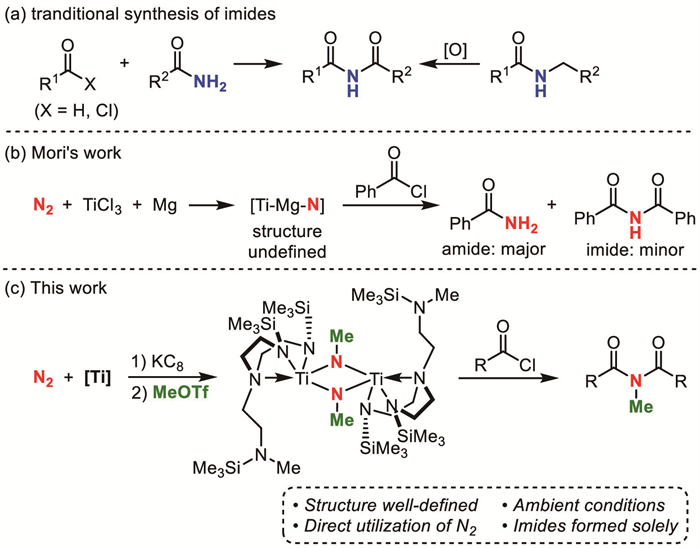
In 2018, Liddle and co-workers reported a dinitrogen-bridged end-on dititanium side-on dipotassium complex bearing a TrenTMS ligand, [{(TrenTMS)Ti}2(μ-η1:η1:η2:η2-N2K2)] (1) [25]. This complex marked the first instance of a homogeneous titanium complex capable of catalytically converting N2 to ammonia. However, such a dinitrogen dititanium dipotassium complex demonstrated a chemical inability in the synthesis of nitrogen-containing organic compounds, presumably attributable to the steric hindrance imposed by the bulky TrenTMS ligands and the pronounced reducing ability of this complex.
As part of our long-term goal of developing catalytic N2 transformations to nitrogen-containing organic compounds, we commenced our study on constructing C-N bonds via dinitrogen dititanium complex 1 (Scheme 2). The reaction of 1 with 4 equiv. of methyl trifluoromethanesulfonate (MeOTf) in pentane under ambient conditions resulted in the generation of orange [{(NMe, TMSNN2TMS)Ti}(μ-NMe)]2 (2) in 31% isolated yield, in which the nitrogen-nitrogen bond of N2 was completely cleaved and mono-methylated. Considering that the dinitrogen moiety underwent reduction in the absence of an external reducing agent, this reaction might be a disproportionation reaction induced by MeOTf. Analysis of the gaseous products in the reaction via gas chromatography-mass spectrometry (GC–MS) confirmed the formation of ethane (Fig. S1 in Supporting information), which was the homo-coupling product of MeOTf [26,27]. Furthermore, 15N-labeling experiments substantiated the production of 15N2, thereby providing a solid evidence for occurrence of the disproportionation reaction. Also, the byproduct (TrenTMS)Ti-OTf (3) was successfully isolated as a yellow solid in 42% yield, representing the form of residual titanium species, which could further be transformed into dinitrogen dititanium complex 1 in 76% NMR yield upon reduction with potassium graphite (KC8).
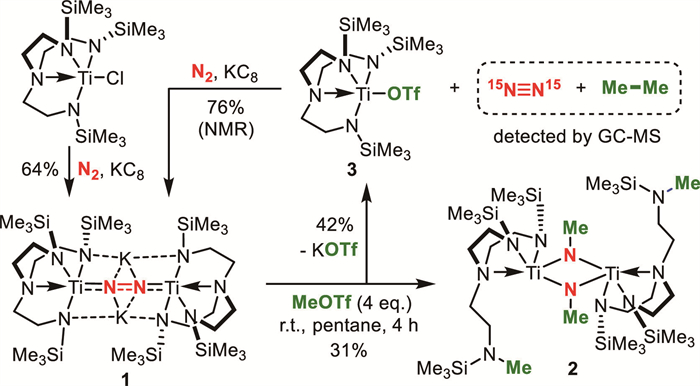
The single-crystal X-ray diffraction (SXRD) analysis of 2 revealed that the interatomic distance between the two nitrogen atoms (N1, N2) extended to 2.602 Å (Fig. 1a), indicative of a fully cleavage of N2 in this complex. Concurrently, the coordination geometry of dinitrogen moiety transformed from an η2 to a μ2 ligand configuration. The bond lengths of Ti1-N1 and Ti1-N2 were 1.913 Å and 1.953 Å, respectively, which were consistent with the range of Ti-N single bonds [28,29]. Owing to the change in coordination modes of titanium and dinitrogen, one side chain of the TrenTMS ligand had dissociated from the titanium center, accompanied by N-methylation of the terminal anionic –(CH2)2N–TMS group. In the 1H NMR spectrum, within the deshielding regions of the two titanium centers, the methyl groups linked to N1 and N2 appeared as a singlet at 4.27 ppm. In contrast, the methyl groups bound to the ligand nitrogen atoms were observed at a chemical shift of 2.40 ppm. Furthermore, through the employment of 15N-labeling experiments, the 15N-enriched product 2-15N was successfully synthesized and its 15N NMR spectrum exhibited a single resonance at 462.22 ppm (Fig. S4 in Supporting information). These spectral observations were consistent with the highly symmetric molecular crystal structure of 2. The SXRD analysis of 3 revealed that the Ti1-O1 bond length was 2.003 Å, which is consistent with the typical range of Ti-O single bonds (Fig. 1b).
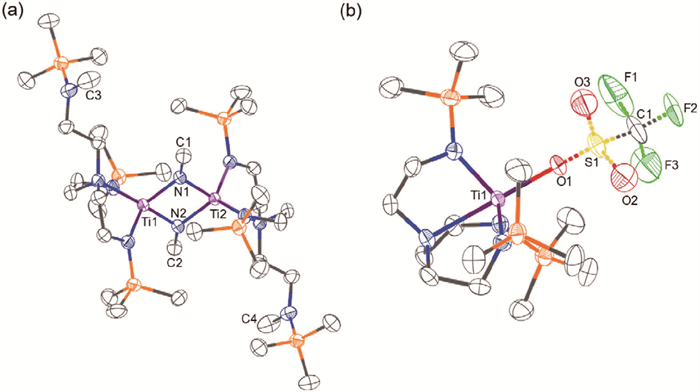
As the N1-N2 bond was completely cleaved and C—N bond formation occurred in complex 2, it was proposed that the reductive capacity of 2 was significantly diminished in comparison to that of complex 1, and therefore it might exhibit considerable nucleophilicity at the nitrogen atoms. As a validation, trimethylsilyl chloride was first employed as an electrophilic reagent to react with complex 2. After reacting in deuterobenzene (C6D6) at 60 ℃ for 12 h, the formation of heptamethyldisilazane (4) was successfully detected by ¹H NMR spectroscopy, with a yield of 54% (Scheme 3).
Next, the scope of electrophilic reagents was expanded to include carbon nucleophiles, aiming for further C—N bond formation to synthesize multi-substituted N-containing organic compounds. Acyl chlorides, as strong electrophilic reagents, are widely utilized in nucleophilic substitution/addition reactions [30]. Through preliminary attempts, it was found that the reaction of 2 with 4-methylbenzoyl chloride (5a) yielded trisubstituted N, 4-dimethyl-N-(4-methylbenzoyl)benzamide (6a) and disubstituted N, 4-dimethylbenzamide (7) in NMR yields of 5% and 13%, respectively (Table 1, entry 1). Extending the reaction time to 12 h resulted in an increase in both yields; however, the disubstituted product 7 remained as the major product (Table 1, entry 2). Raising the temperature to 50 ℃ led to an increase in the yield of 6a, accompanied by a decrease in the yield of 7 (Table 1, entry 3), indicating that there was likely a conversion from the intermediate 7 to product 6a in the reaction. Further raising the temperature resulted in formation of a small amount of 4-methylbenzonitrile as a byproduct (Table 1, entry 4). The increase in the equivalents of 5a had a significant positive effect on the yields of 6a (Table 1, entries 5 and 6). Solvent-screening results indicated that toluene was the optimal solvent (Table 1, entries 6–10). Eventually, when 10 equivalents of 5a was used and the reaction proceeded at 50 ℃ for 12 h, the formation of 7 was completely suppressed and 6a was formed as the sole product in an NMR yield of 61% and isolated yield of 56% (Table 1, entry 11).
 DownLoad:
CSV
DownLoad:
CSV
 |
||||||
| Entry | 5a (equiv.) | T (℃) | t (h) | Solvent (1 mL) | Yield (%)a | |
| 6a | 7b | |||||
| 1 | 4 | 23 | 3 | Toluene | 5 | 13 |
| 2 | 4 | 23 | 12 | Toluene | 8 | 25 |
| 3 | 4 | 50 | 12 | Toluene | 12 | 11 |
| 4c | 4 | 80 | 12 | Toluene | 4 | 25 |
| 5 | 6 | 50 | 12 | Toluene | 20 | 17 |
| 6 | 8 | 50 | 12 | Toluene | 56 | 7 |
| 7 | 8 | 50 | 12 | Hexane | 44 | 6 |
| 8 | 8 | 50 | 12 | Et2O | 39 | 8 |
| 9 | 8 | 50 | 12 | THF | 36 | 16 |
| 10 | 8 | 50 | 12 | DME | 53 | 6 |
| 11 | 10 | 50 | 12 | Toluene | 61(56)d | N.D. |
| a Yields were determined by 1H NMR with 1,3,5-trimethoxybenzene as an internal standard. b The proton in 7 was provided by the silica gel used for quenching the reaction. c 4-Methylbenzonitrile was detected as a byproduct. d Isolated yield was shown. |
||||||
With the optimized reaction conditions in hand, we continued to investigate the substrate scope of acyl chlorides (Scheme 4). Initially, the influence of steric hindrance on the reaction was examined. Benzoyl chloride without any substituent (5b) afforded the desired product 6b in 53% isolated yield. Substitutions with methyl groups at the meta-positions of the acyl chloride had no obvious impact on the reaction yield (6c). However, when a methyl group was introduced at the ortho-position, the yield of 6d was only 26%, indicating that the steric hindrance significantly inhibited the reaction efficiency. Furthermore, the reaction was completely suppressed when using 2,4,6-trimethylbenzoyl chloride of more steric bulkiness as the substrate.
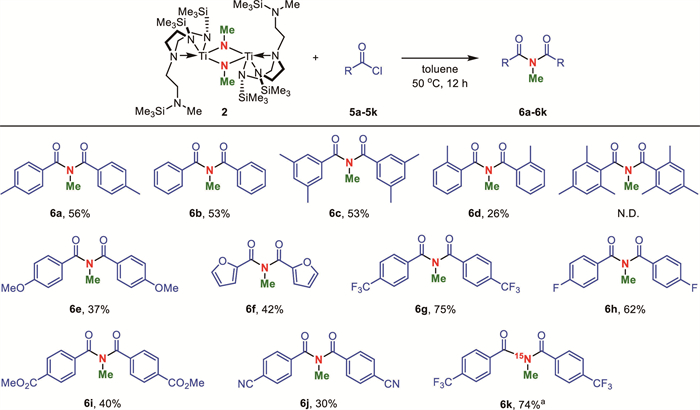
From an electronic perspective, electron-donating substituents were found to be relatively detrimental to the reaction outcome. For instance, when a methoxy group was introduced to the para-position of benzoyl chloride (5e), the yield of product 6e was 37%. Similarly, the use of electron-rich 2-furoyl chloride (5f) in the reaction also resulted in the formation of product 6f in a relatively lower yield of 42%. In contrast, acyl chlorides with non-coordinating electron-withdrawing substituents, such as 4-trifluoromethylbenzoyl chloride (5g) and 4-fluorobenzoyl chloride (5h), were favorable in the reaction, yielding the corresponding imides 6g and 6h in 75% and 62% yields, respectively. These results reflected the pronounced nucleophilic character of complex 2 [31]. For substrates with other electron-withdrawing groups, such as 4-methoxycarbonylbenzoyl chloride (5i) and 4-cyanobenzoyl chloride (5j), the yields of the corresponding products 6i and 6j were not as high as expected. This might be attributed to the coordinating ability of ester and cyano groups with titanium [32-35], which could compete with acyl chlorides for coordination sites on the titanium center. Of note, aliphatic acyl chlorides failed to give the expected products in this reaction.
¹5N-labeled organic compounds play significant roles in biomedicine, agricultural science, and environmental science [36,37]. However, the natural isotopic abundance of 15N is merely 0.365% [38], which restricts the synthetic access to ¹5N-labeled compounds. ¹5N-labeled dinitrogen gas is the most readily available source of ¹5N, the direct use of which holds great potentials to approach ¹5N-labeled organic products. As an illustrative example, the ¹5N-labeled dinitrogen was applied into our protocol and ¹5N-labeled imide 6k was synthesized in 74% isolated yield. The ¹5N NMR spectrum of 6k showed a singlet at 154.06 ppm (Fig. S21 in Supporting information).
In conclusion, a titanium-promoted protocol for the conversion of N2 to N-methylimides was developed, which involved the formation of [{(TrenTMS)Ti}2(μ-η1:η1:η2:η2-N2K2)] from dinitrogen, MeOTf-induced disproportionation and simultaneous N-methylation yielding the titanium-amido complex [{(NMe, TMSNN2TMS)Ti}(μ-NMe)]2, and nucleophilic reaction with acyl chlorides producing N-methylimides, thus forging three C−N bonds from N2. The titanium-amido species was also capable of reacting with TMSCl to yield heptamethyldisilazane, in which the formation of one C−N bond and two N−Si bonds was achieved from N2 by sequential steps. We believe this work provides important insights into direct transformations of N2 into N-containing organic molecules. Further organic reactions using N2 as the nitrogenous source are under explorations in our laboratory.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Rui Hu: Writing – original draft, Validation, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Yidan Qi: Validation, Investigation, Data curation. Xingyu Wang: Validation, Investigation, Data curation. Yunhui Yang: Writing – review & editing, Supervision, Project administration, Data curation, Conceptualization. Congyang Wang: Writing – review & editing, Writing – original draft, Supervision, Resources, Project administration, Investigation, Funding acquisition, Formal analysis, Data curation, Conceptualization.
Financial supports from the National Natural Science Foundation of China (Nos. 22025109, 22371283), the National Key R&D Program of China (No. 2023YFA1507902), CAS Project for Young Scientists in Basic Research (No. YSBR-050) and the State Key Laboratory of Fine Chemicals, Dalian University of Technology (No. KF2102) are gratefully acknowledged. We are also thankful for the help from Engineer Zhenpeng Wang in gas chromatography-mass spectrometry and Dr. Haihan Yan in single-crystal X-ray diffraction analysis.
Supplementary material associated with this article can be found, in the online version, at doi:
S. Kim, F. Loose, P.J. Chirik, Chem. Rev. 120 (2020) 5637–5681. doi: 10.1021/acs.chemrev.9b00705
Z. Lv, J. Wei, W. Zhang, et al., Nat. Sci. Rev. 7 (2020) 1564–1583. doi: 10.1093/nsr/nwaa142
J. Yin, J. Li, G. Wang, et al., J. Am. Chem. Soc. 141 (2019) 4241–4247. doi: 10.1021/jacs.9b00822
Z. Lv, Z. Huang, W. Zhang, Z. Xi, J. Am. Chem. Soc. 141 (2019) 8773–8777. doi: 10.1021/jacs.9b04293
X. Xin, I. Douair, Y. Zhao, et al., J. Am. Chem. Soc. 142 (2020) 15004–15011. doi: 10.1021/jacs.0c05788
J. Yang, M. Peng, D. Zhai, et al., ACS Catal. 12 (2022) 2898–2906. doi: 10.1021/acscatal.1c04435
J. Zeng, J. Su, F. You, J. Zhu, Chin. Chem. Lett. 34 (2023) 107759. doi: 10.1016/j.cclet.2022.107759
Z. Yin, B. Wu, G. Wang, J. Wei, Z. Xi, J. Am. Chem. Soc. 145 (2023) 7065–7070. doi: 10.1021/jacs.3c00266
Y. Chen, X. Shi, D. Huang, J. Wei, Z. Xi, Chin. Chem. Lett. 35 (2024) 109292. doi: 10.1016/j.cclet.2023.109292
W. Sheng, T. Rajeshkumar, Q. Zhao, et al., J. Am. Chem. Soc. 147 (2025) 7203–7208. doi: 10.1021/jacs.4c18519
J.R. Jennings, Catalytic Ammonia Synthesis: Fundamentals and Practice, Springer, 1991.
Z. Zhao, J. Yue, X. Ji, et al., Bioorg. Chem. 108 (2021) 104557. doi: 10.1016/j.bioorg.2020.104557
W. Jiang, Z. Wang, J. Am. Chem. Soc. 144 (2022) 14976–14991. doi: 10.1021/jacs.2c04642
X. Li, Y. Fang, P. Deng, et al., Org. Lett. 13 (2011) 4628–4631. doi: 10.1021/ol2018455
J. Wang, C. Liu, J. Yuan, A. Lei, Chem. Commun. 50 (2014) 4736–4739. doi: 10.1039/c4cc01447b
K.C. Nicolaou, C.J.N. Mathison, Angew. Chem. Int. Ed. 44 (2005) 5992–5997. doi: 10.1002/anie.200501853
M. Mori, Y. Uozumi, M. Shibaki, Tetrahedron Lett. 28 (1987) 6187–6190. doi: 10.1016/S0040-4039(00)61842-7
J.S. Figueroa, N.A. Piro, C.R. Clough, C.C. Cummins, J. Am. Chem. Soc. 128 (2006) 940–950. doi: 10.1021/ja056408j
J.J. Curley, E.L. Sceats, C.C. Cummins, J. Am. Chem. Soc. 128 (2006) 14036–14037. doi: 10.1021/ja066090a
P. Hu, J. Chai, Y. Duan, et al., J. Mater. Chem. A 4 (2016) 10070–10083. doi: 10.1039/C6TA02907H
M.M. Guru, T. Shima, Z. Hou, Angew. Chem. Int. Ed. 55 (2016) 12316–12320. doi: 10.1002/anie.201607426
F. Schendzielorz, M. Finger, J. Abbenseth, et al., Angew. Chem. Int. Ed. 58 (2019) 830–834. doi: 10.1002/anie.201812125
M. Fritz, S. Rupp, C.I. Kiene, et al., Angew. Chem. Int. Ed. 61 (2022) e202205922. doi: 10.1002/anie.202205922
V. Bonatto, R.F. Lameiro, F.R. Rocho, et al., RSC Med. Chem. 14 (2023) 201–217. doi: 10.1039/d2md00204c
L.R. Doyle, A.J. Wooles, L.C. Jenkins, et al., Angew. Chem. Int. Ed. 57 (2018) 6314–6318. doi: 10.1002/anie.201802576
R.T. Smith, X. Zhang, J.A. Rincón, et al., J. Am. Chem. Soc. 140 (2018) 17433–17438. doi: 10.1021/jacs.8b12025
C. Chen, M. Wang, H. Lu, B. Zhao, Z. Shi, Angew. Chem. Int. Ed. 60 (2021) 21756–21760. doi: 10.1002/anie.202109723
S.A. Harris, J.T. Ciszewski, A.L. Odom, Inorg. Chem. 40 (2001) 1987–1988. doi: 10.1021/ic001238r
A. Novak, A.J. Blake, C. Wilson, J.B. Love, Chem. Commun. (2002) 2796–2797.
C.A.G.N. Montalbetti, V. Falque, Tetrahedron 61 (2005) 10827–10852. doi: 10.1016/j.tet.2005.08.031
T.A. Hamlin, M. Swart, F.M. Bickelhaupt, ChemPhysChem 19 (2018) 1315–1330. doi: 10.1002/cphc.201701363
B.M. Chadwick, A.G. Sharpe, Transition metal cyanides and their complexes, in: H.J. Emeléus, A.G. Sharpe (Eds.), Advances in Inorganic Chemistry and Radiochemistry, Academic Press, 1966, pp. 83–176.
T. Carofiglio, C. Floriani, A. Chiesi-Villa, C. Guastini, Inorg. Chem. 28 (1989) 4417–4419. doi: 10.1021/ic00323a026
B. Kubiak, T. Muzioł, M. Jabłonski, A. Radtke, P. Piszczek, Dalton Trans. 53 ´ (2024) 14457–14468. doi: 10.1039/d4dt01710b
Y. Li, N. Chekshin, Y. Lu, J. Yu, Nature 637 (2025) 608–614. doi: 10.1038/s41586-024-08281-4
X. Shi, Q. Wang, C. Qin, et al., Natl. Sci. Rev. 9 (2022) nwac168. doi: 10.1093/nsr/nwac168
J.D. Gouveia, T.L.P. Galvão, K. Iben Nassar, J.R.B. Gomes, npj 2D Mater. Appl. 9 (2025) 8. doi: 10.1038/s41699-025-00529-5
A. Mariotti, Nature 311 (1984) 251–252. doi: 10.1038/311251a0

Scheme 1 (a) Synthesis of imides from amides. (b) Synthesis of imides as minor products from N2. (c) Synthesis of N-methylimides as sole products from N2.

Figure 1 (a) X-ray crystal structure of 2. (b) X-ray crystal structure of 3. Thermal ellipsoids are depicted at the 50% probability. All hydrogen atoms are omitted for clarity.
Table 1. Optimization of reaction conditions.
|
||||||
| Entry | 5a (equiv.) | T (℃) | t (h) | Solvent (1 mL) | Yield (%)a | |
| 6a | 7b | |||||
| 1 | 4 | 23 | 3 | Toluene | 5 | 13 |
| 2 | 4 | 23 | 12 | Toluene | 8 | 25 |
| 3 | 4 | 50 | 12 | Toluene | 12 | 11 |
| 4c | 4 | 80 | 12 | Toluene | 4 | 25 |
| 5 | 6 | 50 | 12 | Toluene | 20 | 17 |
| 6 | 8 | 50 | 12 | Toluene | 56 | 7 |
| 7 | 8 | 50 | 12 | Hexane | 44 | 6 |
| 8 | 8 | 50 | 12 | Et2O | 39 | 8 |
| 9 | 8 | 50 | 12 | THF | 36 | 16 |
| 10 | 8 | 50 | 12 | DME | 53 | 6 |
| 11 | 10 | 50 | 12 | Toluene | 61(56)d | N.D. |
| a Yields were determined by 1H NMR with 1,3,5-trimethoxybenzene as an internal standard. b The proton in 7 was provided by the silica gel used for quenching the reaction. c 4-Methylbenzonitrile was detected as a byproduct. d Isolated yield was shown. |
||||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们



 下载:
下载: